
Abstract
Gaucher disease (GD) is a rare lysosomal storage disease that is caused by mutations in the GBA gene. It is classified into three main phenotypes according to the patient’s clinical presentation. Of these, chronic neuronopathic GD (GD3) is characterized by progressive neurological damage. Understanding the unique neurological manifestations of GD3 has important diagnostic and therapeutic implications. Our article summarizes the neurological symptoms specific to GD3 and related therapeutic advances, and it highlights the relevance of the gene to clinical symptoms, so as to provide a reference for the diagnosis and treatment of GD3.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Gaucher disease (GD) is a rare autosomal recessive lysosomal storage disease that is caused by mutations in the GBA gene on chromosome 1q21, resulting in a decrease in acid β-glucosidase (GBA) activity and the intracellular accumulation of its substrates, glucosylceramides, particularly in macrophages [1]. These pathological “Gaucher cells” usually invade the liver, spleen, lungs, bone marrow, and central nervous system, and they are considered the main factor responsible for the typical symptoms of GD [2, 3]. Based on the presence and severity of primary central nervous system (CNS) disease, GD is classified into three major phenotypes. Type 1 Gaucher disease (GD1) is a non-neuronopathic type, which mainly affects the internal organs without neurological involvement [4, 5]. Neuronopathic Gaucher disease (nGD) includes type 2 Gaucher disease (GD2) and type 3 Gaucher disease (GD3). Of these, GD2 is the most severe form and can present as hydrops fetalis, congenital ichthyosis, hepatosplenomegaly, and thrombocytopenia, leading to death of the affected child in infancy or early childhood [6]. GD3 is a chronic neuronopathic type that can be subdivided into three subtypes according to its common clinical manifestations. Specifically, type 3a (GD3a) has more severe neurological manifestations; type 3b (GD3b) has severe visceral manifestations [7]; and type 3c (GD3c) mainly presents with calcification of heart valves [8]. However, given that there is an overlap between these phenotypes, GD is typically considered a phenotypic continuum. In this article, we explore the unique neurological manifestations of GD3, with the aim to aid practitioners with the diagnosis and staging of GD, so that timely and precise treatment can be provided.
Epidemiology
Estimates of the prevalence of GD per 100,000 population range from 0.02 to 139.0. The prevalence is higher in America than in other regions, including the Middle East, Europe, and Latin America. The highest prevalence is in the population of Ashkenazi Jewish (AJ) descent in North America. nGD with poor prognosis, including GD2 and GD3, has the lowest prevalence [9]. Compared with GD2 and GD3, patients with GD1 have a higher median age at diagnosis, but there is often a delay between first symptoms and diagnosis. The disease has a wide variety of clinical presentations, ranging from little or no symptoms to very severe forms [10]. GD2 is relatively rare and usually begins in infants between 3 and 6 months of age. Children with GD2 usually have systemic and neurological manifestations during the prenatal, perinatal, or first months of life, and they die in infancy [11]. GD3 usually starts before the age of 2 years, but some slower progressing cases may be diagnosed in adulthood. In addition to clinical manifestations similar to those of type 1, there are neurological symptoms that may appear years earlier than other clinical events [10].
In AJ patients, the c.1226A < G (N370S or N409S in the new nomenclature) mutation is the most common mutation, followed by the c.84dupG (84GG) mutation, which is much rarer [12]. Homozygous N370S mutations are the most common genotype in the German–Jewish population, accounting for 70% of all mutations, and N370S mutations can also be found in European, North American, and Israeli patients [13]. In contrast, in Egypt, Korea, and China, C.1448 T > C (L444P or L483P according to the new nomenclature) and C.754 T > A (F252I) are usually associated with GD2 and GD3 [12], and at least one-third of GD patients have the nGD phenotype [14,15,16,17,18]. Globally, the most common genotype of nGD is the L444P mutation, which may often lead to more severe symptoms and can present with more CNS involvement compared with the homozygous N370S mutation [19, 20]. L444P is also the most common mutation in Caucasians of non-AJ ancestry [21]. The C.1342G > C (D409H) variant results in the appearance of the characteristic heart-valve-calcification type, GD3c. Since its discovery in the 1980s, heterozygosity or homozygosity has been observed in patients of different ethnicities, with the highest prevalence in Greece and Spain [22].
Detection methods
Enzymatic assay
The definitive method of diagnosis for GD is an enzymatic assay of GBA activity in peripheral blood leukocytes or skin fibroblasts of patients [23]. A dried blood spots (DBS) method has also been developed for the initial screening of GBA activity [24]. This method requires less sample collection, facilitates sample storage, and allows for more convenient concurrent enzymatic, biomarker, and genetic testing.
Gene diagnosis
The GBA gene is approximately 8 kb in length, consists of 11 exons and 10 introns, and is located on chromosome 1q21 [25]. The L444P, F252I, and RecNil GBA alleles are associated with nGD, and they are more prevalent in Asian populations [26, 27]. According to Wan Lei et al. [28], nGD is associated with the L444P/RecNciI gene in Taiwan. Analyses of corresponding GBA mutations may be able to predict GD phenotypes. For example, people with biallelic N370S GBA mutations are predisposed to developing GD1 [29], whereas those with severe L444P GBA mutations are more likely to manifest nGD [30], and the rare homozygous D409H GBA mutation typically presents with heart valve damage [22]. C.680A > G (N188S), C.1246G > A (G377S), and C.1297G > T (V394L) are more likely to be associated with myoclonic epilepsy [12]. Therefore, preliminary prediction of relevant phenotypes based on genotypes may capture the possible direction of disease development. Based on this notion, genetic testing is recommended for patients who have been diagnosed with GD, so as to ensure more accurate disease classification and develop appropriate treatment plans.
However, there are often differences in symptom presentation between members of the same family even when they have the same genotype, which suggests that genotype–phenotype correlations are not absolute [12] and that additional genetic modifications may be present elsewhere [31]. For several GBA genotypes, considerable variation in disease severity has been reported, even in identical twins. For example, Biegstraaten et al. reported a pair of identical twins of Moroccan origin with the N188S GBA mutation whose neurological manifestations were highly heterogeneous, namely, one had severe visceral involvement, epilepsy, and cerebellar syndrome as predicted, but the other did not have the clinical manifestations of GD but had type 1 diabetes [32]. Three new variants have been identified in a recent study of genetic variants in Mexican and Spanish patients with GD [33]. The continuous discovery of new pathogenic GBA variants [33] and unique genotypes due to atypical mutational mechanisms (including somatic chimerism and a single GBA allele with both variants in cis) [34] has allowed evolving genetic information to confound genotype–phenotype correlations in different GD types. Such a complex pattern of inheritance poses a challenge for genotyping.
Biomarkers
Several biomarkers, such as angiotensin-converting enzyme (ACE), chitotriosidase, and a chemokine, CCL18, have been used as routine tests in the diagnosis of GD [35]. Recent studies have suggested that glucosylsphingosine—a deacylated derivative of glucosylceramide—may be the main pathogenic molecule responsible for nGD. Compared with other biomarkers such as glucosylceramide, glucosylsphingosine is more specifically elevated in patients with nGD [36, 37]. Elevated levels of Lyso-GB1 have been found in patients with GD compared to healthy controls, and levels are generally higher in patients with NGD than in patients with GD1 [38]. A clinical trial showed that glucosylsphingosine level in the cerebrospinal fluid was below the lower limit of quantification in all of the control subjects but was elevated in patients with nGD [39]. Glucosylsphingosine measured in dried blood spots is also highly sensitive and specific for GD [40]. Even in the neonatal period, plasma glucosylsphingosine concentrations are significantly higher in patients with nGD than in those with non-nGD [38]. In a cohort of children with GD, Hurvitz et al. [41] found that glucosylsphingosine level was significantly lower in mild type 1 than in severe type 1, but there was no significant difference in glucosylsphingosine level between severe type I and type 3. In untreated children, glucosylsphingosine level was negatively correlated with platelet counts; after treatment, glucosylsphingosine level was negatively correlated with hemoglobin levels.
According to Saville et al. [38], glucosylsphingosine concentration measured in dried blood papers has prognostic power and may serve as a pharmacodynamic biomarker that decreases sharply after the initiation of enzyme replacement therapy. The extent and severity of disease caused by GBA mutations are related to the degree of GBA deficiency. For example, GBA activity is reduced by about 20–50% in GBA mutation-associated Parkinson disease (PD-GBA), usually more than 90% in nGD, and almost 100% in hydrops fetalis [42]. Reduced levels of GBA activity leads to the accumulation of glucosylsphingosine. In summary, glucosylsphingosine level correlates with the severity and prognosis of GD3 and other GBA mutation-associated diseases, and it may be used as a marker to monitor GD progression and treatment response. However, the elevated concentrations of one or more of these markers are not sufficient to diagnose GD, considering that they may also be elevated in other diseases; therefore, these biomarkers are mostly used as aids in the diagnosis of GD.
Neurological manifestations of GD3
The GD3 phenotype is very heterogeneous, particularly in terms of neurological manifestations. Some patients present with mild neuropathy, having horizontal eye muscle palsy as the only neurological symptom, while others present with a more severe form with different neurological signs including progressive myoclonic epilepsy, ocular lesions, and dementia [43, 44].
Ocular lesions
Neurological lesions in patients with GD3 often involve the eyes, and they are generally referred to as functional ocular abnormalities. The most common functional ocular abnormality is supranuclear gaze palsy (horizontal and vertical) [8]. It includes slowed saccadic velocity, delayed initiation of saccades [45], and reduced accuracy of saccades, which are all typically more severe in the horizontal direction [46]. The saccadic impairment mainly affects the horizontal direction first, followed by the downward direction, and finally impairs upward eye movement [47]. Slowed horizontal saccadic velocity, which is a specific clinical manifestation, has been used to differentiate GD3 from other types of GD [48]. Since horizontal gaze is more severely impaired—to the point that technical measurements are not possible [46]—measurement of the less-impaired vertical eye beat has received much attention as a more sensitive measure [49]. In addition, vertical peak velocity is valuable as a biomarker of neuropathic manifestations for future longitudinal studies, because it correlates more highly with other neurological symptoms [47].
The most common anterior-segment abnormalities among structural ocular abnormalities include pinguecula-like lesions and corneal opacities. Diffuse corneal opacities are less common in patients with GD3 [50], and usually occur in patients with homozygous D409H GBA mutations, which are typically associated with cardiac valve calcification [51, 52]. In addition, posterior segment abnormalities, including vitreous opacities, condensation, and preretinal white dots, are common in patients with GD3. Recent studies have also identified subretinal lesions in severe phenotypes and longer durations [47]. Sam et al. [53] found that patients with GD3 had lower total retinal thickness in all subdomains (fovea, inner and outer rings) than healthy individuals (even after adjusting for sex, age, and spherical equivalent), which may be associated with reduced GBA activity, accumulation of α-synuclein, inhibition of apoptosis, reduced mitochondrial function, and associated oxidative stress. Watanabe [50] and Zhao [54] described tractional detachment of the retina due to strong vitreo-retinal adhesions and massive vitreous opacities in GD3. In cases of retinal detachment, surgical treatment is crucial to preserving vision; otherwise, permanent vision loss may occur. Therefore, in patients with GD, careful ophthalmic examinations, including astigmatic fundus examination, electroretinography, and optical coherence tomography, are necessary to prevent ocular lesions.
Auditory hypoesthesia
Patients with GD3 may also present with auditory pathway damage due to brainstem neuropathy. Auditory brainstem-evoked responses (ABR) provide a noninvasive method for assessing local brainstem function, and they are well suited for evaluating degenerative brainstem diseases. By administering acoustic stimulation, the electrical response to acoustic stimulation is recorded on the scalp from structures at all levels of the auditory pathway, from the cochlea to the brainstem. Given that ABR waveforms reflect synchronous neural activity in the brainstem auditory pathway, progressive deterioration of ABR most likely reflects underlying subclinical brainstem degeneration [55]. The most common finding in ABR is the absence or delay of V waves, followed by the absence or delay of III waves [56]. Bamiou et al. [56] studied the auditory pathway in children with GD3 and found a range of abnormalities, including elevated acoustic reflex, poor medial olivo-cochlear suppression, and poor brainstem-evoked potentials. The results of this cross-sectional study suggested that combined audiometric testing may reflect the severity of neurological damage in patients with GD3. Audiological tests are of great interest for the diagnosis, classification, and prognostic assessment of GD.
Myoclonic epilepsy
Myoclonic epilepsy is a rare manifestation of GD3. Is not considered an independent phenotype with a characteristic age-of-onset and has no predicted rate of clinical progression [57, 58]. It has a cortical origin, and it is known to be associated with a significant increase in the amplitude of somatosensory-evoked potentials or to indicate a defect in cortical inhibitory input [59], which may be an extreme manifestation of the general corticogenic process. Specific mutations in GBA may be associated with the development of myoclonic epilepsy, suggesting that this abnormal enzyme may also have modifying effects on other proteins involved in epilepsy. One study [60] identified three GBA point mutations associated with this disease, namely, V394L, N188S, and G377S. In contrast, the N370S mutation was not found to be associated with myoclonus. In a study of neurological abnormalities in patients with GD1 and GD3, four patients with GD3 experienced spontaneous rapid and repetitive hyperkinetic dystonia-like movement disorders in the face and extremities, and repetitive blepharospasms were also noted [61]. Although it is not possible to determine the association between these mutations and the development of myoclonic epilepsy, any patient carrying one of these three mutations without N370S should be carefully evaluated for the development of the condition.
Cognitive and performative abnormalities
Cognitive dysfunction [62], performative changes, and a tendency toward aggressive behavior have been reported in patients with GD3 [18]. A longitudinal study, conducted over 29 years, assessed 34 multiracial patients (19 males and 15 females) with GD3 using the Wechsler IQ scale [63]. In all of the subjects in the cohort, verbal IQ (VIQ), performance IQ (PIQ), and full-scale IQ (FSIQ) scores were in the below-average to the critical range. Lower PIQ and FSIQ scores were associated with bilateral electroencephalogram (EEG) lateralization and splenectomy, which may indicate a decrease in overall neurological function. Several reports have noted widespread cognitive impairment in patients with GD3 [61, 62, 64]. A previous study showed mild deficits in attention and memory speed in patients with GD1, as evidenced by poor ability to focus attention and slow retrieval of information held in memory [65]. In contrast, cognitive deficits in patients with GD3 usually affect non-verbal abilities and are more widespread. About 60% of patients have below-average intelligence, with particular deficits in processing speed, visuospatial relationships, and perceptual abilities, and have problems performing everyday activities [62]. Another retrospective study reported abnormal behavior in a cohort of 34 Egyptian patients diagnosed with GD3 [66]. Anger and aggressive behaviors were the most common, prominent, and troublesome aberrant behaviors that correlated with lower IQ scores and were more common in males than in females [66]. In female patients, internalizing behaviors such as crying and social withdrawal were associated with EEG abnormalities and seizure scores [66]. Repetitive behaviors have also been associated with EEG and seizure scores. In patients with GD, quantitative scores of cognitive function and abnormal behaviors may help to confirm GD diagnosis and classification and to assess the progression and severity of neuropathy. Careful assessment of cognitive function and abnormal behavior is therefore crucial for GD patients. Cognitive and performative evaluation includes the assessment of general cognitive intellectual abilities, language function, non-verbal memory ability, verbal learning ability, verbal memory, and executive functioning [67]. Computerized cognitive testing using the Mindstreams test system (the Global Assessment Battery [68]) detects incipient neurological involvement, which is of great significance for identifying patients with mild neuropathy and delaying the progression of the disease.
EEG abnormalities
EEG is a widely used noninvasive tool to identify abnormalities in brain function and epileptiform abnormalities that indicate an increased risk of seizures. A longitudinal cohort study retrospectively evaluated 293 EEGs in 67 patients with GD3 [69]. The authors found that more than 90% of the patients with GD3 had at least one EEG abnormality. Epileptiform discharges were found in 54% of the patients, while slowed background activity was observed in 90% of the patients, which was the most consistent finding correlated with neurological involvement. Another study showed that 62% of GD3 patients had EEG abnormalities and seizures, consistent with neurological GD3, and a range of behavioral problems [70]. The most common behaviors were internalizing ones such as anxiety and depression, but they also extended to externalizing ones, such as inattention and behavioral abnormalities [70].
Therefore, the observation of background slowing, epileptiform activity, or seizures in patients with mild or no neurological manifestations may serve as an additional finding to identify CNS involvement. This would enable the assessment of CNS involvement and early intervention to inhibit progression. However, an abnormal EEG does not provide a definitive diagnosis of GD3, given that patients with GD1 may also have other causes of epilepsy. In addition, the abnormalities found in the above studies—background slowing and interval epileptiform activity—tend to occur intermittently. Thus, they are subject to sampling error due to the limited duration of conventional EEG studies. The presence of abnormalities may also be influenced by factors unrelated to the severity of CNS pathology, including waking state, medications, and time of day [69].
These cohort data provide baseline EEG information for the diagnosis of GD3. Although imperfect, they are still useful in the ancillary diagnosis of GD3 and the assessment of the severity of neuropathy [71].
Other neurological manifestations
The earliest descriptions of the relationship between GD and PD were disseminated case reports of GD patients presenting with early onset refractory Parkinson syndrome. Tayebi et al. [72] conducted a cohort study of 17 such patients, including Jews and different ethnicities. These patients had relatively mild manifestations of GD, and they had a relatively early onset of PD symptoms, with a mean age at diagnosis of 48 years. These patients presented typical features, including asymmetric tremor, tonus, decreased movement, and even dementia. A large multicenter study across four continents analyzed 5691 ethnically diverse patients with PD compared with controls and confirmed an overall odds ratio (OR) of 5.43 [73]. Heterozygous variants in the GBA gene are the most important genetic risk factor for PD [74]. The pathogenicity of different variants varies considerably. L444P, classified as a severe variant, has an OR of 10–15 for developing PD, whereas the mild variant N370S has an OR of less than 5 [75,76,77]. However, in specific ethnic groups, this categorization does not seem to make sense. For example, in AJ, the majority of PD patients carrying the GBA variant carry mild N370S, whereas the severe L444P variant is found in about 5% of patients [73].
In addition to dementia secondary to PD, dementia with Lewy bodies (DLB) caused by GBA mutations is gradually being discovered. In a study on Spanish subjects, brain tissue specimens were extracted from patients with pathologically confirmed DLB, and GBA mutations were found in 12–13% of these cases [78]. A study synthesizing the current evidence from clinical studies of GBA-associated DLB confirmed the strong correlation between GBA mutations and DLB, namely, GBA mutation carriers showed a more severe phenotype in the spectrum of DLB disorders, with earlier age of symptom onset, more severe motor and cognitive dysfunction, and more visual hallucinations and sleep disturbances [79].
GD3 can also present with complex cerebellar symptoms such as cerebellar ataxia, intention tremor, and progressive myoclonic epilepsy [73, 80, 81]. In the follow-up of neurological lesions in patients with GD1 and GD3, one-half of the patients with GD3 suffer from trunk and appendicular ataxia, which manifests as an ataxic gait with dyskinesia [61]. Longitudinal studies have shown that neurological symptoms in patients with GD do not progress significantly over time [61, 63].
Neuropathy of GD1
Neurological symptoms may reappear several years after the appearance of visceral symptoms, even in patients initially identified as having GD1 who were subsequently treated. Some patients may present with signs or symptoms that blur the boundaries between clinical types, indicating that they are part of a spectrum of possible phenotypes, ranging from non-neurological to life-threatening neurological lesions. Recent studies have performed ophthalmological and audiological examinations on GD1 patients, and these have shown that some patients with GD1 have neurological manifestations, such as retinal nerve fiber layer (RNFL) damage, visual-evoked potential (VEP) changes, and noise speech perception impairment [82]. Neurological involvement, especially in terms of cognitive ability, is more obvious in GD1 patients. Tullo et al. [82] performed neurological, neuropsychological, and other assessments of 22 GD patients (19 with GD1 and 3 with GD3). The authors found that the patients initially diagnosed with GD1 and GD3 had varying degrees of neurological involvement, including a high incidence of Parkinson motor and non-motor symptoms, cognitive and psychiatric disorders, and impaired short-term and long-term memory. However, GD3 patients often have more extensive cognitive deficits that affect daily activities, while the mild cognitive impairment in GD1 patients is often not perceived by the patients and their families [65]. Neurological deficits in patients with GD1 may be delayed. One study reported that 30.7% of patients with GD1 developed neurological problems in adulthood, including dementia, psychomotor retardation, and Parkinson syndrome [83, 84].
Treatment
Enzyme replacement therapy
Enzyme replacement therapy (ERT) aims to reduce the accumulation of glucosylceramides by supplementing active GBA. Three drugs are currently available for this type of treatment, namely, imiglucerase, velagluserase alfa, and taliglucerase alfa. However, only imiglucerase is currently approved for the treatment of non-neurological symptoms in patients with GD3 in China. Lee et al. [85] analyzed seven patients with GD3 with a homogeneous genetic background (homozygous p.L444P mutation) and treated them from an early stage. The results showed that ERT rapidly restored visceral symptom, but neurological symptoms including horizontal gaze palsy, epilepsy, and mental retardation persisted over the treatment period. Although neurological lesions are a prominent feature of GD3, treatment to relieve neurological symptoms remains an important unmet need in patients with GD3 [43]. In one study, five patients with GD3 who exhibited white vitreous turbidity were treated with ERT, but they still developed or continued to see white shadows [86]. Cognitive performance in adult GD3 patients remained stable or slowly declined while receiving enzyme replacement therapy [46]. However, given that ERT cannot cross the blood–brain barrier or the blood–retinal barrier, it may not be able to reach the retina. Therefore, ERT is usually not effective in treating ocular-related and neurological manifestations of GD. Therefore, scientists have been working on new approaches to treat neurological symptoms in patients with GD3.
Substrate reduction therapy
Substrate reduction therapy (SRT) reduces the accumulation of glucosylceramides by inhibiting glucosylceramide synthase. SRT targets the biosynthetic cycle of glucosylceramides and reduces their loading into the lysosome. Compared with ERT, SRT is more amenable to oral administration and reduces the time required for treatment [87]. In multiple clinical trials, the SRT drug eliglustat improved platelet count, hemoglobin level, liver and spleen volume, and skeleton-related clinical parameters, but it did not improve neurological symptoms in patients with GD3 [49, 88].
Molecular chaperone
Protein misfolding is an important cause of enzyme deficiency. Missense mutations often result in proteins that do not fold efficiently into their native conformations. Such misfolded proteins are recognized by the intracellular quality control system, retrotranslocated into the cytosol, and degraded by the endoplasmic reticulum [89]. Thus, the number of enzymes transported from the endoplasmic reticulum to the lysosome is reduced, leading to a decrease in effective enzyme activity. Normally, small-molecule chaperones contribute to the correct folding of proteins in the endoplasmic reticulum by binding and stabilizing misfolded mutant GBA in the endoplasmic reticulum, and once correctly folded GBA reaches the lysosome, the small molecules dissociate from the nascent unstable protein, thereby relieving enzyme inhibition [90]. Small-molecule chaperones, including iminosugars, ambroxol, and other competitive GBA inhibitors, are under investigation as emerging therapeutic approaches. Some of these molecules have a particular potential for the treatment of neurological symptoms, given their ability to penetrate the blood–brain barrier and promote appropriate enzyme folding and translocation to lysosomes [91]. Early initiation of ambroxol therapy has been reported to prevent neurological damage in some patients with GD3 [92]. In a study of five patients with GD3, treatment with amiloride resulted in increased lymphocyte GBA activity and decreased glucosylsphingosine level in the cerebrospinal fluid, as well as improvements in all neurological symptoms, including myoclonus, seizures, and pupillary light reflex dysfunction [39].
Gene therapy
Gene therapy, which uses viruses as vectors to administer and integrate healthy genes in place of defective ones, has shown early success in animal experiments. In one study, researchers injected recombinant adeno-associated virus vectors (rAAV) encoding GBA into a GBA-knockout mouse model [93]. Treatment with rAAV restored neuronal GBA expression in this mouse model. In addition, a non-integrative approach using Sendai virus delivery was used to establish induced pluripotent stem cells (iPSCs) from fibroblasts of a patient with GD3. Differentiation of iPSCs can be used to generate a variety of complex cell types with a high degree of genetic complexity—a research topic that is still under investigation [94]. As the correlation between GBA mutations and neuropathy in GD is studied in depth, many possibilities for gene therapy will be further explored.
Hematopoietic stem cell transplantation
Allogeneic hematopoietic stem cell transplantation (HSCT) from a healthy donor replaces defective monocytes with hematopoietic stem cells that produce GBA [95] to provide a permanent source of enzymes, improve visceral and skeletal damage [96], and stabilize the progression of neurological disease in patients with GD [97]. Although transplantation of HSCT has been reported to improve associated symptoms, long-term follow-up of nine Swedish and UK nGD patients showed deterioration in neurological function after surgery [97]. HSCT may provide a valuable treatment option for patients with GD3, which is quite inexpensive compared with ERT. However, the mortality rate associated with HSCT and the risk of graft-versus-host disease (GVHD) remain high.
Follow-up and evaluation
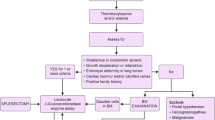
All patients with GD should have a comprehensive baseline evaluation to assess their particular disease pattern. Ongoing monitoring of all patients is important to continually assess the severity and rate of disease progression. Monitoring of patients includes regular clinical, biological, and radiological assessments. For patients with GD, regardless of the subtype at their initial diagnosis, the evaluation of blood biochemistry, biomarkers, visceral damage, and especially the nervous system is indispensable. GD patients with a high incidence of non-motor symptoms can be examined using the Epworth Sleepiness Scale (ESS), a sleep latency test, and dedicated polysomnography studies [82]. The Mini-Mental State Examination (MMSE) and Brief Psychiatric Rating Scale (BPRS) can also be used to assess cognitive impairment and mental disorders, and an episodic memory test (such as the Babcock Story Recall Test) can be used to assess short-term and long-term memory impairment [82]. In addition, extensive neuropsychological evaluation is required (Table 1). As the disease progresses, new neurological manifestations may develop. One patient with GD3b, for example, developed new neurological features, including myoclonic seizures that grew out of epilepsy, and an increasingly rapid cognitive decline, after more than 20 years of ERT treatment [98]. Frequent testing and follow-up allow for early detection of relevant progression in this patient group, leading to timely assessment and adjustment of treatment regimens (Fig. 1).

Therapeutic strategies to enhance acid β-glucosidase activity. Mutations in the GBA gene cause misfolding of acid β-glucosidase (GBA) in the endoplasmic reticulum, resulting in reduced GBA activity. Enzyme replacement therapy introduces exogenous enzymes into the cell to improve the enzyme defect. Substrate reduction therapy targets the reduction of glucosylceramides to prevent substrate accumulation. Gene therapy targets the host genome to restore glucocerebrosidase activity. Molecular chaperones bind to mutant GBA to stabilize and facilitate enzyme translocation to the lysosome. GBA acid β-glucosidase, GluCer glucosylceramide
Summary
Here, we compiled some of the clinical manifestations and treatments for GD3, focusing on the neurological impairments typically present in GD3, which included manifestations such as ocular gaze palsy, retinopathy, auditory impairment, myoclonic epilepsy, and cognitive impairment. To identify patients with GD3, evaluations should always include neuro-ophthalmological examination, audiometric testing, cognitive behavioral testing, and EEG. Evaluation of pulmonary, cardiac, and skeletal involvement is also important and may further suggest manifestations of specific subtypes of GD3. There is increasing evidence of the correlation between GBA mutant phenotypes and clinical manifestations of GD. Compared to GD1, patients with GD3 present earlier and more severe neurological impairments such as cognitive impairment, hearing impairment, and visual abnormalities. Therefore, the importance of gene sequencing for GD diagnosis is becoming more prominent. The discovery of new GBA mutations and in-depth studies of mutation mechanisms and clinical biomarkers are imminent. This will provide a theoretical basis for the development of specific therapeutic approaches that cross the blood–brain or blood–retinal barrier and target GD neuropathy. In addition, many studies have shown varying degrees of neuropathy in patients initially diagnosed with GD1. All patients with confirmed or suspected GD, especially in the nervous system, should be comprehensively evaluated, diagnosed, and treated. Detailed and comprehensive assessment enables clinicians to detect subtle system damage, even unperceived by patients, so as to provide early symptomatic treatment and prevent disease progression. Established therapies such as ERT and SRT may improve visceral involvement in patients with GD, but they are typically ineffective for neuropathy. However, emerging therapies that can cross the blood–brain barrier are being developed and investigated to treat the neurological symptoms of GD. In that context, ongoing treatment, monitoring, and evaluation are essential to improve survival and quality of life in patients with GD.
Data availability
All data supporting the findings of this study are available within the paper.
References
Beavan MS, Schapira AHV (2013) Glucocerebrosidase mutations and the pathogenesis of Parkinson disease. Ann Med 45:511–521. https://doi.org/10.3109/07853890.2013.849003
Grabowski GA, Zimran A, Ida H (2015) Gaucher disease types 1 and 3: phenotypic characterization of large populations from the ICGG gaucher registry. Am J Hematol 90:S12–S18. https://doi.org/10.1002/ajh.24063
Grabowski GA (2008) Phenotype, diagnosis, and treatment of Gaucher’s disease. Lancet 372:1263–1271. https://doi.org/10.1016/s0140-6736(08)61522-6
Gary SE, Ryan E, Steward AM, Sidransky E (2018) Recent advances in the diagnosis and management of Gaucher disease. Expert Rev Endocrinol Metab 13:107–118. https://doi.org/10.1080/17446651.2018.1445524
Goker-Alpan O, Schiffmann R, Park JK et al (2003) Phenotypic continuum in neuronopathic Gaucher disease: an intermediate phenotype between type 2 and type 3. J Pediatr 143:273–276. https://doi.org/10.1067/s0022-3476(03)00302-0
Lal TR, Seehra GK, Steward AM et al (2020) The natural history of type 2 Gaucher disease in the 21st century. Neurology 95:e2119–e2130. https://doi.org/10.1212/wnl.0000000000010605
Schwartz IVD, Göker-Alpan Ö, Kishnani PS et al (2018) Characteristics of 26 patients with type 3 Gaucher disease: a descriptive analysis from the Gaucher outcome survey. Mol Genet Metab Rep 14:73–79. https://doi.org/10.1016/j.ymgmr.2017.10.011
Winter AW, Salimi A, Ospina LH, Roos JCP (2019) Ophthalmic manifestations of Gaucher disease: the most common lysosomal storage disorder. Br J Ophthalmol 103:315–326. https://doi.org/10.1136/bjophthalmol-2018-312846
Castillon G, Chang S-C, Moride Y (2022) Global incidence and prevalence of Gaucher disease: a targeted literature review. J Clin Med 12:85. https://doi.org/10.3390/jcm12010085
Nguyen Y, Stirnemann J, Belmatoug N (2020) Maladie de Gaucher. Rev Prat 70:416–420
Weinreb NJ, Goker-Alpan O, Kishnani PS et al (2022) The diagnosis and management of Gaucher disease in pediatric patients: where do we go from here? Mol Genet Metab 136:4–21. https://doi.org/10.1016/j.ymgme.2022.03.001
Riboldi GM, Fonzo ABD (2019) GBA, Gaucher disease, and Parkinson’s disease: from genetic to clinic to new therapeutic approaches. Cells 8:364. https://doi.org/10.3390/cells8040364
Taddei TH, Kacena KA, Yang M et al (2009) The underrecognized progressive nature of N370S Gaucher disease and assessment of cancer risk in 403 patients. Am J Hemat 84:208–214. https://doi.org/10.1002/ajh.21362
Tajima A, Yokoi T, Ariga M et al (2009) Clinical and genetic study of Japanese patients with type 3 Gaucher disease. Mol Genet Metab 97:272–277. https://doi.org/10.1016/j.ymgme.2009.05.001
Jeong S-Y, Park S-J, Kim HJ (2011) Clinical and genetic characteristics of Korean patients with Gaucher disease. Blood Cells Mol Dis 46:11–14. https://doi.org/10.1016/j.bcmd.2010.07.010
El-Morsy Z, Khashaba MT, Soliman OE-S et al (2011) Glucosidase acid beta gene mutations in Egyptian children with Gaucher disease and relation to disease phenotypes. World J Pediatr 7:326–330. https://doi.org/10.1007/s12519-011-0309-1
Choy FYM, Zhang W, Shi H-P et al (2007) Gaucher disease among Chinese patients: review on genotype/phenotype correlation from 29 patients and identification of novel and rare alleles. Blood Cells Mol Dis 38:287–293. https://doi.org/10.1016/j.bcmd.2006.11.003
Abdelwahab M, Blankenship D, Schiffmann R (2016) Long-term follow-up and sudden unexpected death in Gaucher disease type 3 in Egypt. Neurol Genet 2:e55. https://doi.org/10.1212/nxg.0000000000000055
Bennett LL, Mohan D (2013) Gaucher disease and its treatment options. Ann Pharmacother 47:1182–1193. https://doi.org/10.1177/1060028013500469
Mistry PK, Cappellini MD, Lukina E et al (2010) A reappraisal of Gaucher disease-diagnosis and disease management algorithms. Am J Hematol 86:110–115. https://doi.org/10.1002/ajh.21888
Casta A, Hayden K, Wolf WJ (1984) Calcification of the ascending aorta and aortic and mitral valves in Gaucher’s disease. Am J Cardiol 54:1390–1391. https://doi.org/10.1016/s0002-9149(84)80115-0
Kurolap A, del Toro M, Spiegel R et al (2019) Gaucher disease type 3c: new patients with unique presentations and review of the literature. Mol Genet Metab 127:138–146. https://doi.org/10.1016/j.ymgme.2019.05.011
Charrow J, Esplin JA, Gribble TJ et al (1998) Gaucher disease. Arch Intern Med 158:1754. https://doi.org/10.1001/archinte.158.16.1754
Momosaki K, Kido J, Matsumoto S et al (2018) High-risk screening for Gaucher disease in patients with neurological symptoms. J Hum Genet 63:717–721. https://doi.org/10.1038/s10038-018-0438-7
Horowitz M, Wilder S, Horowitz Z et al (1989) The human glucocerebrosidase gene and pseudogene: structure and evolution. Genomics 4:87–96. https://doi.org/10.1016/0888-7543(89)90319-4
Kim Y-M, Choi J-H, Kim G-H et al (2020) The GBA p.G85E mutation in Korean patients with non-neuronopathic Gaucher disease: founder and neuroprotective effects. Orphanet J Rare Dis. https://doi.org/10.1186/s13023-020-01597-0
Tan E-K, Tong J, Fook-Chong S et al (2007) Glucocerebrosidase mutations and risk of Parkinson disease in Chinese patients. Arch Neurol 64:1056. https://doi.org/10.1001/archneur.64.7.1056
Wan L, Hsu C-M, Tsai C-H et al (2006) Mutation analysis of Gaucher disease patients in Taiwan: high prevalence of the RecNciI and L444P mutations. Blood Cells Mol Dis 36:422–425. https://doi.org/10.1016/j.bcmd.2006.02.001
Hruska KS, LaMarca ME, Scott CR, Sidransky E (2008) Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum Mutat 29:567–583. https://doi.org/10.1002/humu.20676
Koprivica V, Stone DL, Park JK et al (2000) Analysis and classification of 304 mutant alleles in patients with type 1 and type 3 Gaucher disease. Am J Hum Genet 66:1777–1786. https://doi.org/10.1086/302925
Mistry PK, Lopez G, Schiffmann R et al (2017) Gaucher disease: progress and ongoing challenges. Mol Genet Metab 120:8–21. https://doi.org/10.1016/j.ymgme.2016.11.006
Biegstraaten M, van Schaik IN, Aerts JMFG et al (2011) A monozygotic twin pair with highly discordant Gaucher phenotypes. Blood Cells Mol Dis 46:39–41. https://doi.org/10.1016/j.bcmd.2010.10.007
García RS, de Frutos LL, Arreguin EÁ et al (2021) Gaucher disease: identification and novel variants in Mexican and Spanish patients. Arch Méd Res 52:731–737. https://doi.org/10.1016/j.arcmed.2021.05.001
Daykin EC, Ryan E, Sidransky E (2021) Diagnosing neuronopathic Gaucher disease: new considerations and challenges in assigning Gaucher phenotypes. Mol Genet Metab 132:49–58. https://doi.org/10.1016/j.ymgme.2021.01.002
Aerts JM, van Breemen MJ, Bussink AP et al (2008) Biomarkers for lysosomal storage disorders: identification and application as exemplified by chitotriosidase in Gaucher disease. Acta Paediatr 97:7–14. https://doi.org/10.1111/j.1651-2227.2007.00641.x
Rolfs A, Giese A-K, Grittner U et al (2013) Glucosylsphingosine is a highly sensitive and specific biomarker for primary diagnostic and follow-up monitoring in Gaucher disease in a non-jewish, Caucasian cohort of Gaucher disease patients. PLoS ONE 8:e79732. https://doi.org/10.1371/journal.pone.0079732
Dekker N, van Dussen L, Hollak CEM et al (2011) Elevated plasma glucosylsphingosine in Gaucher disease: relation to phenotype, storage cell markers, and therapeutic response. Blood 118:e118–e127. https://doi.org/10.1182/blood-2011-05-352971
Saville JT, McDermott BK, Chin SJ et al (2019) Expanding the clinical utility of glucosylsphingosine for Gaucher disease. J Inherit Metab Dis 43:558–563. https://doi.org/10.1002/jimd.12192
Narita A, Shirai K, Itamura S et al (2016) Ambroxol chaperone therapy for neuronopathic Gaucher disease: a pilot study. Ann Clin Transl Neurol 3:200–215. https://doi.org/10.1002/acn3.292
Savostyanov K, Pushkov A, Mura’vova L, et al (2019) Glucosylfingosine (Lyso-GL1) may be the primary biomarker for screening Gaucher disease in Russian patients. Mol Genet Metab 126:S130. https://doi.org/10.1016/j.ymgme.2018.12.334
Hurvitz N, Dinur T, Becker-Cohen M et al (2019) Glucosylsphingosine (lyso-Gb1) as a biomarker for monitoring treated and untreated children with Gaucher disease. International J Of Molecular Sciences 20:3033. https://doi.org/10.3390/ijms20123033
Abeliovich A, Hefti F, Sevigny J (2021) Gene therapy for Parkinson’s disease associated with GBA1 mutations. J Parkinson’s Dis 11:S183–S188. https://doi.org/10.3233/jpd-212739
Tylki-Szymańska A, Vellodi A, El-Beshlawy A et al (2010) Neuronopathic Gaucher disease: demographic and clinical features of 131 patients enrolled in the international collaborative gaucher group neurological outcomes subregistry. J Inherit Metab Dis 33:339–346. https://doi.org/10.1007/s10545-009-9009-6
Kraoua I, Sedel F, Caillaud C et al (2011) A french experience of type 3 Gaucher disease: phenotypic tdersity and neurological outcome of 10 patients. Brain Develop 33:131–139. https://doi.org/10.1016/j.braindev.2010.02.005
Accardo AP, Pensiero S, Perissutti P (2005) Saccadic analysis for early identification of neurological involvement in Gaucher disease. Ann N Y Acad Sci 1039:503–507. https://doi.org/10.1196/annals.1325.054
Benko W, Ries M, Wiggs EA et al (2011) The saccadic and neurological deficits in type 3 Gaucher disease. PLoS One 6:e22410. https://doi.org/10.1371/journal.pone.0022410
Hopf S, Pfeiffer N, Liesenfeld M et al (2019) A comprehensive monocentric ophthalmic study with Gaucher disease type 3 patients: vitreoretinal lesions, retinal atrophy and characterization of abnormal saccades. Orphanet J Rare Dis. https://doi.org/10.1186/s13023-019-1244-9
Patterson MC, Horowitz M, Abel RB et al (1993) Isolated horizontal supranuclear gaze palsy as a marker of severe systemic involvement in Gaucher’s disease. Neurology 43:1993–1993. https://doi.org/10.1212/wnl.43.10.1993
Schiffmann R, FitzGibbon EJ, Harris C et al (2008) Randomized, controlled trial of miglustat in Gaucher’s disease type 3. Ann Neurol 64:514–522. https://doi.org/10.1002/ana.21491
Watanabe A, Gekka T, Arai K, Tsuneoka H (2016) A case of traction retinal detachment in a patient with Gaucher disease. Ophthalmic Genet 38:273–276. https://doi.org/10.1080/13816810.2016.1193878
Cindik N, Ozcay F, Süren D et al (2009) Gaucher disease with communicating hydrocephalus and cardiac involvement. Clin Cardiol 33:E26–E30. https://doi.org/10.1002/clc.20348
Guemes A, Kosmorsky GS, Moodie DS et al (1998) Corneal opacities in gaucher disease. Am J Ophthalmol 126:833–835. https://doi.org/10.1016/s0002-9394(98)00249-9
Hopf S, Schuster AK, Hennermann JB et al (2021) Retinal thinning in phenylketonuria and Gaucher disease type 3. Graefe’s Arch Clin Exp Ophthalmol 260:1153–1160. https://doi.org/10.1007/s00417-021-05424-5
Zhao T-T, Li H-L, Guo X-J et al (2018) Retinal detachment in a boy with Gaucher disease. Int J Ophthalmol. https://doi.org/10.18240/ijo.2018.09.23
Campbell PE, Harris CM, Vellodi A (2004) Deterioration of the auditory brainstem response in children with type 3 Gaucher disease. Neurology 63:385–387. https://doi.org/10.1212/01.wnl.0000130191.31669.48
Bamiou D-E, Campbell P, Liasis A et al (2001) Audiometric abnormalities in children with Gaucher disease type 3. Neuropediatrics 32:136–141. https://doi.org/10.1055/s-2001-16611
Stone DL, Tayebi N, Orvisky E et al (2000) Glucocerebrosidase gene mutations in patients with type 2 Gaucher disease. Hum Mutat 15:181–188. https://doi.org/10.1002/(sici)1098-1004(200002)15:2%3c181::aid-humu7%3e3.0.co;2-s
Tayebi N, Reissner KJ, Lau EK et al (1998) Genotypic heterogeneity and phenotypic variation among patients with type 2 Gaucher’s disease. Pediatr Res 43:571–578. https://doi.org/10.1203/00006450-199805000-00003
Manganotti P, Tamburin S, Zanette G, Fiaschi A (2001) Hyperexcitable cortical responses in progressive myoclonic epilepsy: a TMS study. Neurology 57:1793–1799. https://doi.org/10.1212/wnl.57.10.1793
Park JK, Orvisky E, Tayebi N et al (2003) Myoclonic epilepsy in Gaucher disease: genotype-phenotype insights from a rare patient subgroup. Pediatr Res 53:387–395. https://doi.org/10.1203/01.pdr.0000049515.79882.94
Machaczka M, Paucar M, Björkvall CK et al (2018) Novel hyperkinetic dystonia-like manifestation and neurological disease course of Swedish Gaucher patients. Blood Cells Mol Dis 68:86–92. https://doi.org/10.1016/j.bcmd.2016.10.011
Goker-Alpan O, Wiggs EA, Eblan MJ et al (2008) Cognitive outcome in treated patients with chronic neuronopathic Gaucher disease. J Pediatr 153:89-94.e4. https://doi.org/10.1016/j.jpeds.2007.12.023
Steward AM, Wiggs E, Lindstrom T et al (2019) Variation in cognitive function over time in Gaucher disease type 3. Neurology 93:e2272–e2283. https://doi.org/10.1212/wnl.0000000000008618
Tantawy AAG, Adly AAM, Abdeen MSED, Salah NY (2020) Cognitive decline and depressive symptoms: early non-motor presentations of parkinsonism among Egyptian Gaucher patients. Neurogenetics 21:159–167. https://doi.org/10.1007/s10048-020-00607-4
Biegstraaten M, Wesnes KA, Luzy C et al (2012) The cognitive profile of type 1 Gaucher disease patients. J Inherit Metab Dis 35:1093–1099. https://doi.org/10.1007/s10545-012-9460-7
Abdelwahab M, Potegal M, Shapiro EG, Nestrasil I (2017) Previously unrecognized behavioral phenotype in Gaucher disease type 3. Neurol Genet 3:e158. https://doi.org/10.1212/nxg.0000000000000158
Elstein D, Guedalia J, Doniger GM et al (2005) Computerized cognitive testing in patients with type I Gaucher disease: effects of enzyme replacement and substrate reduction. Genet Med 7:124–130. https://doi.org/10.1097/01.gim.0000153666.23707.ba
Dwolatzky T, Whitehead V, Doniger G et al (2003) Validity of a novel computerized cognitive battery for mild cognitive impairment. BMC Geriatr. https://doi.org/10.1186/1471-2318-3-4
Poffenberger CN, Inati S, Tayebi N et al (2020) EEG abnormalities in patients with chronic neuronopathic Gaucher disease: a retrospective review. Mol Genet Metab 131:358–363. https://doi.org/10.1016/j.ymgme.2020.10.010
Dunn DW, Austin JK, Perkins SM (2009) Prevalence of psychopathology in childhood epilepsy: categorical and dimensional measures. Dev Med Child Neurol 51:364–372. https://doi.org/10.1111/j.1469-8749.2008.03172.x
Revel-Vilk S, Szer J, Zimran A (2021) Hematological manifestations and complications of Gaucher disease. Expert Rev Hematol 14:347–354. https://doi.org/10.1080/17474086.2021.1908120
Tayebi N (2003) Gaucher disease with parkinsonian manifestations: does glucocerebrosidase deficiency contribute to a vulnerability to parkinsonism? Mol Genet Metab 79:104–109. https://doi.org/10.1016/s1096-7192(03)00071-4
Sidransky E, Nalls MA, Aasly JO et al (2009) Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med 361:1651–1661. https://doi.org/10.1056/nejmoa0901281
Furderer ML, Hertz E, Lopez GJ, Sidransky E (2022) Neuropathological features of Gaucher disease and Gaucher disease with parkinsonism. Int J Mol Sci 23:5842. https://doi.org/10.3390/ijms23105842
Straniero L, Asselta R, Bonvegna S et al (2020) The SPID-GBA study. Neurol Genet 6:e523. https://doi.org/10.1212/nxg.0000000000000523
Zhang Y, Shu L, Sun Q et al (2018) Integrated genetic analysis of racial differences of common GBA variants in Parkinson’s disease: a meta-analysis. Front Mol Neurosci. https://doi.org/10.3389/fnmol.2018.00043
Iwaki H, Blauwendraat C, Leonard HL et al (2019) Genetic risk of Parkinson disease and progression: neurology. Genetics 5:e348. https://doi.org/10.1212/nxg.0000000000000348
Gámez-Valero A, Prada-Dacasa P, Santos C et al (2016) GBA mutations are associated with earlier onset and male sex in dementia with lewy bodies. Mov Disord 31:1066–1070. https://doi.org/10.1002/mds.26593
Gaubert S, Hourregue C, Mouton-Liger F et al (2022) Exploring the link between GBA1 mutations and dementia with lewy, bodies, a mini-review. Neurosci Biobehav Rev 141:104856. https://doi.org/10.1016/j.neubiorev.2022.104856
Alcalay RN, Dinur T, Quinn T et al (2014) Comparison of Parkinson risk in ashkenazi jewish patients with Gaucher disease and GBA heterozygotes. JAMA Neurol 71:752. https://doi.org/10.1001/jamaneurol.2014.313
Biegstraaten M, Mengel E, Maródi L et al (2010) Peripheral neuropathy in adult type 1 Gaucher disease: a 2-year prospective observational study. Brain 133:2909–2919. https://doi.org/10.1093/brain/awq198
Tullo MG, Irelli EC, Caramia F et al (2023) The spectrum of neurological and sensory abnormalities in Gaucher disease patients: a multidisciplinary study (SENOPRO). Int J Mol Sci 24:8844. https://doi.org/10.3390/ijms24108844
McNeill A, Duran R, Proukakis C et al (2012) Hyposmia and cognitive impairment in Gaucher disease patients and carriers. Mov Disord 27:526–532. https://doi.org/10.1002/mds.24945
Alaei M, Jafari N, Rohani F et al (2018) Are there neurological symptoms in type 1 of Gaucher disease. Iran J Child Neurol 12:99–106
Lee N-C, Chien Y-H, Wong S-L et al (2014) Outcome of early-treated type III Gaucher disease patients. Blood Cells Mol Dis 53:105–109. https://doi.org/10.1016/j.bcmd.2014.05.007
Seehra GK, Eghbali A, Sidransky E, FitzGibbon E (2020) White vitreous opacities in five patients with Gaucher disease type 3. Am J Med Genet A 182:808–812. https://doi.org/10.1002/ajmg.a.61479
Futerman AH, Sussman JL, Horowitz M et al (2004) New directions in the treatment of Gaucher disease. Trends Pharmacol Sci 25:147–151. https://doi.org/10.1016/j.tips.2004.01.004
Stirnemann J, Belmatoug N, Camou F et al (2017) A review of Gaucher disease pathophysiology, clinical presentation and treatments. Int J Mol Sci 18:441. https://doi.org/10.3390/ijms18020441
Ellgaard L, Helenius A (2003) Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol 4:181–191. https://doi.org/10.1038/nrm1052
Ron I, Horowitz M (2005) ER retention and degradation as the molecular basis underlying Gaucher disease heterogeneity. Hum Mol Genet 14:2387–2398. https://doi.org/10.1093/hmg/ddi240
Jung O, Patnaik S, Marugan J et al (2016) Progress and potential of non-inhibitory small molecule chaperones for the treatment of Gaucher disease and its implications for Parkinson disease. Expert Rev Proteom 13:471–479. https://doi.org/10.1080/14789450.2016.1174583
Ramadža DP, Zekušić M, Žigman T et al (2021) Early initiation of ambroxol treatment diminishes neurological manifestations of type 3 Gaucher disease: a long-term outcome of two siblings. Eur J Paediatr Neurol 32:66–72. https://doi.org/10.1016/j.ejpn.2021.03.013
Massaro G, Mattar CNZ, Wong AMS et al (2018) Fetal gene therapy for neurodegenerative disease of infants. Nat Med 24:1317–1323. https://doi.org/10.1038/s41591-018-0106-7
Duarte AJ, Ribeiro D, Santos R et al (2019) Induced pluripotent stem cell line (INSAi001-A) from a Gaucher disease type 3 patient compound heterozygote for mutations in the GBA1 gene. Stem Cell Res 41:101595. https://doi.org/10.1016/j.scr.2019.101595
Ito S, Barrett AJ (2013) Gauchers disease—a reappraisal of hematopoietic stem cell transplantation. Pediatr Hematol Oncol 30:61–70. https://doi.org/10.3109/08880018.2012.762076
Somaraju URR, Tadepalli K (2017) Hematopoietic stem cell transplantation for Gaucher disease. Cochrane Database Syst Rev. https://doi.org/10.1002/14651858.cd006974.pub4
Donald A, Björkvall CK, Vellodi A et al (2022) Thirty-year clinical outcomes after haematopoietic stem cell transplantation in neuronopathic Gaucher disease. Orphanet J Rare Dis. https://doi.org/10.1186/s13023-022-02378-7
Elstein D, Abrahamov A, Altarescu G, Zimran A (2013) Evolving features in type 3 Gaucher disease on long-term enzyme replacement therapy. Blood Cell Mol Dis 50:140. https://doi.org/10.1016/j.bcmd.2012.09.008
Acknowledgements
The authors thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
Funding
This work was supported by the Zhejiang Medical and Health Science and Technology Plan Project (Grant Nos. 2021KY1137 and 2023RC290).
Author information
Authors and Affiliations
Contributions
WZ: concept, literature search, methodology, manuscript preparation, manuscript editing, and review. DL: validation, investigation, and resources. YF: literature search and methodology. PH: concept, design, manuscript review, and project administration.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhong, W., Li, D., Fei, Y. et al. A review of type 3 Gaucher disease: unique neurological manifestations and advances in treatment. Acta Neurol Belg 124, 1213–1223 (2024). https://doi.org/10.1007/s13760-024-02493-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13760-024-02493-1