
Abstract
Pompe disease is a glycogen storage disease caused by acid alfa-glucosidase deficiency. Here, we report clinical properties, genetic features of our late-onset Pompe patients. Seven patients were followed during the last 10 years in our institute. The clinical and laboratory findings were reviewed. Neuropsychological evaluation was performed in four patients. Myotonic discharges of paraspinal muscles and denervation potentials were seen in all patients at the diagnosis and were disappeared during follow-up in two. Only one patient, whose MRI showed cerebral atrophy, had attention and executive dysfunction. Compound heterozygous patients with IVS 1-13T>G have a milder disease. One patient who has homozygous IVS 1-13T>G mutation had more severe disease. Two of our patients who had very severe and fatal disease course carry double mutations on both alleles (c.547-39T>G and c.858+5ins7) that previously scored as “unknown” in Erasmus Pompe Center database. Lastly, we found new mutations (c.1209 C>A, 2737dupG) in two patients carrying IVS 1-13T>G in the other allele. Systemic involvements are very rare in late-onset Pompe patients. Similarly, Pompe disease does not cause cognitive impairment in adult population. Homozygous IVS 1-13T>G mutation and c.547-39T>G mutation which are previously noted as “unknown” pathogenicities cause a more severe disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pompe disease is a glycogen storage disease (type II) caused by acid alfa-glucosidase (GAA, acid maltase) deficiency. The mutation of GAA gene located on 17p25.2–17p25.3 causes deficiency of the enzyme activity that results in the intra-lysosomal accumulation of glycogen in skeletal muscles and other tissues [1, 2]. The disease was first described by JC Pompe in 1932 in an 8-month-old girl whose cardiac muscle and other tissues contained glycogen accumulation within vacuoles [3].
The disease recently has been classified into two forms as infantile and late-onset [3]. Infantile form generally manifests in progressive muscle weakness with “floppy infant” appearance and progressive respiratory insufficiency as well as organomegaly. Most infants die within 1 year due to cardiorespiratory insufficiency [4].
Late-onset form includes all milder forms presented after the first year of life. The disease generally presents as progressive proximal muscle weakness with respiratory insufficiency due to diaphragmatic and accessory respiratory muscle involvement. The disease predominantly affects the pelvic girdle muscles and may also cause skeletal abnormalities such as scoliosis and rigid spine [5]. Sensorineural hearing loss, cerebral aneurysms are other rare organ involvements [1]. The electrophysiological hallmark is the myotonic discharges in paraspinal muscles accompanying the myopathic pattern with spontaneous activity [6]. Reduced GAA enzyme activity assay in blood is the screening tool for the disease whereas the diagnosis is confirmed by a second GAA enzyme activity test in another tissue or GAA gene sequencing [7]. Although the cognitive impairment in infantile Pompe patients is a well-known phenomenon there is limited information about adult cognitive profiles with late-onset Pompe disease patients and in our knowledge, only one study showed cognitive impairment mainly in executive functions in nine late-onset Pompe patients [8].
During the last 10 years we diagnosed and followed seven late-onset Pompe patients. Herein we report the clinical properties, newly found mutations, new genotype–phenotype correlations, and enzyme replacement therapy (ERT) outcome of our patients as well as the other rare organ involvements such as cerebral aneurisms and cognitive functions.
Methods
Between 2004 and 2015, we diagnosed and followed seven patients with Pompe disease. Six of these patients referred to our neurology department because of proximal weakness. Only one patient diagnosed before the onset of symptoms, as she was the younger sister of patient 3. The diagnosis was confirmed by reduced GAA enzyme activity assay in dry blood sample (DBS) or in lymphocytes or muscle as well as GAA gene sequencing in all patients. The study was approved by local ethics committee (GO 16/630).
Neurological examination, 6-minute walk test (6MVT), Walton–Gardner-Medwin scale and short form 36 (SF 36) were assessed to evaluate physical activities in daily life and functional capacity of the patients. The electrodiagnostic studies were performed. At minimum; one upper limb and one lower limb motor and sensory nerve conduction studies were obtained. Needle electromyography (EMG) was performed by a concentric needle electrode and consisted of assessing one lower and upper limb proximal–distal sites routinely. Paraspinal muscles were sampled to determine myopathic and myotonic changes. Motor unit potential (MUP) morphology and recruitment, spontaneous activity (fibrillations, positive sharp waves, complex repetitive discharges and myotonic discharges) compound motor action potential amplitudes (CMAP), distal latencies, conduction velocities, F-wave latencies were recorded. Seven patients had initial electrodiagnostic examinations and four of them had follow-up examinations (mean time interval 6 years; range 3–9 years).
Cranial magnetic resonance imaging and angiographies were done for the diagnosis of cerebral aneurysm. Moreover, detailed neuropsychological examinations were performed in four of our patients who accepted the tests (patients 3, 4, 6 and 7). Mini-mental state examination (MMSE), Beck depression scale, Stroop color test, trail making test A and B, Benton line orientation test, Benton’s facial recognition test, months backward test, clock drawing test, auditory verbal learning test (AVLT), Wisconsin card sorting test, visual reproduction subtest of Wechsler’s memory scale (WMS-VR), Boston naming test, digit span forward and backward were administered to evaluate general cognition, depression, attention, memory, executive functions and visuo-spatial abilities. Scores of MMSE, Beck depression scale, digit span forwards and backwards, Boston naming test, Wisconsin card sorting test, WMS-VR, clock drawing test, AVLT, Benton line orientation test, Benton’s facial recognition test were presented as the total number of the correct responses with higher scores indicating better responses. The time of reciting months backward, Stroop test, trail making test A and B were noted in seconds. Performance of a patient was reported as impaired or normal according to cutoff scores of each test.
Transthoracic echocardiography, pulmonary function tests were performed by specialists to determine the systemic complications of Pompe disease.
Results
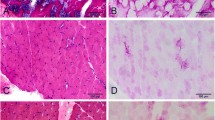
Five of our Pompe patients were female. The mean age of onset was 29.43 (range 17–37 years) and the mean age of diagnosis was 39.5 (range 27–41 years) years (Table 1a). Muscle biopsies were performed from left biceps brachii muscles in six patients and all showed vacuolar myopathy (Fig. 1) but one who showed only type 1 fiber atrophy.

Muscle biopsies of the patients showed vacuoles in the majority of muscle fibers by modified Gomori trichrome staining (a), with increased acid phosphatase activity (b) and glycogen deposition detected by periodic acid-Schiff (c)
Neurological manifestations
The proximal weakness was the first manifestation in all patients except one who had both proximal and distal weakness. The weakness was prominent in lower extremities in all except one patient (patient 1). Respiratory insufficiency was seen in two patients and they needed mechanical ventilation after several years of the diagnosis (after 2 years in patient 1, after 5 years in patient 2). Besides two other patients had decreased forced vital capacities (FVC) without symptoms. Patient 4 had a normal FVC whereas the polysomnographic analysis depicted hypopnea and apnea.
The cranial magnetic resonance imaging (MRI) and magnetic resonance angiography (MRA) were performed and none of these patients had cerebral aneurism. Cranial MRI showed mega cisterna magna, partial empty cella and minimal cerebral atrophy only in one patient (patient 4) but these findings were not related to Pompe disease.
Electrophysiological findings
Myopathic changes as well as myotonic discharges of paraspinal muscles and denervation potentials (complex repetitive discharges, positive sharp wave and fibrillations) were seen in all patients at the diagnosis. Four patients had a follow-up electrophysiological evaluation. Myotonic discharges were disappeared in two of four patients during follow-up (one after 3 years of ERT, the other after 7 years of ERT).
Cognitive functions
Neuropsychological test performance of each case is presented in Table 2. General cognition, language, visuo-spatial functions were normal in all patients. Attention and executive functions were impaired in one patient (patient 4). This case also had a slight impairment of verbal recent memory measured by AVLT.
Other organ involvements
The echocardiography was normal in all patients except one who had minimal tricuspid and mitral insufficiency.
Enzyme replacement treatment (ERT) response and prognosis
Enzyme replacement treatment was administered to six Pompe patients, one of whom stopped the treatment after the second infusion due to allergic reaction (Table 1b). The mean follow-up period was 5.2 (3–8) years. Two of these patients, who were already severely affected, worsened during ERT and died during the follow-up. Furthermore, the neurological examination stayed stable in four patients.
Genotype–phenotype
IVS 1-13T>G (c. 32-13 T>G) mutation, which is the most common mutation of Pompe disease, was homozygous only in one of our patients (patient 6). Three patients were compound heterozygous for IVS. Interestingly, two of these patients with compound heterozygous IVS mutation carry unreported mutations, c.1209C>A and 2737dupG, in the other alleles. Our observation in this small group suggests that patients with homozygous IVS mutation have more severe myopathy than the patients with heterozygous IVS mutation; as the heterozygous patients were still ambulatory whereas the homozygous patient is mostly wheelchair bound after 2 years of disease (Table 1).
Two patients were homozygous for both c. 547-39 T>G and c.858+5ins7. Both of these mutations were reported as “unknown” for their mutagenicity in Erasmus Pompe Center (http://www.pompecenter.nl) database. These two patients had severe disease course with loss of ambulation and respiratory failure and both died within 10 years after the diagnosis.
Discussion
Late-onset Pompe disease, which is a rare metabolic myopathy, has drawn attention after the discovery of enzyme replacement treatment. Although the prominent manifestation is proximal myopathy, it is known that the disease can have several systemic and neurological involvements. Here, we reviewed the clinical and laboratory findings of our late-onset Pompe patients.
Proximal weakness was the first symptom in all patients except one who had both proximal and distal myopathy. Rigid spine was observed in two of our patients. Respiratory failure due to diaphragmatic, intercostal and accessory muscle weakness is a well-known phenomenon in Pompe patients [3]. Accordingly four of our patients had respiratory dysfunction and two of them needed mechanical ventilation. Patient 4 has never complained about dyspnea but his polysomnography depicted central and obstructive sleep apnea syndrome. However, his polysomnography showed minimal improvement after 3 years of ERT.
Several electrophysiological findings such as reduced CMAPs, positive sharp waves, fibrillations, myotonic discharges and myopathic MUPs prominent in proximal limb muscles were reported in Pompe patients [9–11]. Myotonic discharges may be isolated to the paraspinal muscles. Thus, these muscles should be examined in case of suspicion of Pompe disease [12]. We also observed that the myotonic discharges disappeared during the follow-up of ERT in two patients. This finding is in accordance with the previous report of Hobson-Webb et al. [6]. It may be related to the fibrosis and fatty tissue replacement during the course of chronic myopathy. Thus, it should be kept in mind that this finding may be missing in advanced Pompe patients.
Glycogen storage in brain results in mild cognitive deficits in patients with infantile Pompe disease [13]. It is recommended to start the ERT early, as it is shown early treatment results in better motor outcome and cognitive development [14]. A few available autopsy data of untreated infants indicate that glycogen is stored in the anterior horn cells of the spinal cord, the brain stem, the thalamus, the cerebellum and the cerebral cortex [15–17]. Brain abnormalities including delay in myelination, white matter changes might be observed on neuroimaging studies [18]. However, there is limited information about adult cognitive profiles with late-onset Pompe disease patients and in our knowledge, only one study showed cognitive impairment mainly in executive functions in nine late-onset Pompe patients [8]. We could not confirm these data in our limited patient group. Only one of our patients (patient 4) had mild dysfunction in attention, executive functions and memory domains when compared to age, education adjusted healthy controls. Moreover, this unique patient had several brain MRI pathologies such as mega cisterna magna, partial empty cella and minimal cerebral atrophy.
So far more than 500 mutations were identified in GAA gene, which is located on 17q21–23. IVS 1-13T>G mutation is a highly prevalent mutation found in 70% of the Caucasian compound heterozygous patients [19]. This mutation generally leads a milder form of the disease [19–21]. Accordingly three of our patients were compound heterozygous owing one IVS 1-13T>G mutation and they have a milder disease with lower Walton–Gardner-Medwin scale compared to other patients. However, two of these patients, who are sisters, have a second nonsense mutation as E888X 2662G>T previously reported as a cause of severe infantile and late-onset Pompe disease [22–25].
Homozygous IVS 1-13T>G Pompe patients is very rare and Musumeci et al. reported that homozygous status is not so mild as previously supposed [26]. We agree with them as our unique patient with homozygous IVS 1-13T>G mutation has a more severe disease than our compound heterozygous patients.
Remarkably, two of our patients had a very severe and fatal disease course with loss of ambulation and necessity of mechanical ventilation. Both of them carry double mutations on both alleles (c.547-39T>G and c.858+5ins7) that previously scored as “unknown” in Erasmus Pompe Center database (http://www.pompecenter.nl). The alteration c.858+5ins7 in intron 4 was previously reported with high frequency in Brazilian population and considered as a non-pathogenic polymorphism [27]. Therefore, c.547-39T>G mutation should be responsible for the disease in these two patients and results in a severe phenotype.
We also determined two new mutations as c.1209C>A and 2737dupG, which cause Pompe disease.
Enzyme replacement treatment (ERT) became available during the follow-up of our patients. Six of our patients received ERT. Only one patient decided not to continue to the treatment after the second infusion due to allergic reaction. Two of our patients were severely affected when they started ERT thus they did not benefit from the treatment. The other four patients on ERT have steady muscle strength and respiratory function.
In summary, our observation in this small group showed that the systemic involvements as well as neurological complications other than myopathy are very rare in late-onset Pompe patients. Similarly, Pompe disease does not cause cognitive impairment in adult population. Homozygous IVS 1-13T>G mutation causes a more severe disease than compound heterozygosity. Moreover, c.547-39T>G mutation which is previously noted as “unknown” pathogenicity may cause a severe Pompe phenotype.
References
Filosto M, Todeschini A, Cotelli MS, Vielmi V, Rinaldi F, Rota S, Scarpelli M, Padovani A (2013) Non-muscle involvement in late-onset glycogenosis II. Acta Myol 32(2):91–94
Montalvo AL, Bembi B, Donnarumma M, Filocamo M, Parenti G, Rossi M, Merlini L, Buratti E, De Filippi P, Dardis A, Stroppiano M, Ciana G, Pittis MG (2006) Mutation profile of the GAA gene in 40 Italian patients with late onset glycogen storage disease type II. Hum Mutat 27(10):999–1006. doi:10.1002/humu.20374
Di Rocco M, Buzzi D, Taro M (2007) Glycogen storage disease type II: clinical overview. Acta Myol 26(1):42–44
Van den Hout JM, Kamphoven JH, Winkel LP, Arts WF, De Klerk JB, Loonen MC, Vulto AG, Cromme-Dijkhuis A, Weisglas-Kuperus N, Hop W, Van Hirtum H, Van Diggelen OP, Boer M, Kroos MA, Van Doorn PA, Van der Voort E, Sibbles B, Van Corven EJ, Brakenhoff JP, Van Hove J, Smeitink JA, de Jong G, Reuser AJ, Van der Ploeg AT (2004) Long-term intravenous treatment of Pompe disease with recombinant human alpha-glucosidase from milk. Pediatrics 113(5):e448–e457
Schuller A, Wenninger S, Strigl-Pill N, Schoser B (2012) Toward deconstructing the phenotype of late-onset Pompe disease. Am J Med Genet Part C Semin Med Genet 160C(1):80–88. doi:10.1002/ajmg.c.31322
Hobson-Webb LD, Dearmey S, Kishnani PS (2011) The clinical and electrodiagnostic characteristics of Pompe disease with post-enzyme replacement therapy findings. Clin Neurophysiol 122(11):2312–2317. doi:10.1016/j.clinph.2011.04.016
American Association of N, Electrodiagnostic M (2009) Diagnostic criteria for late-onset (childhood and adult) Pompe disease. Muscle Nerve 40(1):149–160. doi:10.1002/mus.21393
Borroni B, Cotelli MS, Premi E, Gazzina S, Cosseddu M, Formenti A, Gasparotti R, Filosto M, Padovani A (2013) The brain in late-onset glycogenosis II: a structural and functional MRI study. J Inherit Metab Dis 36(6):989–995. doi:10.1007/s10545-013-9601-7
Engel AG (1970) Acid maltase deficiency in adults: studies in four cases of a syndrome which may mimic muscular dystrophy or other myopathies. Brain 93(3):599–616
Engel AG, Seybold ME, Lambert EH, Gomez MR (1970) Acid maltase deficiency: comparison of infantile, childhood, and adult types. Neurology 20(4):382
Kassardjian CD, Engel AG, Sorenson EJ (2015) Electromyographic findings in 37 patients with adult-onset acid maltase deficiency. Muscle Nerve 51(5):759–761. doi:10.1002/mus.24620
Barohn RJ, McVey AL, DiMauro S (1993) Adult acid maltase deficiency. Muscle Nerve 16(6):672–676. doi:10.1002/mus.880160614
Spiridigliozzi GA, Heller JH, Kishnani PS (2012) Cognitive and adaptive functioning of children with infantile Pompe disease treated with enzyme replacement therapy: long-term follow-up. Am J Med Genet Part C Semin Med Genet 160C(1):22–29. doi:10.1002/ajmg.c.31323
Ebbink BJ, Aarsen FK, van Gelder CM, van den Hout JM, Weisglas-Kuperus N, Jaeken J, Lequin MH, Arts WF, van der Ploeg AT (2012) Cognitive outcome of patients with classic infantile Pompe disease receiving enzyme therapy. Neurology 78(19):1512–1518. doi:10.1212/WNL.0b013e3182553c11
Gambetti P, DiMauro S, Baker L (1971) Nervous system in Pompe’s disease. Ultrastructure and biochemistry. J Neuropathol Exp Neurol 30(3):412–430
Mancall EL, Aponte GE, Berry RG (1965) Pompe’s disease (diffuse glycogenosis) with neuronal storage. J Neuropathol Exp Neurol 24:85–96
Martini C, Ciana G, Benettoni A, Katouzian F, Severini GM, Bussani R, Bembi B (2001) Intractable fever and cortical neuronal glycogen storage in glycogenosis type 2. Neurology 57(5):906–908
Chien YH, Lee NC, Peng SF, Hwu WL (2006) Brain development in infantile-onset Pompe disease treated by enzyme replacement therapy. Pediatr Res 60(3):349–352. doi:10.1203/01.pdr.0000233014.84318.4e
Kroos MA, Pomponio RJ, Hagemans ML, Keulemans JL, Phipps M, DeRiso M, Palmer RE, Ausems MG, Van der Beek NA, Van Diggelen OP, Halley DJ, Van der Ploeg AT, Reuser AJ (2007) Broad spectrum of Pompe disease in patients with the same c.-32-13T->G haplotype. Neurology 68(2):110–115. doi:10.1212/01.wnl.0000252798.25690.76
Boerkoel CF, Exelbert R, Nicastri C, Nichols RC, Miller FW, Plotz PH, Raben N (1995) Leaky splicing mutation in the acid maltase gene is associated with delayed onset of glycogenosis type II. Am J Hum Genet 56(4):887–897
Huie ML, Chen AS, Tsujino S, Shanske S, DiMauro S, Engel AG, Hirschhorn R (1994) Aberrant splicing in adult onset glycogen storage disease type II (GSDII): molecular identification of an IVS1 (-13T→G) mutation in a majority of patients and a novel IVS10 (+1GT→CT) mutation. Hum Mol Genet 3(12):2231–2236
Wan L, Lee CC, Hsu CM, Hwu WL, Yang CC, Tsai CH, Tsai FJ (2008) Identification of eight novel mutations of the acid alpha-glucosidase gene causing the infantile or juvenile form of glycogen storage disease type II. J Neurol 255(6):831–838. doi:10.1007/s00415-008-0714-0
Liu X, Wang Z, Jin W, Lv H, Zhang W, Que C, Huang Y, Yuan Y (2014) Clinical and GAA gene mutation analysis in mainland Chinese patients with late-onset Pompe disease: identifying c.2238G>C as the most common mutation. BMC Med Genet 15:141. doi:10.1186/s12881-014-0141-2
McCready ME, Carson NL, Chakraborty P, Clarke JT, Callahan JW, Skomorowski MA, Chan AK, Bamforth F, Casey R, Rupar CA, Geraghty MT (2007) Development of a clinical assay for detection of GAA mutations and characterization of the GAA mutation spectrum in a Canadian cohort of individuals with glycogen storage disease, type II. Mol Genet Metab 92(4):325–335. doi:10.1016/j.ymgme.2007.07.006
Chien YH, Lee NC, Huang HJ, Thurberg BL, Tsai FJ, Hwu WL (2011) Later-onset Pompe disease: early detection and early treatment initiation enabled by newborn screening. J Pediatr 158(6):1023–1027 e1021. doi:10.1016/j.jpeds.2010.11.053
Musumeci O, Thieme A, Claeys KG, Wenninger S, Kley RA, Kuhn M, Lukacs Z, Deschauer M, Gaeta M, Toscano A, Glaser D, Schoser B (2015) Homozygosity for the common GAA gene splice site mutation c.-32-13T>G in Pompe disease is associated with the classical adult phenotypical spectrum. Neuromuscul Disord 25(9):719–724. doi:10.1016/j.nmd.2015.07.002
Turaca LT, de Faria DO, Kyosen SO, Teixeira VD, Motta FL, Pessoa JG, Rodrigues ESM, de Almeida SS, D’Almeida V, Munoz Rojas MV, Martins AM, Pesquero JB (2015) Novel GAA mutations in patients with Pompe disease. Gene 561(1):124–131. doi:10.1016/j.gene.2015.02.023
Acknowledgements
The authors would like to thank Dr. İrsel Tezer for polysomnographic analysis of patient 4.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Nothing to declare.
Ethical standards
The study was approved by local ethics committee (GO 16/630).
Informed consent
The patients have given their informed consent.
Rights and permissions
About this article

Cite this article
Bekircan-Kurt, C.E., Güneş, H.N., Yildiz, F.G. et al. New mutations and genotype–phenotype correlation in late-onset Pompe patients. Acta Neurol Belg 117, 269–275 (2017). https://doi.org/10.1007/s13760-016-0738-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13760-016-0738-7