
Abstract
A combined headspace solid phase extraction-dispersive liquid–liquid microextraction method was developed for the extraction of residual solvents (dichloromethane, chloroform, methanol, ethanol, acetone, 2-propanol, 1-butanol, tetrahydrofuran, and pyridine) from herbal laxative medicines. The headspace solid phase extraction procedure was performed by a home-made device. For this purpose, the sample was placed into the extraction device and after dilution with sodium chloride solution, the analytes were forced to inter the headspace of samples and adsorbed onto the sorbent. After extraction, the analytes were eluted by a suitable solvent and more concentrated by a lighter than water organic solvent-based dispersive liquid–liquid microextraction. The enriched analytes were determined by gas chromatography-flame ionization detector. The validation parameters confirmed good sensitivity (limits of detection, 0.59–0.94 ng g−1) and repeatability (relative standard deviations, ≤ 3.9%), broad linear ranges and high extraction recovery (89–98%). The method was successful used in determination of the analytes in herbal laxative medicines and only methanol and dichloromethane were found in three samples.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Based on the literature, residual solvents (RSs) analysis in pharmaceutical products is a significant subject due to their potential hazard to human as they are toxic in many cases (Dwivedi, 2002; Grodowska and Parczewski, 2010). Usually, synthesis of drugs from medicinal herbs consists of several steps and different solvents are employed in each step. Some utilized solvents are not completely removed by the pragmatic manufacturing techniques and their residuals may be existed in the final product (Kolář et al. 2002; Nojavan et al. 2005; Ghaderi et al. 2016; Nemati et al. 2015). The presence of residue of solvents may affect the quality of final product like drug crystals, stability, wettability, odor, and taste. Therefore, quality control of pharmaceutical products is needed regarding to the amount and the type of the possible residual solvents (Feng, et al. 2016). Typically, RSs are divided into three major groups based on to their potential toxicity and maximum residue limits in the final products. Class 1 consists of the solvents that should be avoided in pharmaceutical preparations due to their high toxicity. Class 2 are the solvents that should be limited in final products and class 3 consists of the solvents with a relatively low toxicity (Tripartite harmonisedguideline Q3C Impurities, Residual Solvents, 1997).
Despite to the fact that RSs are volatile organic compounds, they can be determined by gas chromatography (GC) (Jószai et al. 2021), the complicated matrix of herbal medicines restricts the determination of RSs, directly (Angel Salatti-Dorado et al. 2019). On the other hand their low concentration in the samples requires performing an isolation and preconcentration prior to determination. Headspace sampling-based methods like headspace solid phase microextraction (HS-SPME) (Coran et al. 2001) and single drop microextraction (SDME) (Tamen and Vishnikin, 2021) can be good candidates to isolate volatile compounds. Although these methods are beneficial approaches for the analysis of RSs but they have several defects. In SPME, extraction of the analytes is done onto a fiber that is fragile and expensive. In many cases SPME fibers have carry over effect (Coran et al. 2001b; Legrand et al. 2003). HS-SDME is another method in which a single drop of an organic solvent is contacted with the headspace of sample solution and the analytes are dissolved in the drop (Yu et al. 2010; Mehravar et al. 2020). Classically, the drop is hanged from the tip of a needle and it is obvious that keeping the drop in the needle tip is not easy. To overcome these drawbacks several attempts have been done to use microextraction methods in the analysis of RSs (Farajzadeh et al. 2010). Dispersive liquid–liquid microextraction (DLLME) is one of the known sample preparation approaches that benefits from high extraction efficiency and low toxicity. This method is based on a ternary component solvent system in which a mixture of extraction and disperser solvents is rapidly injected into an aqueous sample containing analytes by a syringe. A cloudy solution (water/disperser solvent/extraction solvent) is formed. It has been used in the extraction of different compounds (Kalyniukova et al. 2022; Jing et al. 2022; Mohebbi et al. 2018; Sorouraddin et al. 2018). However this method is not suitable for direct analysis of solid samples and its combination with another step is advised.
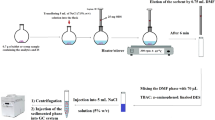
In the present study, development of a combined method of headspace solid phase extraction (HS-SPE) with DLLME was considered for isolation of RSs from herbal medicine tablets prior to their quantification by GC-flame ionization detector (FID). For performing the HS-SPE step a home-made extraction device was used in which a few amounts of a commercial sorbent were used. The device was designed so that the adsorbent was contacted with the headspace of the sample solution easily and effectively. Subsequently, the extracted analytes were concentrated by DLLME to reach low detection limits. The DLLME step was done by a lighter than water organic solvent due to its safeness compared to solvents with higher density than water.
Materials and methods
Material and solutions
All of the analytes including dichloromethane, chloroform, methanol, ethanol, acetone, 2-propanol, 1-butanol, tetrahydrofuran, and pyridine were purchased from Merck (Darmstadt, Germany). The compounds purity was extra-pure. Sodium chloride, sodium hydroxide, and HCl (37% w/v) were obtained from Dr. Mojallali Co. (Tehran, Iran). The used dispersive and extraction solvents and sorbents (n-octanol, n-decanol, dimethylformamide (DMF), octadecyl silane (ODS), primary secondary amine (PSA), amino, dimethyl sulfoxide (DMSO), and n-hexanol) were all purchased from Merck. For preparation of stock solution of the studied compounds, proper volumes of analytes were taken and dissolved in DMF at a concentration of 2000 mg L−1 (each analyte) and it was used for the preparation of daily working solutions.
Instrumentation
Chromatographic determination of the analytes was performed by an Agilent GC-FID (6850, CA, USA) instrument. The GC injection port was adjusted at 300 °C and it was worked in spilt mode at a ratio of 1:5. The detector fuel and oxidant were hydrogen (35 mL min−1) and oxygen (350 mL min−1), respectively, and its temperature was set at 300 °C. The analytes separation was done by an HP-1 capillary column (60 m × 0.53 mm i.d., and film thickness of 5.0 µm). The column oven temperature was initially set at 35 °C for 5 min and then raised to 250 °C at 8 °C min–1. Finally, the column temperature was maintained at 250 °C for 3 min. The GC mobile phase was helium (99.999%) at a flow rate of 1 mL min−1.
Real samples
The method ability in determination of the studied analytes was tested by analyzing five laxative medicines produced by different companies. They were bought from local pharmacies in Tabriz City (East Azarbaijan, Iran).
Extraction procedure
HS-SPE
This step was done in the home-made extraction device used in our pervious study (Afshar Mogaddam et al. 2022). For this purpose, 0.7 g of the powdered herbal medicine was poured into a vial and then spiked with the analytes (at a concentration of 1 mg kg−1, each analyte) and internal standard (IS) (ethyl acetate, 2 mg kg−1). Then, 5 mL sodium chloride solution (7.5%, w/v) was used. Then extraction vessel tube was filled with 25 mg ODS and placed on top of the sample solution. The device was placed on a heater/ stirrer for 6 min (300 rpm) at 65 °C. During this time, the analytes were interred into the sample solution headspace and then were adsorbed onto the ODS surface. In the following, the analytes were desorbed by 0.75 mL DMF after removing the tube from the vessel. Then, the total of the elution solvent was collected into a glass test tube and used in the following microextraction procedure as a disperser.
DLLME
The elution solvent obtained from the previous step was mixed with 140 µL n-octanol and the mixture was injected quickly into 2.5 mL NaCl solution (2.5%, w/v) placed into a 10-mL conical bottom glass test tube. The obtained mixture was centrifuged for collection of n-octanol on top of the solution at 5000 rpm for 3 min. Then, the aqueous phase was removed by a glass syringe and n-octanol was conducted to the conical section of glass test tube. Finally, 1 µL of the organic phase was injected into the GC-FID system.
Results and discussion
Study of HS-SPE step parameters
Influence of adsorbent type and amount
In this study, efficiency of the method is highly related to type of the adsorbent used in SPE step. The sorbent is placed on top of the solution, into the tube of the home-made device, and adsorbs the analytes from headspace of the sample solution. To achieve the best sorbent different commercial adsorbents including ODS, NH2, PSA, and activated carbon were tested at a constant amount. The experimental results showed that activated carbon could not be used in the extraction procedure due to the fact that it did not separated from elution solvent during elution of the analytes. The obtained signals for the analytes in the presence of the other sorbents are shown in Fig. 1A. The data confirm the usefulness of ODS compared to the other sorbents.

Selection of sorbent type A and amount B. A Extraction conditions: sample, 0.7 g powdered herbal medicine tablet spiked with the analytes at a concentration of 1.0 mg kg–1 (each analyte) and IS at a concentration of 2 mg kg–1; sorbent amount, 50 mg; stirring time (speed), 5 min (200 rpm); sample solution temperature, 75 °C; eluent type (volume), DMF (1.0 mL); and extraction solvent type (volume) in DLLME, n-hexanol (170 µL). The error bars indicate maximum and minimum of three experiments. B Extraction conditions: are the same as those used in Fig. 1A, except ODS was selected as the sorbent
After selection of the sorbent type, its amount should be optimized. It is apparent that low amounts of ODS has no enough sites to adsorb the analytes. However at high amount the sorbent, the analytes desorption cannot be occurred easily. In this context, the effect of ODS amount on the method efficiency was studied in the range of 25–150 mg and the analytical signals regarding peak area ratio were constant at this range. Lastly, 25 mg ODS was used in the extraction step.
Investigation of eluent solvent type and volume
The adsorbed analytes must be eluted by a proper organic solvent for more concentration in the following DLLME step. Elution of the analytes by a water-miscible solvent can be suitable as it can be used in DLLME as a dispersive solvent. To avoid interference of the disperser peak with the analytes peaks, high boiling point solvents including DMF and DMSO were tested as possible elution solvent at a fixed volume (0.25 mL). The data (Fig. 2A) obtained from the experimental tests show that DMF is more proper than DMSO and it was opted to use in the next tests.

The elution solvent volume may change the method efficacy during elution of the analytes from the adsorbent surface and their preconcentration in DLLME. The previously published methods emphasized that elution of the analytes from an adsorbent surface can be effectively performed by increasing the elution solvent volume. However when the elution solvent is used in DLLME as the dispersive solvent, high elution solvent volumes increase the analytes solubility in the aqueous phase used in DLLME step and this is not interest in this method. In this approach, a series of experiments was done by different volumes of DMF in the range of 0.25–1.50 mL. The data (Fig. 2B) show that 0.75 mL of DMF leads to higher analytical signals compared to the other used volumes and it was used in the following experiments.
Optimization of stirring time and speed
In this investigation, the analytes must be interred into the headspace of the sample solution to adsorb onto the sorbent surface. This phenomenon can be accelerated by stirring the sample solution via a magnetic stirrer. It is apparent that the stirring speed and time can affect the method efficacy. For this purpose, the method efficacy was tested by stirring the sample solution at different speeds (100–400 rpm) and the experimental results (Fig. 3A) depict that increasing the stirring speed has an enhancing effect on the method efficiency up to 300 rpm and it reaches to a constant value at higher speeds. So, 300 rpm was selected as the optimum stirring speed.

Investigation of stirring speed A and time B. A Extraction conditions: are the same as those used in Fig. 2B, except 0.75 mL of DMF was used as the elution solvent. B Extraction conditions: are the same as those used in Fig. 3A, except 300 rpm was selected as the optimum stirring speed
In the following, in order to find the optimum stirring time, different experiments were done by altering the stirring time in the range of 2–7 min and the results (Fig. 3B) show that the method efficiency is constant at the times ≥ 5 min and due to this outcome, 5 min was selected at the optimum stirring time.
Optimization of sample solution temperature
Enhancing the sample solution temperature may have a positive effect on the extraction of the analytes. Performing the method in an elevated temperature facilitates the analytes iterance into the sample solution headspace and accelerates their adsorption. Study of sample solution temperature effect was done by performing the method on the heated sample solutions in the range of 45–75 °C. The experimental results (Fig. 4) show that increasing the sample solution temperature enhances the method efficacy up to 65 °C and then reaches to a constant value. Thus, the future experiments were done by the solutions adjusted at 65 °C.

Optimization of sample solution temperature Extraction conditions: are the same as those used in Fig. 3B, except 5 min was selected as the stirring time.
Salt addition
In extraction methods, an agent which decreases the analytes solubility in the sample solution may increase the method efficiency. Salt addition is a facile strategy to follow this goal due to the fact that salt addition enhances the sample solution ionic strength and decreases the organic compounds solubility into aqueous phase. In this approach, different concentrations of NaCl were dissolved in the sample solution (0–10%, w/v) and after performing the method highest analytical signals were obtained in the presence of 7.5%, w/v, NaCl. Thus, 7.5%, w/v, NaCl was used in all experiments.
Investigation of effective parameters in DLLME step
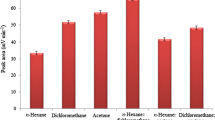
In DLLME, selection of extraction solvent is a crucial parameter that has a direct effect on the method efficacy. Physicochemical properties of the extraction solvent such as immiscibility with aqueous phase, density, melting point, viscosity, chromatographic behavior, and toxicity are the major factors that should be considered for solvent selection. Between these factors, toxicity of the solvent has attracted distinguished attentions in the recent years and DLLME approaches were done by safer solvent like deep eutectic solvents, ionic liquids, and organic solvents having lower density than water. Lower density than water organic solvents usage is most convenient than the other mentioned solvents due to their more accessibility, cheapness, and low viscosity. Because of these factors, this method was tested by those solvents and several experiments were done in the presence of n-octanol, n-hexanol, and n-decanol. The experiments were done at different volumes of each solvent to adjust the final collected organic phase volume at the same volume (10 ± 0.5 µL). Among the investigated solvents n-octanol was more appropriate than the other solvents and it was selected to use in the other tests (Fig. 5A).

Optimization of effective parameters in DLLME. A Extraction solvent selection Extraction conditions: are the same as those used in Fig. 4, except 65 °C and 7.5%, w/v, were selected as the optimum temperature of sample solution and NaCl solution concentration, respectively. B Ionic strength study. Extraction conditions: are the same as those used in Fig. 5A, except 140 µL n-octanol was selected as the extraction solvent.
In order to obtain high extraction recovery for the analytes, diverse volumes of n-octanol (140–230 µL) were subjected to the same procedure. It is notable that the final collected phase volume changed from 10 to 48 µL by altering the initial volume of n-octanol in the mentioned range. The data showed that the analytical signals decreased due to dilution of the analytes in the collected phase. Consequently, 140 µL was selected for the other tests.
Capability of salt addition in enhancing the DLLME step efficacy was studied by dispersing the dispersive and extraction solvents mixture into NaCl solutions at different concentrations (0–10%, w/v). From the results in Fig. 5B it can be concluded that salt addition has an increasing effect on the method efficacy at 2.5%, w/v, and it was selected for the other tests.
Method validation
Validation of the method was followed based on International Council Harmonization recommendations and the values are presented in Table 1. The plotted calibration curves were linear in wide ranges with a correlation coefficient ≥ 0.996. Calculation of limits of detection (LOD) and quantification (LOQ) were done considering \(\frac{{{\text{Signal}}}}{{{\text{Noise}}}}\) = of 3 and 10, and they were in the ranges of 0.59–0.94 and 1.9–3.1 ng g−1, respectively. Repeatability of the method in the day and different days was studied by calculation of relative standard deviations (RSDs) for repeated analysis at a concentration of 5 ng g−1. The RSD values were ≤ 3.2 and ≤ 3.9% for intra and inter-day precisions, respectively. To calculate the method extraction recovery (ER), the extraction procedure was done on a spiked sample solution and percent of the analytes migrated into n-octanol was calculated. The amount of analyte transferred into the organic phase was divided by its initial amount and multiplied to 100. The data showed that ERs were in the range of 89–98%.
Real samples analysis
The method under the optimized conditions was done on seven herbal tablets to confirm the method usability in determination of the analytes. The obtained chromatograms showed that dichloromethane was found in two samples at the concentrations of 13 ± 4 and 22 ± 4 ng g−1. Also methanol was found in one sample at a concentration of 10.00 ± 0.08 ng g−1. The rest of samples were free of the analytes. In the following the samples were spiked with the analytes at two concentrations of 20 and 60 ng g−1 (each analyte) and the method was performed them. The analyte of concentration was found in the samples after performing the method, divided by the initial concentrations and multiplied to 100. The obtained relative recoveries (Table 2) confirm the method accuracy and the absence of matrix effect. Typical GC-FID chromatogram of an unspiked samples after performing the method is shown in Fig. 6.

Typical GC-FID chromatogram of A standard solution of the analytes at a concentration of 25 mg L−1 (each analyte) B spiked sample # 3 (in Table 2) with the analytes at a concentration of 60 ng g−1 (each analyte), and C unspiked sample after performing the introduced method
Comparison of the method with other approaches
The proposed method from the views of various aspects such as RSD, LOD, LR, and ER was compared with the other methods reported in the literature. The data are summarized in Table 3. The LODs of the proposed method are better than the other methods. The proposed method has wide LRs. The RSDs% of the method are comparable with the other methods. The ER values for the method were comparable or better than with other approaches. The results show that the present method is a fast, easy, sensitive, and reliable analytical method and can be used to measure the analytes residues in herbal medicine samples.
Conclusions
An effective, accurate, and simple extraction procedure based on HS-SPE-DLLME has been introduced for the simultaneous determination of RSs in herbal medicines, and the extracted analytes were determined by GC-FID system. The HS-SPE step was done in a home-made extraction vessel which provided facile and efficient extraction of the analytes. After that the following DLLME was done by a lighter than water organic solvent due to its low toxicity. This extraction system has been employed for fast and impressive preconcentration of the analytes with high ERs. The results indicated that the suggested method had good repeatability, wide linear ranges, low LODs, and LOQs, and short analysis time. The obtained satisfactory results proved that this method can be as a suitable alternative to the previously reported methods.
Data availability
Not applicable.
Abbreviations
- RS :
-
Residual solvent
- GC :
-
Gas chromatography
- DLLME :
-
Dispersive liquid–liquid microextraction
- FID :
-
Flame ionization detector
- HS-SPE :
-
Headspace solid phase extraction
- RSD :
-
Relative standard deviation
- ER :
-
Enrichment recovery
- LOD :
-
Limit of detection
- LOQ :
-
Limit of quantification
References
Afshar Mogaddam MR, Altunay N, Tuzen M, Katin KP, Nemati M, Lotfipour F (2022) Headspace µ–solid phase extraction of 1,4–dioxane and 2–methyl–1,3–dioxolane from shampoo samples in a home–mode device and large volume injection of deep eutectic solvent: Theoretical and experimental studie. Microchem J 173:107040
Angel Salatti-Dorado J, Gonzalez-Rubio S, García-Gomez D, Lucena R, Cardenas S, Rubio S (2019) A high thermally stable oligomer-based supramolecular solvent for universal headspace gas chromatography: proof-of-principle determination of residual solvents in drugs. Anal Chim Acta 1046:132–139
Coran SA, Giannellini V, Furlanetto S, Bambagiotti-Alberti M, Pinzauti S (2001) Improving gas chromatographic determination of residual solvents in pharmaceuticals by the combined use of headspace solid-phase microextraction and isotopic dilution. J Chromatogr A 915:209–216
Dwivedi AM (2002) Residual solvent analysis in pharmaceuticals. Pharm Technol 26(11):42–46
Farajzadeh MA, Goushjuii L, Djozan D, Kompani Mohammadi J (2012) Dispersive liquid–liquid microextraction combined with gas chromatography for extraction and determination of class 1 residual solvents in pharmaceuticals. J Sep Sci 35:1027–1035
Farajzadeh MA, Dehghani H, Yadeghari A, Khoshmaram L (2017) Extraction and preconcentration of residual solvents in pharmaceuticals using dynamic headspace–liquid phase microextraction and their determination by gas chromatography–flame ionization detection. Biomed Chromatogr 31:e3788
Feng XZ, Han GC, Qin J, Yin S, Chen Z (2016) Determination of residual solvents in linezolid by static headspace GC. J Sep Sci 54(4):487–491
Ghaderi F, Nemati M, Siahi-Shadbad MR, Valizadeh H, Monajjemzadeh F (2016) DSC kinetic study of the incompatibility of doxepin with dextrose: application to pharmaceutical preformulation studies. J Therm Anal Calorim 123:2081–2209
Grodowska K, Parczewski A (2010) Organic solvents in the pharmaceutical industry. Acta Pol Pharm Res Drug 67:12–19
Heydari R (2012) Residual solvents determination in pharmaceuticals by static headspace-gas chromatography and headspace liquid phase microextraction gas chromatography. Anal Lett 45:1875–1884
Jing X, Huang X, Zhang Y, Wang M, Xue H, Wang X, Jia L (2022) Cyclodextrin-based dispersive liquid–liquid microextraction for the determination of fungicides in water, juice, and vinegar samples via HPLC. Food Chem 367:130664
Jószai I, Vékei N, Bajnai D, Kertész I (2021) Trencsényi G A generic gas chromatography method for determination of residual solvents in PET radiopharmaceuticals. J Pharm Biomed Anal 207:114425
Kalyniukova A, Tomášková I, Pešková V, Pastierovič F, Samek M, Balogh J (2022) Development of a novel dispersive liquid-liquid microextraction for the determination of ergosterol in roots and various fungi samples. Microchem J 174:107095
Kolář P, Shen JW, Tsuboi A, Ishikawa T (2002) Solvent selection for pharmaceuticals. Fluid Ph Equilibria 194:771–782
Legrand S, Dugay J, Vial J (2003) Use of solid-phase microextraction coupled with gas chromatography for the determination of residual solvents in pharmaceutical products. J Chromatogr A 999:195–201
Mehravar A, Feizbakhsh A, Mohsen Sarafi AH, Konoz E, Faraji H (2020) Deep eutectic solvent-based headspace single-drop microextraction of polycyclic aromatic hydrocarbons in aqueous samples. J Chromatogr A 1632:461618
Mohebbi A, Farajzadeh MA, Yaripour S, Mogaddam MRA (2018) Determination of tricyclic antidepressants in human urine samples by the three-step sample pretreatment followed by HPLC-UV analysis: an efficient analytical method for further pharmacokinetic and forensic studies. Excli J 17:952
Nemati M, Ghaderi F, Siahi-Shadbad MR, Valizadeh H, Monajjemzadeh F (2015) Physicochemical evaluation and non-isothermal kinetic study of the drug-excipient interaction between doxepin and lactose. Powder Technol 286:845–855
Nojavan S, Ghassempour A, Bashour Y, Darbandi MK, Ahmadi SH (2005) Determination of residual solvents and investigation of their effect on ampicillin trihydrate crystal structure. J Pharm Biomed Anal 36(5):983–988
Proceedings of international conference on harmonisation of technical requirements for registration of pharmaceuticals for human use (ICH), Tripartite harmonisedguideline Q3C impurities. Residual Solvents, 1997.
Sorouraddin SM, Farajzadeh MA, Hassanyani A, Afshar Mogaddam MR (2016) Combination of homogenous liquid–liquid extraction and dispersive liquid–liquid microextraction for extraction and preconcentration of amantadine from biological samples followed by its indirect determination by flame atomic absorption spectrometry. RSC Advances 6(110):108603–108610. https://doi.org/10.1039/C6RA16572A
Tamen AE, Vishnikin A (2021) In-vessel headspace liquid-phase microextraction. Anal Chim Acta 1172:338670
Yu Y, Chen B, Shen C, Cai Y, Xie M, Zhou W, Chen Y, Li Y, Duan G (2010) Multiple headspace single-drop microextraction coupled with gas chromatography for direct determination of residual solvents in solid drug product. J Chromatogr A 1217:5158–5164
Acknowledgements
The authors express their thanks to the Research Council of Tabriz University of Medical Science for financial support as grant number of 64800.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that there is has no conflict of interest.
Ethical approval
This article does not contain any studies with human or animals.
Informed consent
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Hassani Aliabad, A., Monajjemzadeh, F., Afshar Mogaddam, M. et al. Combination of headspace solid phase extraction with lighter than water organic solvent-based dispersive liquid-liquid microextraction for the extraction of residual solvents from herbal laxative medicine prior to gas chromatography-flame ionization detection. Chem. Pap. 76, 6451–6460 (2022). https://doi.org/10.1007/s11696-022-02332-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11696-022-02332-8