
Abstract
Nintedanib, a triple tyrosine kinase inhibitor of vascular endothelial growth factor receptor, platelet-derived growth factor receptor, and fibroblast growth factor receptor, has been used in idiopathic pulmonary fibrosis and adenocarcinoma in advanced non-small cell lung cancer. Although vascular endothelial growth factor inhibitors have been reported to cause endothelial injury and glomerular microangiopathy, nintedanib-induced glomerular microangiopathy has not been reported. A 68-year-old man with a history of primary aldosteronism, idiopathic pulmonary fibrosis, and pleomorphic carcinoma of the lung developed proteinuria and leg edema after nintedanib initiation. Kidney biopsy revealed prominent endothelial and mesangial injury. Proteinuria improved after nintedanib withdrawal. To the best of our knowledge, this is the second case report of nintedanib-induced glomerular microangiopathy. Although the incidence of nephropathy among patients receiving nintedanib is unknown at this moment, we recommend monitoring urinary protein excretion and blood pressure in patients receiving nintedanib and performing kidney biopsy to determine any histopathological change.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Inhibiting tumor angiogenesis mediated by vascular endothelial growth factor (VEGF) has become a main stream in current cancer treatment. With the expanding use of VEGF inhibitors, their nephrotoxicity causing endothelial injury and glomerular microangiopathy has been reported [1].
Nintedanib, a triple tyrosine kinase inhibitor of VEGF receptor, platelet-derived growth factor (PDGF) receptor, and fibroblast growth factor (FGF) receptor, has been used in idiopathic pulmonary fibrosis and adenocarcinoma in advanced non-small cell lung cancer, offering substantial benefit [2,3,4]. Although nintedanib also inhibits VEGF signaling pathway, there have been no reports on endothelial injury and glomerular microangiopathy [1].
We herein report a patient with pulmonary fibrosis who developed proteinuria due to nintedanib-induced glomerular microangiopathy, which has been rarely reported.
Case presentation
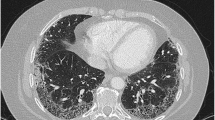
A 68-year-old man with a history of primary aldosteronism, idiopathic pulmonary fibrosis, and pleomorphic carcinoma of the lung was referred to our department because of severe proteinuria and leg edema. He had smoked 50 cigarettes per day for 48 years. He had undergone partial lung resection to remove a tumor 2 years prior to the referral and had been treated with nintedanib (300 mg/day) to halt the progression of pulmonary fibrosis for 10 months (Fig. 1). Medications, except nintedanib, included eplerenone (100 mg/day) and amlodipine (2.5 mg/day). Four months prior to the initiation of nintedanib, his serum creatinine was 0.78 mg/dL; furthermore, neither hematuria nor proteinuria was evident. One week after nintedanib initiation, his dipstick urine suddenly showed proteinuria of 2+ and hematuria of 1+, and he complained of nausea and diarrhea, which was controlled by symptomatic treatment. In addition, his blood pressure increased, so the dose of eplerenone had been titrated from 50 to 100 mg/day.

Clinical images of the present case. (A) Lung tumor on the right upper lobe was evident on chest computed tomography 2 years prior to the referral. (B) Pulmonary fibrosis is observed on bilateral lower part prior to nintedanib initiation
Upon referral, his blood pressure was 126/87 mmHg, heart rate was 86 beats/min, and SpO2 was 95% in room air. His body weight was 67 kg, having increased by 5 kg within 1 year.
The initial laboratory tests showed the presence of hypoalbuminemia (2.5 g/dL) and proteinuria (protein-to-creatinine ratio: 4.1 g/gCr), leading to the diagnosis of nephrotic syndrome. The patient’s laboratory results are shown in Table 1. The patient was hospitalized for kidney biopsy at 10 months after the onset of proteinuria. His kidney biopsy showed patchy tubular atrophy and interstitial enlargement, but no tubulitis was seen. Of the 21 glomeruli, one was globally sclerotic. Other glomeruli showed mild mesangial proliferation and widely expanded subendothelial area occupied by hyaline-like materials with some huge subendothelial deposition, some of which were positive on periodic acid-Schiff (PAS) staining, while others were negative. Focal mesangiolysis and double contour were also observed in some glomeruli (Fig. 2). Immunofluorescence staining showed only moderate IgM deposition in huge subendothelial depositions without any other positive staining. Electron microscopy showed electron-dense deposits (EDDs) in subendothelial and mesangial areas. Foot-process effacement was not noticeable. Histologic diagnosis was glomerular microangiopathy with subendothelial deposition. Since our patient’s proteinuria was observed only 1 week after nintedanib initiation, we highly suspected that his nephrotic syndrome was likely caused by nintedanib. Therefore, we withdrew nintedanib after the kidney biopsy and treated the nephrotic syndrome with furosemide (20 mg/day) and trichlormethiazide (1 mg/day). The respiratory physician also agreed to withdraw nintedanib and planned close monitoring for pulmonary symptoms. Proteinuria improved gradually, and 3 months after nintedanib withdrawal, the serum albumin increased to > 3 g/dL (3.4 g/dL). His edema and hypertension improved, and furosemide, trichlormethiazide, and amlodipine were discontinued, and the dose of eplerenone was reduced to 25 mg/day. At 4 months after nintedanib withdrawal, urine protein decreased to < 1.0 g/gCr (0.3 g/gCr). Hematuria also improved, and 1 month after nintedanib withdrawal, urine red blood cells were not found thereafter. During the 2-year follow-up, the patient’s urine protein remain unchanged, ranging from 0.3 to 0.9 g/gCr, although there have been no recurrences or nephrotic syndrome. Figure 3 summarizes the clinical course of the present case.

Histological findings of the kidney biopsy performed after the onset of proteinuria. (A) Mild mesangial proliferation and widely expanded subendothelial area occupied by hyaline-like materials with some huge subendothelial deposition (arrows). (B) Mesangiolysis (arrows) and double contour. (C) Blue-tinged subendothelial deposition (arrows) suggesting that the depositions are infiltrates of plasma. (D) Red-tinged subendothelial deposition (arrows) suggesting that the deposition is plasma itself. Electron microscopy finding showing unremarkable foot process effacement (E) and electron-dense deposits (arrows) in subendothelial and mesangial areas (F)

Clinical course of the present case regarding serum albumin and urine protein levels before and after nintedanib treatment. The period of nintedanib and timing of kidney biopsy are shown
Discussion
Although many cases of VEGF inhibitor-induced nephropathy have been reported, cases of nephropathy caused by nintedanib have been scarce [4]. A case of anti-glomerular basement membrane glomerulonephritis following nintedanib treatment has been the only reported kidney involvement associated with nintedanib [5]. To the best of our knowledge, this is the second case report of nintedanib-induced glomerular microangiopathy [6]. We confirmed that the proteinuria in the present case had been triggered by nintedanib, because (1) neither proteinuria nor hematuria had been identified by medical checkups performed before nintedanib treatment, (2) urinary abnormalities appeared immediately after nintedanib initiation, and (3) proteinuria improved and hematuria disappeared after nintedanib cessation.
Recently, cancer therapy has achieved marked advances, which are mainly attributed to the development of targeted therapy; furthermore, several types of nephropathy caused by those therapies have been reported as a complication of targeted therapy. The well-known complication of anti-VEGF drugs, such as bevacizumab, ranibizumab, sunitinib, and sorafenib, is endothelial injury which comprises hypertension, proteinuria, thrombotic microangiopathy (TMA), and renal TMA [7,8,9].
Nintedanib is a multi-targeted receptor tyrosine kinase inhibitor that works on key angiogenesis pathways including PDGF, VEGF, and basic FGF [5, 10]. VEGF signaling in the kidney plays a key role in endothelial cell proliferation, migration, and permeability [4]. VEGF is released from podocyte and binds to its receptor on glomerular endothelial cells [4]. VEGF signaling between the podocyte and glomerular endothelial cells is tightly regulated, and VEGF inhibition results in renal endothelial injury, manifested primarily as proteinuria, hypertension, renal-specific TMA, minimal change disease, and focal segmental glomerulosclerosis [4, 10].
The result of our patient’s kidney biopsy showed expansion of subendothelial area, mesangiolysis, and double contour, all of which were compatible with endothelial injury. There were no typical findings for TMA such as fibrin, platelet thrombi, and fragmented erythrocytes in the intracapillary space. Endothelial injury without typical TMA findings in our patient was similar to the morphological changes in other anti-VEGF therapy-induced glomerulopathy [11].
The atypical finding in the present case compared with other anti-VEGF-induced glomerulopathy case was huge EDDs in the subendothelial space, which was only IgM positive and involved mesangial areas. Since other immunoglobulins were negative, we thought that these EDDs were infiltrated from the plasma as a result of severe endothelial injury, not immune complex formation. The lack of glomerular sclerosis also excluded the possibility of IgM nephropathy. Although only one report showed the association of EDDs and anti-VEGF inhibitor in which massive paramesangial deposits were found during the treatment by bevacizumab, EDDs in that report were IgA positive [12]. The reason why these wide ranges of histological findings were seen in the present case remains unclear. One possible explanation is that nintedanib also inhibits PDGF. PDGF regulates proliferation, migration, and extracellular matrix production of mesangial cells [13], and in animal models, inhibition of PDGF increased the vulnerability of kidney to glomerular damage and mesangial insufficiency in the ensuing repair process [14]. PDGF plays a critical role in repairing glomerular damage by proliferating mesangial cells. Therefore, the mesangiolysis seen in our case might have been a result of ineffective repairing, which was caused by PDGF inhibition of nintedanib. Considering these effects of VEGF and PDGF on kidney, we hypothesize that both anti-VEGF and anti-PDGF effects have caused these multiple findings from endothelial to mesangial damage and led to proteinuria. In our case, foot-process effacement was unnoticeable, while the patient had massive proteinuria (4.1 g/gCr) and hypoalbuminemia (2.5 g/dL), which were consistent with the criteria for nephrotic syndrome. Since hyperlipidemia was not seen in our patient and there were no etiologies of primary nephrotic syndrome, we concluded that the unremarkable foot-process effacement in our patient was because his proteinuria was caused by secondary nephrotic syndrome due to glomerular microangiopathy.
In contrast to our case, the previous case of nintedanib-induced glomerulopathy which was reported by Inoue et al. presented proteinuria 3 years after nintedanib initiation, and the highest proteinuria level was less severe (1.7 g/gCr) than that of our case [6]. Histologically, there is similarity in that expansion of subendothelial area and double contour were seen in both cases. However, the expansion of subendothelial area seen in our case was more extensive than that in the previous case. The reason of this difference has yet to be known, but our patient had been a heavy smoker, which might have caused smoking-related glomerulopathy, leading to severer endothelial injury [15].
In cancer patients, there can be many causes of proteinuria, such as membranous nephropathy, minimal change disease, and cancer-related TMA, all of which are refractory until the cancer is treated. On the contrary, if the proteinuria is caused by anti-cancer drug-induced nephropathy, it might be easy to solve the urinary abnormality by withdrawing the drug or changing to a second-line regimen. In this regard, we recommend performing kidney biopsy to detect histological changes in the case of urinary abnormality, which can give us critical diagnostic information. A kidney biopsy is an invasive method, and we should consider the risk and benefit in terms of prognosis, complications, and existence of second-line regimen for cancer patients.
In conclusion, we described a patient with lung cancer and pulmonary fibrosis who developed proteinuria and leg edema after nintedanib initiation. Since we found the improvement of proteinuria after nintedanib cessation, we considered that the clinical course is compatible with nintedanib-induced glomerular microangiopathy, which, to our knowledge, has been reported only in one case [6]. Although the incidence of nintedanib-induced glomerular microangiopathy among patients receiving nintedanib might be uncommon, we still recommend monitoring proteinuria and kidney function in patients receiving nintedanib and considering to perform kidney biopsy in case of proteinuria or kidney dysfunction, which would be of use to elucidate the histopathological change caused by nintedanib.
References
Richeldi L, Costabel U, Selman M, Kim DS, Hansell DM, Nicholson AG, Brown KK, Flaherty KR, Noble PW, Raghu G, Brun M, Gupta A, Juhel N, Klüglich M, du Bois RM. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med. 2011;365:1079–87.
Richeldi L, du Bois RM, Raghu G, Azuma A, Brown K, Ulrich C, Cottn V, Klaherty K, Hansell D, Inouei Y, Kim DS, for the INPULSIS Trial Investigators, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2071–82.
Reck M, Kaiser R, Mellemgaard A, Douillard JY, Orlov S, Krzakowski M, von Pawel J, Gottfried M, Bondarenko I, Liao M, Gann CN, Barrueco J, Gaschler-Markefski B, LUME-Lung Study Group. Novello S Docetaxel plus nintedanib versus docetaxel plus placebo in patients with previously treated non-small-cell lung cancer (LUME-Lung 1): a phase 3, double-blind, randomised controlled trial. Lancet Oncol. 2014;15:143–55.
Estrada CC, Maldonado A, Mallipattu SK. Therapeutic inhibition of VEGF signaling and associated nephrotoxicities. J Am Soc Nephrol. 2019;30:187–200.
Ismail I, Nigam S, Parnham A, Srinivasa V. Anti-glomerular basement membrane glomerulonephritis following nintedanib for idiopathic pulmonary fibrosis: a case report. J Med Case Rep. 2017;11:214.
Inoue D, Nishi H, Honda K, Ishii T, Abe H, Sato M, Nangaku M. Renal thrombotic microantiopathy during nintedanib treatment for idiopathic pulmonary fibrosis. Clin Nephrol. 2020;93:47–50.
Wu S, Kim C, Baer L, Zhu X. Bevacizumab increases risk for severe proteinuria in cancer patients. J Am Soc Nephrol. 2010;21:1381–9.
Eremina V, Jefferson JA, Kowalewska J, Hochster H, Haas M, Weisstuch J, Richardson C, Kopp JB, Kabir MG, Backx PH, Gerber HP, Ferrara N, Barisoni L, Alpers CE, Quaggin SE. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med. 2008;358:1129–36.
Robinson ES, Matulonis UA, Ivy P, Berlin ST, Tyburski K, Penson RT, Humphreys BD. Rapid development of hypertension and proteinuria with cediranib, an oral vascular endothelial growth factor receptor inhibitor. Clin J Am Soc Nephrol. 2010;5:477–83.
Hilberg F, Roth GJ, Krssak M, Kautschitsch S, Sommergruber W, Tontsch-Grunt U, Garin-Chesa P, Bader G, Zoephel A, Quant J, Heckel A, Rettig WJ. BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res. 2008;68:4774–822.
Pfister F, Amann K, Daniel C, Klewer M, Büttner A, Büttner-Herold M. Characteristic morphological changes in anti-VEGF therapy-induced glomerular microangiopathy. Histopathology. 2018;73:990–1001.
Yahata M, Nakaya I, Sakuma T, Sato H, Aoki S, Soma J. Immunoglobulin A nephropathy with massive paramesangial deposits caused by anti-vascular endothelial growth factor therapy for metastatic rectal cancer: a case report and review of the literature. BMC Res Notes. 2013;6:450.
Floege J, Eitner F, Alpers CE. A new look at platelet-derived growth factor in renal disease. J Am Soc Nephrol. 2008;19:12–23.
Nakagawa T, Izumino K, Ishii Y, Oya T, Hamashima T, Jie S, Ishizawa S, Tomoda F, Fujimori T, Nabeshima Y, Inoue H, Sasahara M. Roles of PDGF receptor-beta in the structure and function of postnatal kidney glomerulus. Nephrol Dial Transpl. 2011;26:458–68.
Salvatore SP, Troxell ML, Hecox D, Sperling KR, Seshan SV. Smoking-related glomerulopathy: expanding the morphologic spectrum. Am J Nephrol. 2015;41:66–72.
Funding
None.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have declared that no conflict of interest exists.
Ethics approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
Informed consent was obtained from the patient in the case report.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Hasegawa, M., Uehara, A., Suzuki, T. et al. Nintedanib-induced glomerular microangiopathy: a case report. CEN Case Rep 9, 295–300 (2020). https://doi.org/10.1007/s13730-020-00474-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13730-020-00474-w