
Abstract
S-adenosyl-l-homocysteine hydrolase of Plasmodium falciparum (PfSAHH) has been reported as a potential drug target against malaria. A series of aristeromycin derivatives and analogs were designed and tested for inhibition of PfSAHH. 2-Fluoroaristeromycin has been reported as a potential inhibitor of PfSAHH. Here, we have performed the molecular dynamics simulation study of 2-Fluoroaristeromycin with PfSAHH with 15-ns simulation time to evaluate the dynamic perturbation of inhibitor in the binding site of PfSAHH in docked complex. This indicates that the complex structure of PfSAHH-2-Fluoroaristeromycin is stable after 10 ns of simulation. MD results indicate that Leu53, His54, Thr56, Glu58, Cys59, Asp134, Glu200, Lys230, Leu389, Leu392, Gly397, Hip398, Met403, and Phe407 are the key residues in the binding pocket of PfSAHH that interacts with the inhibitor 2-Fluoroaristeromycin. Earlier studies have reported Cys59 of PfSAHH as a selective residue to design potential and specific inhibitor of PfSAHH. Simulation study also indicates the role of Cys59 in binding interaction with inhibitor. The MD simulation of PfSAHH-2-Fluoroaristeromycin complex reveals the stable nature of docking interaction. The result provides a set of guidelines for the rational design of potential inhibitors of PfSAHH.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
S-Adenosyl-l-homocysteine hydrolase (SAHH) catalyzes the breakdown of S-Adenosyl-l-homocysteine (SAH), which arises after S-adenosylmethionine-dependent methylation, into adenosine and homocysteine. The SAHH enzyme activity is required for biological methylations and metabolic pathway of sulfur-containing amino acids. Because of the essential roles of SAHH for living cells, inhibitors of SAHH are expected to be antimicrobial drugs, especially for viruses and malaria parasite. The methylation of DNA, RNA, and proteins plays essential roles in epigenetic control, virus replication, and cell differentiation. In mammals, the S-adenosylmethionine-dependent methylation process is controlled by SAHH. SAHH hydrolyzes SAH to adenosine and homocysteine, and may be used as a potential drug target for malaria, cancer, and viral disease (Hudec et al. 2013). Plasmodium falciparum is responsible for the millions of death each year. There is a need to search drugs with novel modes of action due to the emergence of new strains of plasmodium that are resistant to the conventional drug therapy. The potential inhibitor of pfSAHH can be designed by the structural comparison of the active site of PfSAHH with HsSAHH. Previous studies have shown that active site residue Cys59 in PfSAHH, and Thr60 in HsSAHH play an important role in selective binding of inhibitor (Tanaka et al. 2013). SAHH has become a pharmacological target for inhibition, as its blockade can affect cellular methylation of proteins, phospholipids, DNA, RNA, and small molecules (Chiang 1998).
Loss of SAHH function can result in inhibition of cellular methyltransferase enzymes. It has been reported that acetylation of Lys (408) of SAHH results in perturbations in the C-terminal hydrogen bonding patterns, a region associated with NAD binding. These results suggest that increased acetylation of SAHH may influence cellular methylation (Wang et al. 2014). Nucleoside analogs results in selective inhibition of recombinant pfSAHH, by 2-position substituted adenosine analogs. Similar selectivity was reported in the growth inhibition assay of cultured cells (Nakanishi 2007). For drug designing, it is recommended to select target that is present in the parasite, but absent in humans. Even if the drug target is common in parasite and human, slight structural differences can be utilized to design selective inhibitor. PfSAHH has capability to utilize 3-deaza-adenosine (DZA) as an alternative substrate in contrast to the HsSAHH, it has a unique inability to substitute 3-deaza-(±) aristeromycin (DZAri) for adenosine (Bujnicki et al. 2003). Single substitution (Thr60-Cys59) between the HsSAHH and malarial PfSAHH results in differential interactions with the nucleoside analogs. This difference in the active site may be considered for the development of novel inhibitors of PfSAHH.
It was reported that 7-deaza-5′-noraristeromycin was designed to inhibit S-adenosyl-l-homocysteine hydrolase, but it has shown no effect on this enzyme (Seley et al. 1997). 5′-Noraristeromycin and its enantiomer have shown a wide range of antiviral effects (Das et al. 2002). Inhibitory activity of 2-Modified aristeromycin derivatives and their related analogs were evaluated against HsSAHH and PfSAHH. It was observed that 2-Fluoroaristeromycin causes potential inhibition of PfSAHH selectively (Ando et al. 2008a). 4′-Modified noraristeromycin (NAM) analogs, 4′-sulfo-, 4′-sulfamoy, 4′-azido, and 4′-amino-NAM, were also synthesized. The inhibitory activities of these analogs against PfSAHH and HsSAHH were investigated (Ando et al. 2008b). The inhibitory role of noraristeromycin derivatives possessing 2- or 8-position-modified adenine or 8-aza-7-deazaadenine was tested against PfSAHH and HsSAHH (Kitade et al. 2002). The oral administration of noraristeromycin could completely prevent the occurrence of experimental arthritis in collagen-induced arthritis. In addition, noraristeromycin has shown excellent pharmacokinetics in mice, and oral administration of noraristeromycin could suppress the production of TNFα (Ito et al. 2014).
Studies have shown that noraristeromycin and some curcumin derivatives interact with the largest cavity of pfSAHH, which is also site of binding for Nicotinamide-Adenine-Dinucleotide (NAD) and Adenosine (ADN). Molecular docking reveals that curcumin and its derivatives have higher binding affinity for pfSAHH than noraristeromycin. Both inhibitors curcumin and noraristeromycin share common residues for binding at the same site of pfSAHH (Singh et al. 2013). Ribavirin is a well-known antiviral drug. Ribavirin is structurally similar to AND, and it causes inactivation of HsSAHH and Trypanosoma cruzi SAHH (TcSAHH). Ribavirin may be used as a lead to designing selective inhibitor of PfSAHH and SAHH of other pathogens (Cai et al. 2007). In the past years, Analogs and derivatives of aristeromycin have been reported as potential inhibitor of PfSAHH, but no effective therapy has been developed yet. Meanwhile, consistent progress has been made in discovering more inhibitors and drug targets against malaria. However, there is still lack of potential drug in the market that can effectively use for the treatment of malaria.
2 Material and method
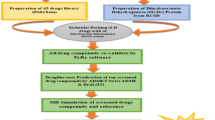
2.1 Docking
Molegro Virtual Docker (MVD) 2007.2.0.0 was used for docking study of PfSAHH with 2-Fluoroaristeromycin. MVD requires a 3-D structure of both protein and ligand. MVD performs flexible docking and the optimal geometry of ligand is determined during the docking of the ligand with protein (Thomsen and Christensen 2006). The candidates with the best conformational and energetic results were selected. MVD calculate the interaction energies between ligands and protein from the 3D structures of the protein and ligands. In the docking studies, flexibility of the protein has not been taken into consideration. Therefore, the MD simulation of docked complex with desmond program was performed to confirm binding mode of ligand in the binding cavity of PfSAHH enzyme.
Docking study of 2-Fluoroaristeromycin with PfSAHH was also performed using the AutoDock Tool (ADT v 1.5.6) (Morris et al. 2009). All water molecules were removed from the protein structure. The hydrogen is added to the structure of PfSAHH and partial atomic charges were assigned. A three-dimensional grid box (dimensions: 126 × 64 × 54 unit in number of grid points, grid spacing: 0.408 Å) centered at the ligand defining the search space was used. Thr54 and Asp134 residues of PfSAHH were defined as reference residue for docking as the reference residue. Twenty runs of Lamarckian Genetic Algorithm were set to optimize the ligand–protein interactions. The results were clustered according to the root-mean-standard deviation values, and ranked by the binding free energy. Five top poses were observed. Interactions of one best pose with PfSAHH were visualized by Maestro of Schrodinger.
2.2 Molecular dynamics simulation
Molecular dynamics (MD) simulations for docking complex were performed using Desmond 3.1 with 15-ns (nanoseconds) simulation time (Shaw 2008). The molecular dynamics of this system were studied using the force field OPLS_2005 (Kaminski and Friesner 2001). The initial coordinates for the MD calculations were taken from the docking experiments. Water molecules were placed to the PfSAHH-2-Fluoroaristeromycin docking complex with the simple point-charge (SPC) water model. The dimensions for each orthorhombic water box were set to 10 Å × 10 Å × 10 Å, which has covered the entire surface of PfSAHH-2-Fluoroaristeromycin complex by the solvent model, and Na+ counter ions were added in system for neutralization to balance the net charges of the systems. The full system of 60,615 atoms was simulated through the multistep MD protocols of Maestro v9.3.515. Maximum 2000 iterations were performed for full system minimization with restraints on solute. The docking complex was simulated for a simulation time of 15 ns with a time step of 2 fs. Trajectories after every 4.8 ps were recorded. The Shake algorithm was set for all hydrogen atoms and the cutoff for van der Waals (VDW) radius was 9 Å (Andersen 1983). The Nose–Hoover thermostat method was used to maintain the temperature of system at 300 K with a relaxation time of 1 ps. MD trajectory analyses and visualization of protein–ligand complex were also carried out (Bowers et al. 2006). The equilibrium phase was investigated by examining the stability of the temperature, energy, and the RMSD of the side chain, backbone atoms.
3 Results and discussion
3.1 Docking of 2-Fluoroaristeromycin with PfSAHH using MVD
Total six-known inhibitors of PfSAHH are reported in the literature with their IC50 values for both targets PfSAHH as well for HsSAHH. Ic50 value of 2-Fluoroaristeromycin (CHEMBL405186) for target PfSAHH and HsSAHH is 1980 and 47,200 nM, respectively. Among all known inhibitors, Ic50 values are in favor of 2-Fluroaristeromycin. Docking results have also indicated the high affinity of 2-Fluoroariteromycin for PfSAHH enzyme than HsSAHH. The 2-Fluoroariteromycin was docked with single chain (A) of PfSAHH (PDB: 1V8B). The largest cavity with volume (285.184 Å3) was specified as binding site during docking. Finally, top five poses of ligand were returned as the output of docking and the best pose (conformation) of 2-Fluoroaristeromycin that forms a low energy complex with PfSAHH was chosen for visualizing the mode of interaction. Best pose (00) of 2-Fluoroaristeromycin forms a low energy complex with a MolDock score of −145.29 kcal/mol. A very favorable H-bond score of −25.81 kcal/mol was contributed by Thr56, Glu58, Asp134, Glu200, Thr201, Lys230, Asp234, Thr396, and Gly397 residues of PfSAHH with 12 hydrogen bonds. Other than H-bonding residues, Leu53, His54, Met403, and Phe407 were also involved in the interaction. Docking energy, H-bond score, No. of H-bonds, and interacting residues for all five poses of 2-Fluoroaristeromycin are given in Table 1.
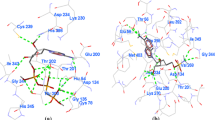
Second pose (01) of 2-Fluoroaristeromycin has shown a MolDock score of −140.66 and the H-bond score of −21.32 kcal/mol. The second pose has shown interaction with Leu53, His54, Thr56, Glu58, Asp134, Glu200, Thr201, Lys230, Asp234, Asn235, Thr396, Gly397, His398, and Phe407 residues. Among them, eleven residues Thr56, Glu58, Asp134, Glu200, Thr201, Lys230, Asp234, Asn235, Thr396, Gly397, and His398 form total 11 H-bonds with the inhibitor. It is interesting to note that second pose (01) has favored two additional residues Asn235 and His398 in H-bonding interaction, but still the total contribution in H-bonding has reduced. Binding mode, interacting residues, and H-bonding residues for the first two best poses of 2-Fluoroaristeromycin with PfSAHH have been shown in Fig. 1. For all the poses of 2-Fluoroaristeromycin, a similar set of residues were seen in interaction with a few variation in the H-bonding residues, total dock score, and H-bond score. The MolDock score for five different poses of 2-Fluoroaristeromycin varies from −145.29 to −129.26 kcal/mol. Similarly, the variation in the number of H-bond (12-8) and H-bond score from −25.81 to −19.52 kcal/mol was also observed. Stability of docked complex for the pose (00) of 2-Fluoroaristeromycin was further analyzed through the MD simulations using Desmond. MD simulations are accredited closer to the physiological environmental conditions, and it reveals better binding conformations for docked complex.

a Interaction of 2-Fluoroaristeromycin (00) with PfSAHH. b Interaction of 2-Fluoroaristeromycin (01) with PfSAHH, residues are labeled with its position and hydrogen bonding are shown by green dashed line (color figure online)
3.2 Docking of 2-Fluoroaristeromycin with PfSAHH using AutoDock Tool
Docking study of 2-Fluoroaristeromycin with PfSAHH was performed to explore the binding interaction of inhibitor. To compare the docking results and interaction, docking was performed using the MVD and AutoDock Tool. MVD ranks the poses on the basis of MolDock score, whereas AutoDock ranks the poses using binding free energy. Binding free energy for the best pose (pose1) of 2-Fluoroaristeromycin with PfSAHH was found of −5.89 kcal/mol. This pose1 interacts with Leu53, His54, Thr56, Glu58, Cys59, Cys78, Asp134, Glu200, Thr201, Asn225, Lys230, Asp234, Asn235, Leu389, Leu392, Thr396, Gly397, His398, Met403, Ser406, and Phe407 residues of PfSAHH (Fig. 2). Pose1 forms 8 hydrogen bonds with five residues Asp134, Thr201, Lys230, Thr396, and His398. Other poses have also shown interaction with the similar set of residues. Result indicates that all five poses have shown interaction nearly at the same binding site. Results obtained by AutoDock support the conclusion drawn from docking of 2-Fluoroaristeromycin with PfSAHH.

Interaction of 2-Fluoroaristeromycin (pose1) with PfSAHH. Coloring scheme used for representing residues and type of interaction is labeled
In MVD docking, top scoring pose of 2-Fluoroaristeromycin interacts with Leu53, His54, Thr56, Glu58, Asp134, Glu200, Thr201, Lys230, Asp234, Thr396, Gly397, Met403, and Phe407, and nine residues forms 12 hydrogen bonds with Thr56, Glu58, Asp134, Glu200, Thr201, Lys230, Asp234, Thr396, and Gly397. Here, AutoDock results indicate interaction with Leu53, His54, Thr56, Glu58, Cys59, Cys78, Asp134, Glu200, Thr201, Asn225, Lys230, Asp234, Asn235, Leu389, Leu392, Thr396, Gly397, His398, Met403, and Ser406, and six residues Asp134, Glu200, Thr201, Lys230, Thr396, and His398 involved in the formation of 7 hydrogen bonds (Table 2). Results by AutodDock show the interaction of few additional residues that are Cys59, Cys78, Asn225, Asn235, Leu389, Leu392, His398, and Ser406. Most of the residues were commonly reported in interaction by both MVD and AutoDock, and supports the binding interaction of 2-Fluoroaristeromycin with the same site of PfSAHH. For the best pose, docking by MVD indicates some additional residues Thr56, Glu58, Asp234, and Gly397 in hydrogen bonding. In the comparative docking analysis, a little variation in the number of hydrogen bonds and H-bonding residues were also observed. A few differences in the docking result might be due to separate algorithm and docking parameters used by both the tools. Results indicate that both the tools report the binding of 2-Fluoroaristeromycin at the same binding site of PfSAHH.
3.3 Molecular dynamics simulation of docking complex
The stability of PfSAHH-2-Fluoroaristeromycin complex was evaluated through 15-ns MD simulation. The simulations provide exact binding interaction of the docking complex with system embedded with water molecules, temperature, and pressure. In the MD simulations, solvating water molecules were considered for evaluating the stability of protein-inhibitor complex in contrast to the molecular docking approach. RMSD and energy fluctuation of the PfSAHH-2-Fluoroaristeromycin complex in each trajectory was calculated with respect to the simulation time. The backbone and side chain root-mean-square fluctuations (RMSF) of each residue were monitored for consistency. The inter-molecular interaction of 2-fluroaristeromycin-PfSAHH was assessed for the stability of the docking complex. The complex PfSAHH-2-Fluoroaristeromycin was originated in all proper binding poses, and different RMSDs for all residues, back bone, C-alpha, and side chain were measured. The acceptable RMSD value is (<2 Å). RMSD values of the protein all residues, C-alpha, side chain, and backbone atoms during the production phase relative to the starting structures were determined and plotted. To explore the dynamic stability of docking complex, RMSD from the starting structure was analyzed. The RMSD for back bone was in between 2.0 and 3.8 Å up to 10 ns. After 10-ns decrease in RMSD of back bone was recorded up to 0.8 Å (Fig. 3a). The mean RMSD for back bone atom was 2.651 Å with a standard deviation of 0.686 Å. Similarly, the mean of RMSD for side chain was 3.298 Å with a standard deviation of 0.678 Å (Fig. 3b). In starting phase, the RMSD for side chains was above 2.0 Å, but after a simulation time of 12 ns, it decreases gradually and lies between 0.8 and 1.5 Å. This indicates that at starting phase of simulation, high perturbations were seen in side chains, but after the simulation time of 12 ns, the fluctuations in side chains were minimized and the system gets stabilized. The mean of RMSD for all residues was 3.072 Å with a standard deviation of 0.681 Å (Supplementary Fig. 1a). Initially, the RMSD for all residues was in between 2.5 and 4 Å, but after a simulation time of 10 ns, it decreases gradually and lies between 1 and 2 Å. The mean of RMSD for C-alpha was 2.613 Å with a standard deviation of 0.678 Å (Supplementary Fig. 1b). Here also, initially, the RMSD for C-alpha was above 2.0 Å, but after a simulation time of 10 ns, it decreases gradually and reaches to 0.8 Å.

The result of MD simulation for PfSAHH-2-Fluoroaristeromycin complex. (a) RMSD (Å) vs. time (ps) of the backbone atoms. (b) RMSD (Å) vs. time (ps) of the side chain atoms
The RMSD for all residues, back bone, side chain, and C-alpha was initially high, but after a simulation time of nearly 10 ns, the RMSD values decreases and reaches to a 0.8 Å. Simulation pattern from RMSD indicates that initially the 2-Fluoroaristeromycin ligand was unstable in the cavity of PfSAHH, but later on it gets stabilized in the binding site of PfSAHH and resulted in a stable complex. The RMSF values of all residues, backbone C-alpha, and side chain atoms were assessed for each residue of PfSAHH. 90 % of backbone residue fluctuations were within the range of 1.0–3.0 Å which is acceptable up to a certain limit (Fig. 4a). Fluctuations within 1–3 Å are acceptable for small proteins (Carugo and Pongor 2001; Carugo 2007). More fluctuations in side chain atoms of each residue were seen as compared to backbone, all residue, and C-alpha fluctuations (Fig. 4b). All residue fluctuations of 80 % of residues were observed within the range of 1.5–3.0 Å; a very high fluctuation of 8 Å and lower fluctuation up to 0.8 Å were also observed for few residues (Supplementary Fig. 2a). A similar pattern of fluctuations were seen for C-alpha atoms of each residue of PfSAHH (Supplementary Fig. 2b). Residues participating in binding site and involved in docking (Leu53, His54, Thr56, Glu58, Asp134, Glu200, Thr201, Lys230, Asp234, Asn235, Thr396, Gly397, His398, and Phe407) or lying nearby region has shown comparatively lower all residues, backbone, C-alpha, and side chain fluctuations than other residues.

The result of MD simulation for PfSAHH-2-Fluoroaristeromycin complex. (a) RMSF (Å) of the backbone atoms for each residue. (b) RMSF (Å) of the side chain atoms for each residue. (c) Energy peak (E_P) of the docked complex along with pressure (P), temperature (T), and volume (V) of the system during simulation time (15 ns)
The RMSF data have shown that high fluctuations occur during binding of the inhibitor 2-fluroroaristermycin with enzyme PfSAHH. It can be also concluded here that binding of the inhibitor causes complete disruption of the original shape of the cavity, thus accomplishing the goal of complete inhibition of enzyme activity. Maximum fluctuations of 7.5 Å for c-alpha and backbone atoms were seen at the c-terminal region from position 465–470. Maximum side chain fluctuations of 8.5 Å were observed in the same region. This broad range of RMSDs and RMSFs reflects major structural rearrangement in the docking complex during simulation time. The energy plot showed that the energy of the system was consistent during 15-ns MD simulations run with a major change in the energy of the system at initial state (Fig. 4c). Initially, the energy of the system was a high but just after few picoseconds (ps) simulation energy gets minimized. It is important to notice that local minima and local maxima energy stages were also seen, but the energy of the docked system was consistent globally during 15-ns simulation.
The binding site of PfSAHH consists of six hydrophobic (Leu53, Cys59, Leu392, Leu389, Met403, and Phe407), three negatively charged (Glu58, Asp134 and Glu200), two polar (Hie54 and Thr56), two positively charged (Lys230 and Hip398), and Gly397. The PfSAHH-2-Fluoroaristeromycin complex has shown six H-bond interaction, three π-π stacking, and four water bridges. The backbone atom of Hip398 forms a hydrogen bond with the nitrogen of 2-Fluoroaristeromycin. The side chain atom of Thr56, Asp134, and Glu200 contributes one, one, and two hydrogen bonds, respectively. The Asp134 also contributes one more hydrogen bond through water bridges. The aromatic residues Hie54 and Phe407 participate in π-π stacking interaction with the fluorinated aromatic ring. The residue Hie54 has shown one additional π-π stacking interaction with another aromatic ring. Thus, both aromatic rings have shown an important role in binding, due to its ability to participate in π-π stacking interactions and H-bonding. It is interesting to note that these two rings of 2-Fluoroaristeromycin are packed by or near to major residues of binding site. Four water bridges, three with oxygen atom, and one with nitrogen atom were also observed in PfSAHH-2-Fluoroaristeromycin complex.
Docking and MD simulation studies reveal that the binding of 2-Fluoroaristeromycin with PfSAHH is stable. MVD docking indicates that the Leu53, His54, Thr56, Glu58, Asp134, Glu200, Thr201, Lys230, Asp234, Thr396, Gly397, Met403, and Phe407 residues of PfSAHH are involved in interaction with inhibitor 2-Fluoroaristeromycin. AutoDock has shown the involvement of Leu53, His54, Thr56, Glu58, Cys59, Cys78, Asp134, Glu200, Thr201, Asn225, Lys230, Asp234, Asn235, Leu389, Leu392, Thr396, Gly397, His398, Met403, Ser406, and Phe407 in binding interaction. MD simulation results reveal the interaction of 2-Fluoroaristeromycin with Leu53, His54, Thr56, Glu58, Cys59, Asp134, Glu200, Lys230, Leu389, Leu392, Gly397, His398, Met403, and Phe407 of PfSAHH (Fig. 5). Docking and simulation results indicate the same conclusion about the binding interaction of 2-Fluoroaristeromycin with PfSAHH. Interaction result of 2-Fluoroaristeromycin obtained by AutoDock is more closer to the set of residues observed in interaction by simulation study. Residues Cys59, Leu389, Leu392, and His398 that were observed in interaction with simulation study, were also predicted by AutoDock. It is important to note that earlier experimental studies have reported Cys59 of PfSAHH as a selective residue to design potential and specific inhibitor of PfSAHH. The last trajectory of the MD simulation indicates interaction of 2-Fluoroaristeromycin with a similar set of residues. The simulation results support the stability and docked binding site information obtained from docking study. Compared with molecular docking results, MD simulation results of inhibitor 2-Fluoroariteromycin have shown a similar binding mode, and also validate the reliability of active conformations obtained by MVD. This study reveals that inhibitor 2-Fluoroaristeromycin binds with the cavity of PfSAHH by displacing the position and orientation of binding residues, and finally inhibits the enzymatic activity of PfSAHH.

PfSAHH-2-Fluoroaristeromycin complex in the last trajectory of 15-ns MD simulation showing different types of interactions
4 Conclusion
The MD simulation study reveals that the PfSAHH-2-Fluoroaristeromycin complex shows the good stability in the physiological environmental conditions. Finally, the docked result indicates better binding orientations of 2-Fluoroaristeromycin with PfSAHH. RMSD, RMSF, and potential energy data from the MD simulation of docked complex is in favor of good stability of 2-Fluroaristeromycin in binding cavity of PfSAHH. RMSD plot revealed that the complex was relatively less stable during starting phase, but gets stabilized after 10 ns simulation. Therefore, 2-Fluroaristeromycin can be used as a lead molecule for searching more potential and stable inhibitors based on pharmacophore.
References
Andersen HC (1983) Rattle: a “velocity” version of the shake algorithm for molecular dynamics calculations. J Comput Phys 52(1):24–34
Ando T, Iwata M, Zulfiqar F, Miyamoto T, Nakanishi M, Kitade Y (2008a) Synthesis of 2-modified aristeromycins and their analogs as potent inhibitors against Plasmodium falciparum S-adenosyl-l-homocysteine hydrolase. Bioorg Med Chem 16(7):3809–3815
Ando T, Kojima K, Chahota P, Kozaki A, Milind ND, Kitade Y (2008b) Synthesis of 4′-modified noraristeromycins to clarify the effect of the 4′-hydroxyl groups for inhibitory activity against S-adenosyl-l-homocysteine hydrolase. Bioorg Med Chem Lett 18(8):2615–2618
Bowers KJ, Chow E, Xu H, Dror R O, Eastwood M P, Gregerson BA, Klepeis JL, Kolossvary I, Moraes M A, Sacerdoti FD, Salmon JK, Shan Y, Shaw D E (2006) Scalable algorithms for molecular dynamics simulations on commodity clusters. In: Proc. ACM/IEEE conf. on supercomputing (Tampa, FL, 2006)
Bujnicki JM, Prigge ST, Caridha D, Chiang PK (2003) Structure, evolution, and inhibitor interaction of S-adenosyl-l-homocysteine hydrolase from Plasmodium falciparum. Proteins 52(4):624–632
Cai S, Li QS, Borchardt RT, Kuczera K, Schowen RL (2007) The antiviral drug ribavirin is a selective inhibitor of S-adenosyl-l-homocysteine hydrolase from Trypanosoma cruzi. Bioorg Med Chem 15(23):7281–7287
Carugo O (2007) Statistical validation of the root-mean-square-distance, a measure of protein structural proximity. Protein Eng Des Sel 20(1):33–37
Carugo O, Pongor S (2001) A normalized root-mean-square distance for comparing protein three-dimensional structures. Protein Sci 10(7):1470–1473
Chiang PK (1998) Biological effects of inhibitors of S-adenosylhomocysteine hydrolase. Pharmacol Ther 77(2):115–134
Das SR, Schneller SW, Balzarini J, De Clercq E (2002) A mercapto analogue of 5′-noraristeromycin. Bioorg Med Chem 10(2):457–460
Hudec R, Hamada K, Mikoshiba K (2013) A fluorescence-based assay for the measurement of S-adenosylhomocysteine hydrolase activity in biological samples. Anal Biochem 433(2):95–101
Ito M, Hamano T, Komatsu T, Asamitsu K, Yamakawa T, Okamoto T (2014) A novel IKKα inhibitor, noraristeromycin, blocks the chronic inflammation associated with collagen-induced arthritis in mice. Mod Rheumatol 24(5):775–780
Kaminski GA, Friesner RA (2001) Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J Phys Chem 105:6474–6487
Kitade Y, Kozaki A, Miwa T, Nakanishi M (2002) Synthesis of base-modified noraristeromycin derivatives and their inhibitory activity against human and Plasmodium falciparum recombinant S-adenosyl-l-homocysteine hydrolase. Tetrahedron 58:1271–1277
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 30(16):2785–2791
Nakanishi M (2007) S-adenosyl-l-homocysteine hydrolase as an attractive target for antimicrobial drugs. Yakugaku Zasshi 127(6):977–982
Seley KL, Schneller SW, Rattendi D, Bacchi CJ (1997) (+)-7-Deaza-5′-noraristeromycin as an anti-trypanosomal agent. J Med Chem 40(4):622–624
Shaw DE (2008) Research desmond molecular dynamics system, version 3.1. New York
Singh DB, Gupta MK, Singh DV, Singh SK, Misra K (2013) Docking and in silico ADMET studies of noraristeromycin, curcumin and its derivatives with Plasmodium falciparum SAH hydrolase: a molecular drug target against malaria. Interdiscip Sci 5(1):1–12
Tanaka N, Umeda T, Kusakabe Y, Nakanishi M, Kitade Y, Nakamura KT (2013) Structural biology for developing antimalarial compounds. Yakugaku Zasshi 133(5):527–537
Thomsen R, Christensen MH (2006) MolDock: a new technique for high-accuracy molecular docking. J Med Chem 49:3315–3321
Wang Y, Kavran JM, Chen Z, Karukurichi KR, Leahy DJ, Cole PA (2014) Regulation of S-adenosylhomocysteine hydrolase by lysine acetylation. J Biol Chem 289(45):31361–31372
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Singh, D.B., Dwivedi, S. Docking and molecular dynamics simulation study of inhibitor 2-Fluoroaristeromycin with anti-malarial drug target PfSAHH. Netw Model Anal Health Inform Bioinforma 5, 16 (2016). https://doi.org/10.1007/s13721-016-0124-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13721-016-0124-7