
Abstract
Giant cell tumor of bone (GCTB) is a locally aggressive and rarely metastasizing tumor. GCTB is characterized by the presence of unique giant cells and a recurrent mutation in the histone tail of the histone variant H3.3, which is encoded by H3F3A on chromosome 1. GCTB accounts for ~ 5% of primary bone tumors. Although GCTB exhibits an indolent course, it has the potential to develop aggressive behaviors associated with local recurrence and distant metastasis. Currently, complete surgical resection is the only curative treatment, and novel therapeutic strategies are required. Patient-derived cancer cell lines are critical tools for basic and pre-clinical research. However, only a few GCTB cell lines have been reported, and none of them are available from public cell banks. Therefore, we aimed to establish novel GCTB cell lines in the present study. Using curetted tumor tissues of GCTB, we established two cell lines and named them NCC-GCTB2-C1 and NCC-GCTB3-C1. These cells harbored a typical mutation in histones and exhibited slow but constant growth, formed spheroids, and had invasive capabilities. We demonstrated the utility of these cell lines for high-throughput drug screening using 214 anticancer agents. We concluded that NCC-GCTB2-C1 and NCC-GCTB3-C1 cell lines were useful for the in vitro study of GCTB.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Giant cell tumor of bone (GCTB) is a locally aggressive and rarely metastasizing tumor. GCTB is histologically characterized by the presence of reactive multi-nuclear osteoclast-like giant cells and neoplastic mononuclear stromal cells [1]. It is genetically characterized by a mutation in the histone tail of the histone variant H3.3, which is encoded by H3F3A on chromosome 1, leading to G34W substitutions [2,3,4]. GCTB accounts for ~ 5% of all primary bone tumors [5, 6] and occurs in young adults between the ages of 20 and 40 years [7]. GCTB has a slightly higher prevalence in females than in males, and certain studies have reported a female-to-male ratio of 1.3–1.5:1.0 [8, 9]. The standard treatment for GCTB consists of surgical tumor removal, which includes extensive curettage with or without local adjuvants for en bloc resection and amputation [10]. GCTB has the potential to develop aggressive behaviors; local recurrence is often observed, ranging from 27 to 65% for isolated curettage, 12–27% for curettage with local adjuvants, and 0–12% for en bloc resection [10,11,12,13,14]. Moreover, pulmonary metastases occur in 2.1–6.6% of patients with advanced or recurrent GCTB [15,16,17]. The receptor activator of the NF-κB ligand (RANKL) pathway plays a key role in the pathogenesis of GCTB [18, 19], and a human monoclonal antibody against RANKL, denosumab, inhibits the activation and differentiation of osteoclast-like giant cells, causing osteolytic damage [20, 21]. The US Food and Drug Administration and European Medicines Agency approved denosumab in 2013 and 2014, respectively, as a neoadjuvant drug therapy for advanced GCTB, based on the results of two phase II trials [22, 23]. Preoperative denosumab treatment with curettage may be a risk factor for recurrence of GCTB [24,25,26]. Although radiotherapy used to be recommended when complete excision or curettage is impractical, a close association of secondary sarcomatous transformation in the region of GCTB following radiotherapy were reported [27], and its application is restricted to the case where the treatments with RANKL inhibitor is difficult for a long term. Altogether, the most appropriate use for denosumab is currently the subject of discussion [28], and the development of novel therapeutic strategies based on the understating of molecular biology of features of GCTB has been required.
Patient-derived cancer cells, which recapitulate the genotype and phenotype of original tumor cells, have considerable potential to accelerate the understanding of mechanisms underlying the etiology and progression of diseases and have contributed to the progress of cancer research since its inception [29]. These cell lines have enabled the study of the functional significance of genetic mutations, and recurrent mutations in the histone tail of the histone variant H3.3, encoded by H3F3A, have been addressed using GCTB cell lines [30, 31]. The cell lines are also useful for screening the antitumor effects of compounds toward the novel discovery of oncology drugs. Additionally, the integration of drug response and genetic data has led to the discovery of predictive biomarkers in multiple cancers [32,33,34,35,36,37,38,39,40]. However, due to the scarcity of patients with GCTB, only 11 cell lines have been established according to Cellosaurus [41]. GCTB is a clinically complex disease, and additional cell lines are required to develop novel therapies for this disease.
Here, we report two novel GCTB cell lines: NCC-GCTB2-C1 and NCC-GCTB3-C1. These cell lines were established from curetted tumor tissues from two patients with GCTB. To demonstrate the utility of these cell lines, we characterized their proliferation, spheroid formation, and invasion capabilities. Additionally, to demonstrated the utility of these cell lines in drug screening, we examined the anti-proliferative effects of anticancer agents.
Materials and methods
Patient history
The ethical committee of the National Cancer Center approved the use of clinical materials for this study, and written informed consent was obtained from the patient donors in this study.
Case 1
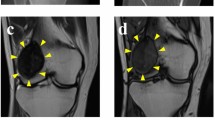
The patient was a 70-year-old man with GCTB. The patient complained of discomfort in the left knee and had visited another hospital previously. X-ray, computed tomography (CT), and magnetic resonance imaging (MRI) detected the bone tumor at the proximal tibia (Fig. 1A–D); thus, the patient was referred to the National Cancer Center Hospital (NCC, Tokyo, Japan). Based on needle biopsy results, GCTB was diagnosed. As there was no evidence of metastasis, the tumor was curetted. A part of the curetted tumor was used to establish the cell line described in this study. The tumor showed conventional GCTB histology, including uniform, non-atypical short spindle cell proliferation, and evenly distributed osteoclast-like giant cells. There was no evidence of malignancy (Fig. 1E). Together with the immunostaining findings that were positive for anti-histone H3.3 G34W in the needle biopsy specimen (Fig. 1F), the tumor was confirmed to be GCTB. 2 years after surgery, there was no evidence of local recurrence or distant metastasis.

Clinical and pathological data for case 1. X-ray and computed tomography show an osteolytic and expansive lesion in the proximal tibia (A, B). Magnetic resonance imaging indicates the mass that showed low intensity in the T1-weighted image (C) and a mix of low and high intensity in the T2-weighted image (D). HE staining shows the typical appearance of giant cell tumor of bone (GCTB) (e). The tumor cells were diffusely positive for anti-histone H3.3 G34W (F)
Case 2
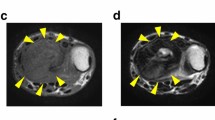
The patient was a 20-year-old woman with GCTB. The patient complained of pain in the left knee and had visited another hospital previously. X-ray and CT scans showed a bone tumor at the proximal tibia (Fig. 2A, B); thus, the patient was referred to the NCC. MRI detected a bone tumor, and its contrast was enhanced using gadolinium (Fig. 2C, D). During open biopsy, the bone tumor was identified as GCTB. As there was no evidence of metastasis, the tumor was curetted. A part of the curetted tumor was used to establish the cell line described in this study. Histologically, the curetted specimen largely showed necrosis, with only a small amount having viable fibroxanthomatous tissue. There was no evidence of malignancy in this specimen or in a previous biopsy specimen that showed conventional GCTB histology (Fig. 2E). Together with the immunostaining findings that were positive for anti-histone H3.3 G34W in the open biopsy specimen (Fig. 2F), the tumor was confirmed as GCTB. 1 year after surgery, there was no evidence of local recurrence or distant metastasis.

Clinical and pathological data of case 2. X-ray and computed tomography show an osteolytic and expansive lesion in the proximal tibia (A, B). Magnetic resonance imaging indicates a homogeneous mass with low intensity in the T1-weighted image (C), which was enhanced by the use of gadolinium (D). HE staining shows predominantly fibrohistiocytic tissue, compatible with GCTB with secondary change (E). The tumor cells were diffusely positive for anti-histone H3.3 G34W (F)
Histological analysis
Histological examination was performed on 4 μm-thick sections from a representative paraffin-embedded tumor sample. Sections were deparaffinized and stained with hematoxylin and eosin (HE).
Immunohistochemistry
Immunohistochemistry was performed using deparaffinized tumor samples. The slides were exposed to 3% hydrogen peroxide for 20 min to block endogenous peroxidase activity. Preparations were pretreated to obtain heat-induced epitope retrieval. The primary antibody used was anti-histone H3.3 G34W (RM263, dilution 1:1000, RevMab Bioscience, South San Francisco, CA, USA). The slides were incubated for 1 h at room temperature and subsequently labeled with the Envision system (Dako).
Cell culture preparation
Primary cells were established using curetted tumor tissues obtained at the time of surgery [42]. In brief, the tumor tissue was dissected into small pieces using scissors and incubated with 1 mg/mL collagenase type II (Worthington Biochemical Corporation, Lakewood, NJ, USA). The status of cell proliferation was monitored via microscopy, and when the cells reached sub-confluence, they were washed with PBS (-) (Gibco, Grand Island, NY, USA) and treated with 0.25% Trypsin–EDTA solution. The cells were then detached and transferred to a tissue culture plate. The cells were maintained in DMEM/F12 supplemented with GlutaMAX (both Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Gibco), 100 U penicillin G, and 100 µg/mL streptomycin (Gibco) at 37 °C in a humidified atmosphere with 5% CO2.
Authentication and quality control of the established cell line
Authentication and quality control of the established cell lines was performed as reported previously [42]. In brief, DNA from the tumor tissue and established cell lines was extracted using AllPrep DNA/RNA Mini kits (Qiagen, Hilden, Germany). Short tandem repeat (STR) analysis was performed for 10 loci using the GenePrint 10 system (Promega, Madison, WI, USA) and a 3500xL Genetic Analyzer (Thermo Fisher Scientific, Waltham, MA, USA). The STR pattern was analyzed using the GeneMapper software (Thermo Fisher Scientific) and matched to the data in the public cell banks using a function of Cellosaurus [41] with a standard match threshold of 80% [43]. Mycoplasma contamination was examined via the DNA fragmentation of mycoplasma using the e-Myco Mycoplasma PCR Detection Kit (Intron Biotechnology, Gyeonggi-do, Korea). The amplified DNA fragments were separated by agarose gel electrophoresis and stained with Midori Green Advanced Stain (Nippon Genetics, Tokyo, Japan).
Mutation analysis
Genomic DNA was extracted from tumor tissue or cultured cells using the DNeasy Blood and Tissue kit (Qiagen, Venlo, Netherlands). Genomic DNA (50 ng) was used for PCR amplification of H3F3A, performed with the forward primer, H3F3A_F (5′-TAAAGCACCCAGGAAGCAAC-3′), and reverse primer, H3F3A_R (5′-CAAGAGAGACTTTGTCCCATTTTT-3′), using KOD-Plus-Neo DNA polymerase (TOYOBO, Osaka, JAPAN). The PCR cycling conditions were: hot-start denaturation at 95 °C for 5 min, followed by 35 cycles of denaturation at 95 °C for 30 s, annealing at 55 °C for 30 s, polymerization at 72 °C for 45 s, and a final incubation for 10 min at 72 °C. The PCR products were purified with ExoSAP-IT (Affymetrix, Santa Clara, CA) and direct sequencing was performed using a BigDye v3.1 Cycle Sequencing Kit (Applied Biosystems) on the Applied Biosystems 3130xL instrument using the GENEWIZ platform (South Plainfield, NJ, USA).
Single nucleotide polymorphism (SNP) array
Chromosomal aberrations were examined using SNP array genotyping, which was conducted using the Infinium OmniExpressExome-8 v1.4 BeadChip (Illumina, San Diego, CA, USA) following the manufacturer’s instructions. Genomic DNA was extracted from tumor cells and normal white blood cells and amplified. The amplified DNA was reacted with the array slides in an iScan system (Illumina). Log R ratios and B allele frequencies were calculated using Genome Studio 2011.1 + cnvPartition v3.2.0 (Illumina) and KaryoStudio Data Analysis Software version 1.0 (Illumina), respectively. Log R ratios and B allele frequencies indicate the normalized signal intensity and normalized ratio of the B allele to the total of both A and B alleles, respectively. Copy number analysis was performed using ASCAT (version 2.1), taking into account non-neoplastic cell infiltration and tumor aneuploidy, and its results showed integral allele-specific copy number profiles for the tumor cells. Amplification was defined as copy number ≥ 5 (for diploid tumors, with ASCAT ploidy, 2.7n) or ≥ 9 (for tumors with evidence of whole-genome duplication, with ASCAT ploidy ≥ 2.7n). Homozygous deletions were considered if there were zero copies in the tumor cells.
Real-time cell analyzer (RTCA) growth assay
The RTCA (xCELLigence, Agilent Technologies, Santa Clara, CA, USA) instrument was used to assess the potential for proliferation of the established cells according to the manufacturer’s instructions. Briefly, background measurements were obtained from the wells by adding 100 μL of DMEM/F12 supplemented with 10% FBS to the E-16 plates (Agilent Technologies). Subsequently, RTCA Software Package 2.0 was used to calibrate the plates. Cells were plated at a density of 10,000 cells/well using DMEM/F12 supplemented with 10% FBS to a final volume of 100 μL and incubated at 37 °C and 5% CO2 in the RTCA cradle. The impedance signals were recorded every 15 min for 72 h and plotted as a function of time after cell seeding. The population doubling time was calculated based on the growth curve. All measurements were performed in duplicate.
Spheroid formation assay
The spheroid formation capability was assessed as described in a previous study [42]. In brief, the tumor cells were plated onto round-bottomed low-attachment 96-well plates (96-well Clear Round Bottom Ultra Low Attachment Microplate; Corning, Inc., Corning, NY, USA) at a concentration of 1.0 × 104 cells/well. After 72 h, spheroid formation was microscopically confirmed and subjected to paraffin sectioning using iPGell (Genostaff, Tokyo, Japan). The sectioned spheroids were stained with HE and analyzed via microscopy.
RTCA invasion assay
To examine the potential for invasion, we used an RTCA according to the manufacturer’s instructions, and the procedure is described in our previous study [29]. Briefly, Matrigel at a concentration of 9.3 mg/mL (BD Biosciences, Franklin Lakes, NJ, USA) was layered onto the membrane in the upper chamber, and 4 × 104 cells were seeded onto it. MG63 osteosarcoma cells (JCRB; Ibaraki Osaka, Japan) were used as controls [30]. The medium used to maintain the cells was added to the lower chamber. Next, the cells on the Matrigel-coated membrane migrated to the bottom chamber and adhered to the electronic sensors on the underside of the membrane. The attached cells influenced the electrical impedance of the electronic sensors. The invasion capability of the cells was estimated based on the positive correlation between the impedance and number of cells. The impedance was monitored every 15 min for 72 h and plotted as a function of time after seeding.
Alkaline phosphatase (ALP) activity
ALP activity in the cell lysate was assayed using an ALP assay kit (Takara Bio Inc Cat# MK301, Shiga, JAPAN) following the instruction. After removing the culture medium, the cell layers were washed twice with physiological salt solution and lysed in 500 μL of Tris base buffer.
Screening for the antitumor effects of anticancer agents
To demonstrate the utility of the established cell lines for drug screening, the anti-proliferative effects of 214 oncology drugs (Supplementary Data Table 1) were examined using the Bravo automated liquid handling platform (Agilent Technologies) as described previously [42]. In brief, the cells were seeded onto a 384-well plate (Thermo Fisher Scientific, Fair Lawn, NJ, USA) at a concentration of 1 × 104 cells/well in DMEM/F12 supplemented with 10% FBS. The next day, oncology drugs (Selleck Chemicals, Houston, TX, USA; Supplementary Table 1) were added at a fixed concentration of 10 µM, and the cells were maintained for 72 h. Inhibitory effects of the oncology drugs were assessed using the CCK-8 reagent according to the manufacturer’s protocol (Dojin-do, Kumamoto, Japan). The response readout was calculated relative to the DMSO control in terms of the percentage of relative growth inhibition.
The anti-proliferative effects were also examined using multiple concentrations to determine the IC50 value, which is the concentration required to inhibit cell growth by 50% relative to control cells. Cell suspensions (1 × 104 cells) were dispensed into each well, and the agents were added to the 384-well plates at various concentrations. The plates were incubated for 72 h, and cell viability was assessed using the CCK-8 assay. The readout was plotted against the concentrations of agents and examined using the GraphPad Prism 4.2 software (GraphPad Software Inc., San Diego, CA, USA). The response readout was calculated as a percentage of the relative growth inhibition compared to the DMSO control. The analysis was performed in duplicate. The cells were incubated at 37 °C in a humidified atmosphere containing 5% CO2.
Results
Establishment and authentication of NCC-GCTB2-C1 and NCC-GCTB3-C1 cell lines
We established two GCTB cell lines using curetted tumor tissues from two patients with GCTB and named the cell lines NCC-GCTB2-C1 and NCC-GCTB3-C1. The two cell lines were cultured for over 30 and 40 passages, respectively, for more than 1 year. To authenticate the established cell line, we examined 10 microsatellite sites (STRs) and found that these STRs were identical between the original tumor tissues and cell lines (Table 1, Supplementary Fig. 1). A database search revealed that the STR patterns of NCC-GCTB2-C1 and NCC-GCTB3-C1 cells did not match those for existing cell lines deposited in public cell banks. These results indicated that NCC-GCTB2-C1 and NCC-GCTB3-C1 cell lines were novel. NCC-GCTB2-C1 and NCC-GCTB3-C1 cells were negative for mycoplasma contamination, as no mycoplasma-specific DNA was found in the cell-conditioned medium (data not shown).
Genetic characterization of NCC-GCTB2-C1 and NCC-GCTB3-C1 cells
The G34W mutation, which is typical of GCTB, was detected in NCC-GCTB2-C1 and NCC-GCTB3-C1 cells (Fig. 3). Both NCC-GCTB2-C1 and NCC-GCTB3-C1 cells were positive for staining with antibodies against histone H3.3 G34W (Fig. 3A, B). The G34W mutation in these two cell lines was confirmed by Sanger sequencing (Fig. 3C, D). We examined the SNPs of NCC-GCTB2-C1 and NCC-GCTB3-C1 cells (Fig. 4). We did not detect multiple allelic amplifications or deletions in either NCC-GCTB2-C1 (Fig. 4A) or NCC-GCTB3-C1 cells (Fig. 4B).

Mutation in NCC-GCTB2-C1 and NCC-GCTB3-C1 cells. Immunohistochemical staining indicates the presence of a mutation in H3F3A in NCC-GCTB2-C1 (A) and NCC-GCTB3-C1 cells (B). Sequencing data for H3F3A showing the mutation peak in both NCC-GCTB2-C1 (C) and NCC-GCTB3-C1 cells (D)

Study of the single nucleotide polymorphism (SNP) array. Allele-specific copy number analysis revealed variations in the DNA copy number. SNP genotyping copy number profiles are displayed for NCC-GCTB2-C1 (A) and NCC-GCTB3-C1 cells (B). The upper panel shows log R and the lower panel B shows the allele frequency (BAF). The plot represents log R, and BAF was determined using the ASCAT algorithm
Phenotypic characteristics of NCC-GCTB2-C1 and NCC-GCTB3-C1 cells
Both NCC-GCTB2-C1 (Fig. 5A) and NCC-GCTB3-C1 (Fig. 5B) cells exhibited spindle-shaped morphology under culture conditions. NCC-GCTB2-C1 and NCC-GCTB3-C1 cells showed the ability to form spheroids when seeded on low-attachment substrates (Fig. 5C, D). The spheroids of NCC-GCTB2-C1 cell line comprised with multinucleate cells, which are not observed in the original tumor tissues of GCTB. The NCC-GCTB3-C1 cell line spheroids also consisted of heterogenous tumor cells, while obvious multinucleate cells were not observed.

Morphology of NCC-GCTB2-C1 and NCC-GCTB3-C1 cells. NCC-GCTB2-C1 (A) and NCC-GCTB3-C1 (B) cells had apparently a large cytoplasm. NCC-GCTB2-C1 (C) and NCC-GCTB3-C1 (D) cells formed spheroids when seeded in 96-well low-attachment microplates. NCC-GCTB2-C1 cells had multi-nuclear giant cells
NCC-GCTB2-C1 and NCC-GCTB3-C1 cell lines did not exhibit alkaline phosphatase activity (data not shown).
NCC-GCTB2-C1 and NCC-GCTB3-C1 cell lines showed constant proliferation, with population doubling times of 64 h and 26 h, respectively, based on the growth curves of these cells (Fig. 6A, B). Furthermore, these cells displayed invasive ability in vitro, which increased when a higher number of cells were seeded onto the upper chamber (Fig. 6C, D).

Growth and invasion of NCC-GCTB2-C1 and NCC-GCTB3-C1 cells. RTCA growth assay of NCC-GCTB2-C1 (A) and NCC-GCTB3-C1 (B) cells, based on which the doubling time was calculated. The results of the RTCA invasion assay of NCC-GCTB2-C1 (C) and NCC-GCTB3-C1 (D) cells, indicated that the cells had invasion capabilities
Sensitivity to anticancer agents
The anti-proliferative effects of 214 oncology drugs on NCC-GCTB2-C1 and NCC-GCTB3-C1 cells were examined using a high-throughput screening approach. In addition to these cells, NCC-GCTB1-C1 cells, which we have established previously [42], were included in the drug screening. First, the effects of oncology drugs at a uniform concentration of 10 μM (Supplementary Tables 1 and 2) were examined, and the drugs were ranked according to the survival rate after treatment. Second, the IC50 values were generated (Table 2), and the growth curves of eight oncology drugs having lower IC50 values in the top 10 commonly used cell lines are shown in Fig. 7.

Growth suppressive effects of oncology drugs on GCTB cells. The anti-proliferative effects of the eight oncology drugs on NCC-GCTB1-C1, NCC-GCTB2-C1, and NCC-GCTB3-C1 cells are shown. The IC50 values are summarized in Table 2
Discussion
GCTB exhibits heterogeneous clinical behavior [44, 45], and curative therapy is limited to en bloc excision at the early stages of lesion development [46]. Because GCTB is a relatively rare disease and due to the paucity of adequate cancer models, clinical studies with a statistically significant number of cases and convincing pre-clinical studies that may drive clinical trials have not been extensively performed. We aimed to improve this situation by generating novel GCTB cell lines.
We successfully established two novel GCTB cell lines and found that they were suitable for in vitro functional studies; they grew constantly and had invasive capabilities, suggesting the feasibility of intervention studies using these cell lines. We investigated the spheroids of NCC-GCTB2-C1 and NCC-GCTB3-C1 cells. First, we immunohistochemically confirmed the presence of mutation in histone H3.3 G34W, which was observed in the monolayer-cultured cells, suggesting that the genetic information was preserved in the tumor cells when formed spheroids. The complex components, such as giant cells with multiple nuclei became noticeable when NCC-GCTB2-C1 cells formed spheroids. It is also noticeable that the multinucleated cells observed in the spheroids of NCC-GCTB2-C1 are histologically different from those observed in GCTB. In addition, the NCC-GCTB2-C1 and NCC-GCTB3-C1 cells in the spheroids had obvious nuclear atypia. In contrast, the original tumors did not have nuclear atypia and the clinical outcome of the donor patients was favorable so far. Thus, it is worth further investigating the molecular backgrounds of the spheroid formation and development along with studying on the mechanisms that generate distinct morphologies in the two cell lines.
We demonstrated the utility of our cell lines for drug development using oncology drug screening. The identified drugs are candidates for further investigation. We found that no drugs showed drastic anti-proliferative effects on all three cell lines. It is unclear whether the different sensitivities among the cells arose due to intra- or inter-tumor heterogeneity. We found that romidepsin markedly suppressed the growth of the three cell lines. Romidepsin is a histone deacetylase (HDAC) inhibitor that is approved for the treatment of cutaneous T-cell lymphoma [47]. When cells were treated with a fixed concentration of 10 μM romidepsin, the survival rates were 0%, 5.8%, and 1.0% in NCC-GCTB1-C1, NCC-GCTB2-C1, and NCC-GCTB3-C1 cells, respectively (Supplementary Table 2). In contrast, the survival rates after treatment with other HDAC inhibitors, such as belinostat and vorinostat, were 3.6%, 30.9%, and 5.6%, and 29.7%, 56.9%, and 37.7% for NCC-GCTB1-C1, NCC-GCTB2-C1, and NCC-GCTB3-C1 cells, respectively (Supplementary Table 2). The molecular background of the specificity of HDAC inhibitors in GCTB cells is worth investigating. Although the cell lines are advantageous over other cancer models and are suitable for use in high-throughput screening, they have inherent limitations because they were maintained under artificial conditions. The results of the drug screening should be further validated in other cancer models, such as patient-derived xenografts. Although our results should be validated in a larger number of cases using different methods, the results of the present study indicate the utility of our cell lines.
Considering the clinical variations of GCTB, we need to establish additional patient-derived cancer models to recapitulate the in vivo behavior of GCTB. Because of the rarity of GCTB occurrence and the variety of available cancer models, such as cell lines, organoids, and xenografts, model establishment requires multi-institutional collaboration. Moreover, multi-institutional collaboration should facilitate the best use of the established cancer models.
References
Liede A, Bach BA, Stryker S, et al. Regional variation and challenges in estimating the incidence of giant cell tumor of bone. J Bone Joint Surg Am. 2014;96:1999–2007.
Behjati S, Tarpey PS, Presneau N, et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat Genet. 2013;45:1479–82.
Presneau N, Baumhoer D, Behjati S, et al. Diagnostic value of H3F3A mutations in giant cell tumour of bone compared to osteoclast-rich mimics. J Pathol Clin Res. 2015;1:113–23.
Cleven AH, Höcker S, Briaire-de Bruijn I, Szuhai K, Cleton-Jansen AM, Bovée JV. Mutation analysis of H3F3A and H3F3B as a diagnostic tool for giant cell tumor of bone and chondroblastoma. Am J Surg Pathol. 2015;39:1576–83.
Eckardt JJ, Grogan TJ. Giant cell tumor of bone. Clin Orthop Relat Res. 1986. https://doi.org/10.1097/00003086-198603000-00006.
McGrath PJ. Giant-cell tumour of bone: an analysis of fifty-two cases. J Bone Joint Surg Br Vol. 1972;54:216–29.
Bridge JA, Neff JR, Mouron BJ. Giant cell tumor of bone. Chromosomal analysis of 48 specimens and review of the literature. Cancer Genet Cytogenet. 1992;58:2–13.
Harness NG, Mankin HJ. Giant-cell tumor of the distal forearm. J Hand Surg. 2004;29:188–93.
Moon JC, Kim SR, Lee YC, Chung MJ. Multiple pulmonary metastases from giant cell tumor of a hand. Am J Med Sci. 2012;343:171–3.
Errani C, Ruggieri P, Asenzio MA, et al. Giant cell tumor of the extremity: A review of 349 cases from a single institution. Cancer Treat Rev. 2010;36:1–7.
Balke M, Schremper L, Gebert C, et al. Giant cell tumor of bone: treatment and outcome of 214 cases. J Cancer Res Clin Oncol. 2008;134:969–78.
Becker WT, Dohle J, Bernd L, et al. Local recurrence of giant cell tumor of bone after intralesional treatment with and without adjuvant therapy. J Bone Joint Surg Am. 2008;90:1060–7.
Kivioja AH, Blomqvist C, Hietaniemi K, et al. Cement is recommended in intralesional surgery of giant cell tumors: a Scandinavian Sarcoma Group study of 294 patients followed for a median time of 5 years. Acta Orthop. 2008;79:86–93.
Algawahmed H, Turcotte R, Farrokhyar F, Ghert M. High-speed burring with and without the use of surgical adjuvants in the intralesional management of giant cell tumor of bone: a systematic review and meta-analysis. Sarcoma. 2010;2010:1–5.
Chan CM, Adler Z, Reith JD, Gibbs CP Jr. Risk factors for pulmonary metastases from giant cell tumor of bone. J Bone Joint Surg Am. 2015;97:420–8.
Dominkus M, Ruggieri P, Bertoni F, et al. Histologically verified lung metastases in benign giant cell tumours–14 cases from a single institution. Int Orthop. 2006;30:499–504.
Wang J, Liu X, Yang Y, et al. Pulmonary metastasis of giant cell tumour: a retrospective study of three hundred and ten cases. Int Orthop. 2021;45:769–78.
Huang L, Xu J, Wood DJ, Zheng MH. Gene expression of osteoprotegerin ligand, osteoprotegerin, and receptor activator of NF-kappaB in giant cell tumor of bone: possible involvement in tumor cell-induced osteoclast-like cell formation. Am J Pathol. 2000;156:761–7.
Atkins GJ, Bouralexis S, Haynes DR, et al. Osteoprotegerin inhibits osteoclast formation and bone resorbing activity in giant cell tumors of bone. Bone. 2001;28:370–7.
Branstetter DG, Nelson SD, Manivel JC, et al. Denosumab induces tumor reduction and bone formation in patients with giant-cell tumor of bone. Clin Cancer Res. 2012;18:4415–24.
Kostenuik PJ, Nguyen HQ, McCabe J, et al. Denosumab, a fully human monoclonal antibody to RANKL, inhibits bone resorption and increases BMD in knock-in mice that express chimeric (murine/human) RANKL. J Bone Mineral Res. 2009;24:182–95.
Chawla S, Henshaw R, Seeger L, et al. Safety and efficacy of denosumab for adults and skeletally mature adolescents with giant cell tumour of bone: interim analysis of an open-label, parallel-group, phase 2 study. Lancet Oncol. 2013;14:901–8.
Thomas D, Henshaw R, Skubitz K, et al. Denosumab in patients with giant-cell tumour of bone: an open-label, phase 2 study. Lancet Oncol. 2010;11:275–80.
Errani C, Tsukamoto S, Leone G, et al. Denosumab may increase the risk of local recurrence in patients with giant-cell tumor of bone treated with curettage. J Bone Joint Surg Am. 2018;100:496–504.
Scoccianti G, Totti F, Scorianz M, et al. Preoperative denosumab with curettage and cryotherapy in giant cell tumor of bone: Is there an increased risk of local recurrence? Clin Orthop Relat Res. 2018;476:1783–90.
Sano K, Suehara Y, Okubo T, et al. Preoperative denosumab treatment with curettage may be a risk factor for recurrence of giant cell tumor of bone. J Orthop Surg (Hong Kong). 2020;28:2309499020929786.
Boriani S, Sudanese A, Baldini N, Picci P. Sarcomatous degeneration of giant cell tumours. Ital J Orthop Traumatol. 1986;12:191–9.
Lipplaa A, Dijkstra S, Gelderblom H. Challenges of denosumab in giant cell tumor of bone, and other giant cell-rich tumors of bone. Curr Opin Oncol. 2019;31:329–35.
Sharma SV, Haber DA, Settleman J. Cell line-based platforms to evaluate the therapeutic efficacy of candidate anticancer agents. Nat Rev Cancer. 2010;10:241–53.
Lim J, Park JH, Baude A, et al. The histone variant H3.3 G34W substitution in giant cell tumor of the bone link chromatin and RNA processing. Sci Rep. 2017;7:13459.
Fellenberg J, Sähr H, Mancarella D, et al. Knock-down of oncohistone H3F3A-G34W counteracts the neoplastic phenotype of giant cell tumor of bone derived stromal cells. Cancer Lett. 2019;448:61–9.
Barretina J, Caponigro G, Stransky N, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–7.
Garnett MJ, Edelman EJ, Heidorn SJ, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–5.
Basu A, Bodycombe NE, Cheah JH, et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell. 2013;154:1151–61.
Seashore-Ludlow B, Rees MG, Cheah JH, et al. Harnessing connectivity in a large-scale small-molecule sensitivity dataset. Cancer Discov. 2015;5:1210–23.
Rees MG, Seashore-Ludlow B, Cheah JH, et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat Chem Biol. 2016;12:109–16.
Haverty PM, Lin E, Tan J, et al. Reproducible pharmacogenomic profiling of cancer cell line panels. Nature. 2016;533:333–7.
Iorio F, Knijnenburg TA, Vis DJ, et al. A landscape of pharmacogenomic interactions in cancer. Cell. 2016;166:740–54.
Behan FM, Iorio F, Picco G, et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature. 2019;568:511–6.
Townsend EC, Murakami MA, Christodoulou A, et al. The public repository of xenografts enables discovery and randomized phase II-like trials in mice. Cancer Cell. 2016;29:574–86.
Bairoch A. The Cellosaurus, a cell-Line knowledge resource. J Biomol Tech. 2018;29:25–38.
Noguchi R, Yoshimatsu Y, Ono T, et al. Establishment and characterization of NCC-GCTB1-C1: a novel patient-derived cancer cell line of giant cell tumor of bone. Hum Cell. 2020;33:1321–8.
Capes-Davis A, Reid YA, Kline MC, et al. Match criteria for human cell line authentication: where do we draw the line? Int J Cancer. 2013;132:2510–9.
Turcotte RE. Giant cell tumor of bone. Orthop Clin North Am. 2006;37:35–51.
Karpik M. Giant Cell Tumor (tumor gigantocellularis, osteoclastoma)—epidemiology, diagnosis, treatment. Ortop Traumatol Rehabil. 2010;12:207–15.
Niu X, Zhang Q, Hao L, et al. Giant cell tumor of the extremity: retrospective analysis of 621 Chinese patients from one institution. J Bone Joint Surg Am. 2012;94:461–7.
Prince HM, Dickinson M. Romidepsin for cutaneous T-cell lymphoma. Clin Cancer Res. 2012;18:3509–15.
Acknowledgements
We appreciate the technical support provided by Mesdames Yu Kuwata and Rina Sasaki (Division of Rare Cancer Research, National Cancer Center). We would like to thank Drs. F Nakatani, S. Iwata, E Kobayashi, M Nakagawa, T Komatsubara, M Saito, C Sato (Department of Musculoskeletal Oncology, National Cancer Center Hospital), Drs. T Shibayama, and H Tanaka (Department of Diagnostic Pathology, National Cancer Center Hospital) for sampling tumor tissue specimens from curetted materials. (Division of Rare Cancer Research). We appreciate the technical support provided by Ms. Y Shiotani, Mr. N Uchiya, and Dr. T Imai (Central Animal Division, National Cancer Center Research Institute). We would also like to thank Editage (www.editage.jp) for their help with English language editing and their constructive comments on the manuscript. This research was technically assisted by the Fundamental Innovative Oncology Core at the National Cancer Center This research was supported by the Japan Agency for Medical Research and Development (Grant number 20ck0106537h0001).
Funding
This research was supported by the Japan Agency for Medical Research and Development (Grant number 20ck0106537h0001).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
The ethical committee of the National Cancer Center approved the use of clinical materials for this study (approval number 2004-050).
Informed consent
Written informed consent for publication was provided by the patients.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yoshimatsu, Y., Noguchi, R., Tsuchiya, R. et al. Establishment and characterization of novel patient-derived cell lines from giant cell tumor of bone. Human Cell 34, 1899–1910 (2021). https://doi.org/10.1007/s13577-021-00579-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13577-021-00579-z