
Abstract
Colletotrichum truncatum is one of the most economically important fungal pathogen causing anthracnose disease in pre and post-harvest stages of many crops worldwide. Little information is available in the literature on the genetic analysis and demographic history of this fungal pathogen. In the present study nucleotide sequence data of internal transcribed spacer (ITS) region were analyzed for C. truncatum isolates infecting chili and other crops worldwide to determine a metageographic pattern of distribution and evolution of the species. Levels of differentiation (genetic distances and F ST values) among sequences of C. truncatum from 23 countries were minimal suggesting the global occurrence of a large and geographically undifferentiated population. Only 11 haplotypes were detected among 98 isolates from 24 geographically distant populations of C. truncatum. Predominant haplotype H1 which occupied a central position in the median joining network was inferred to be ancestral haplotype as it was detected at a high frequency and was shared by multiple populations. Phylogeographic pattern of the species with worldwide presence and predominance of single haplotype suggests human mediated dispersal through domestication and introduction of host plants in different parts of the world, and might have played a significant role in structuring the populations of this devastating pathogen.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The genus Colletotrichum is an important plant pathogenic fungus associated with anthracnose disease of crops including cereals, legumes, vegetables, perennial crops and tree fruits grown in tropical, subtropical and temperate regions of the world (Sutton 1992; Cai et al. 2009; Hyde et al. 2009). Among Colletotrichum species, Colletotrichum truncatum (capsici) (Syd.) Butler & Bisby first reported from the Coimbatore of Madras Presidency [now Tamil Nadu], India on chili (Capsicum annum var. frutescens Kuntze,Solanaceae) as Vermicularia capsici by Sydow (1913) is the most aggressive and commonly infects chili and papaya (Rampersad 2013; Than et al. 2008). The fungus takes heavy toll of the crop as it attacks chili plants at different growth stages causing seedling rot in nursery and die back and fruit rot at adult plant stage. The pre and post-harvest pathogenic behavior of the pathogen accounts for huge economic losses incurred by the growers and affect chili and papaya production world over (Bosland and Votava 2003; Hadden and Black 1989; Rampersad 2013). Anthracnose disease has been reported to cause significant losses to the tune of 491.67 million US dollars in India, whereas these estimates are 100 million US dollars in Korea (Garg et al. 2013; Park and Kim 1992) on chili and other solanaceous crops though it also infects a wide range of other broad-leaved plants. Presently, it is widely distributed in different regions of the world and has been recorded on 121 plant genera distributed in 45 plant families from different parts of the world (Farr et al. 2007).
The internal transcribed spacers (ITS) region of the nuclear rDNA repeat unit is by far the most commonly studied region of the genome to resolve taxonomy at the genus and species levels in fungi (Nilsson et al. 2009). The ITS1 and ITS2 spacers show a high rate of evolution and have proved to be robust evolutionary markers for determining intra- and inter-specific relationships (Glass and Donaldson 1995). The rDNA sequences exhibit variation within species (Ganley and Kobayashi 2007), which can manifest itself by difference in length due to insertion or deletion (indels of single or several bases) or by nucleotide substitutions with no change in overall number of base pairs. Several workers have used rDNA sequences to reconstruct phylogeny between distantly related taxa (Berbee and Taylor 2001; James et al. 2006; Karol et al. 2001; Medina et al. 2001; Soltis and Soltis 2000; Woese et al. 1990). The sequence analysis of ITS regions has proved useful in studying phylogenetic relationships and also accepted as a universal DNA barcode marker for fungi including Colletotrichum species (Photita et al. 2005; Schoch et al. 2012; Sreenivasaprasad et al. 1996).
C. truncatum possesses wide host range and different host-limited populations of the pathogen display high pathologic variability (Afanador-Kafuri et al. 2003; Than et al. 2008, Torres-Calzada et al. 2013, Montri et al. 2009). Sharma et al. (2005) have shown existence of five races in chili infecting populations of C. truncatum in Himachal Pradesh. The population structure of C. truncatum has not so far been explored properly with reference to its pathogenic variability due to absence of well-defined differential set. Its wide host range is indicative of selection pressure on the different hosts rather than pathogen which seem to evolve much easily due to presence of diverse populations on different hosts. The analysis of genetic diversity among various host-limited populations of the pathogen from different parts of the world could yield useful information on their evolutionary behaviour as well as facilitate the formulation of effective management strategies based on host resistance.
Knowledge of population genetic structure, levels of intra species divergence, gene flow among wide geographic populations of the species help in the understanding of biogeographic history, evolutionary and adaptive potential of the pathogenic species (McDermott and McDonald 1993; McDonald 1997; Rampersad et al. 2013). Information on variation in the species at wide geographical level is a prerequisite for the development of disease management strategies i.e. identification of resistance sources, pesticide use, predicting resistance breakdown, development and deployment of disease resistant varieties and in streamlining cultural practices (McDonald and Linde 2002; Rampersad et al. 2013). Presently, metageographic population study of Colletotrichum and related pathogen in the literature is scanty. The objective of the present study was to (i) compare the genetic and phylogenetic relationships between C. truncatum isolates from 23 countries and (ii) elucidation of demographic history based on ITS -locus sequence data.
Materials and methods
Collection of diseased samples, isolation of Colletotrichum isolates and their maintenance
Diseased fruits of chilies with ripe fruit rot symptoms were collected from different locations of nine districts of Himachal Pradesh during 2007–08 (Table S1). Pure culture of each isolate was raised from disease samples using standard methodology on Mathur’s medium (Sharma et al. 2005). Small bits of infected tissues were surface sterilized in 0.1 % solution of mercuric chloride for 10–15 s and washed thrice in sterilized water under laminar air flow. The bits were dried under two folds of sterilized filter paper and transferred to Mathur’s medium slants. The tubes were incubated in BOD Incubator at 23 ± 1 °C for 7–8 days. Fungal cultures were purified by single spore isolation technique and colonies arising from single spores were multiplied on Mathur’s medium for further studies. Each isolate was transferred to live host after 3–4 sub-cultures to avoid loss of virulence. In all 20 isolates originating from diverse chili production areas of Himachal Pradesh were used in the study.
DNA extraction
Total genomic DNA of twenty C. truncatum isolates was extracted using CTAB method (Murray and Thompson 1980) with minor modifications. Fungal mycelium was grown in conical flasks containing 150 ml of potato dextrose broth inoculated with mycelium from 7 day old cultures and incubated at 25 °C in an orbital incubator shaker (100 rpm) for 6–7 days. Mycelia were harvested by filtration through double layers of filter paper, dried between two layers of sterilized filter paper in laminar air flow cabinet and stored at −20 °C for further use. The amount of DNA was quantified by recording the absorbance at 260 nm wavelength using UV/VIS spectrophotometer (Bio rad SmartSpec 3000) and stored at −80 °C (Deep Freezer Labtech®, Daihan Labtech Co. Pvt. Ltd.) for further use.
PCR amplification and rDNA sequencing
The rDNA amplification was carried out in 0.2 ml PCR tube with 50 μl reaction volume containing 5 μl of 10× buffer (20 mM Tris HCl, pH 8.0, 50 mM KCl ) and 3 μl of 1.5 mM MgCl2, 4 μl dNTP mix (0.2 mM each) (MBI Fermentas), 0.4 μl of Taq polymerase (Merck Biosciences, India, 5 U/μl), 4 μl of DNA template (20 ng), 1 μl each of ITS1 and ITS4 primers (10 μM) (ITS1: 5’-TCCGTAGGTGAACCTGCGG-3’, ITS4: 5’-TCCTCCGCTTATTGATATGC-3’; (White et al. 1990) and 31.6 μl of sterilized distilled water. Amplifications were performed in Gene-Amp PCR system 9700 (Applied Biosystems, USA) programmed for initial denaturation at 95 °C for 2 min followed by 30 cycles at 95 °C for 1 min, 60 °C for 30 s, 72 °C for 1 min and a final extension at 72 °C for 10 min. The PCR product was separated in 2 % (w/v) agarose gel using TAE buffer (40 mM Tris-acetate, 1 mM EDTA) and visualized using ethidium bromide (0.5 μg/ml) staining. The gel images were captured using Alphaimager 2200 gel documentation system (Alphaimager, USA). Amplified PCR products (~500 bp) were freeze dried and were custom sequenced using same upstream and downstream primers (Xcelris Labs Limited, India).
Data analysis
Twenty ITS sequences of C. truncatum generated in the present study were merged with 78 ITS sequences of C. truncatum retrieved from NCBI GenBank for combined analysis (Table 1). All 98 sequences were first aligned using ClustalW as implemented in MEGA 5.1 (Tamura et al. 2011) and analyzed for genetic and phylogenetic relationships between C. truncatum isolates in MEGA 5.1, including Colletotrichum fructi (GU227844), Colletotrichum lineola (GU227838) and Colletotrichum lindemuthianum (GU227800) as out groups. Genetic distances within and between populations of C. truncatum were calculated using the Kimura two-parameter (K2P) method (Kimura 1980). The neighbour-joining (Saitou and Nei 1987) procedure was used for phylogeny reconstruction and confidence levels for the groups were computed by bootstrap analysis with 1000 replications (Felsenstein 1985). Median joining network among sequence variants of C. truncatum was constructed using NETWORK ver. 4.6 (Bandelt et al. 2000) to study the evolutionary relationships among the haplotypes. Descriptive statistics (number of haplotypes, haloptype and nucleotide diversity) were calculated in DNASP ver. 5.0 (Librado and Rozas 2009).
Demographic history
In order to determine whether C. truncatum populations underwent recent population expansions, we calculated mismatch distributions and compared these with predicted distributions from models of population expansion (Rogers 1995). Mismatch distributions were calculated in DNASP ver. 5.0 (Librado and Rozas 2009). All parameters were tested against the expected values under the hypothesis of a recent population expansion based on 1000 bootstrap replicates. Tajimas’ D ad Fu’s F S were calculated to test for neutrality (Tajima 1989; Fu 1997).
Results
Nucleotide information
The 98 ITS sequences comprising of test isolates and available in the Genbank used in the genetic analysis of C. truncatum were trimmed to 461 bp before analysis. The average nucleotide composition of the ITS sequences of C. truncatum was 25.2 % T, 23.4 % A, 25 % G, 26.5 % C and 51.5 % G + C content with 13 variable sites including 12 singleton sites and 1 parsimony informative sites at which more than one isolates showed mutation at same the locus.
Genetic diversity and Phylogenetic analysis
A total of 11 haplotypes were identified in 98 ITS gene sequences. The most common and predominant haplotype H1 was found in sequences from 21 countries except Bangladesh and Nepal. The number of haplotypes per population ranged from 1 to 4. Whereas, overall haplotype diversity (Hd) and nucleotide diversity (π) was 0.297 ± 0.060 and 0.00090 ± 0.00021, respectively. The analyses suggest that overall C. truncatum populations retain a very low level of haplotypes and nucleotide diversity in the ITS region of rDNA. C. truncatum isolates did not form any group based on haplotypes and nucleotide diversity. Population pairwise F ST analysis (only analyzed for populations with atleast two sample sequences) revealed that most populations did not differ genetically (Table 2). Genetic analysis showed no sign of population differentiation in C. truncatum, thus no population structure was evident in isolates representing 23 countries.
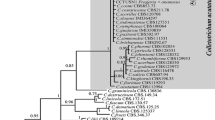
The phylogenetic analysis clustered all the C. truncatum sequences from different countries in one clade irrespective of origin and host of the isolates (Fig. 1). The genetic distance (K2P) between in-group ranged between 0.000–0.005 (within C. truncatum isolates) and in outgroup species it varied from 0.057–0.062, 0.065–0.070 and 0.093–0.096 between C. truncatum/ C. fructi, C. truncatum/ C. lineola and C. truncatum/ C. lindemuthianum, respectively (Table S2), though C. fructi was more closely related to C. truncatum on the phylogenetic tree.

Phylogenetic tree based on ITS sequences of C. truncatum with C. fructi, C. lineola and C. lindemuthianum as out groups using the neighbor-joining method and confidence level calculated with the bootstrap (1000 replicates)
Median joining network of haplotypes and Demographic history
A median joining (MJ) network reconstructed from 98 sequences of ITS region of rDNA revealed no major divergence lineage (Fig. 2) in C. truncatum. All 10 (H2-H11 except H1) haplotypes were connected with predominant haplotype (H1) by single or maximum of two mutation steps. MJ network reconstructed for evolutionary relationships among haplotypes showed a star-like shape, characteristic of population expansion (Slatkin and Hudson 1991). Haplotype H1 positioned in the center of the network was found at higher frequency and was shared by all geographically widespread populations of C. truncatum (Fig. 2) except those from Bangladesh and Nepal. Haplotype H5, H6, H9, H10 and H11 showed maximum two step mutational divergence among haplotypes from the ancestor or original haplotype (H1). Three missing haplotypes were detected in the present analysis including two in South India and one in Mexico.

Median joining network of rDNA haplotypes of C. truncatum populations. Size of the circle is related with frequency of haplotypes. Colours indicate the proportion of individuals sampled in different populations within the study area
Mismatch distribution analysis revealed a unimodal mismatch graph (Fig. 3), characteristic of recent demographic expansion for C. truncatum population. This pattern is consistent with the star-like shape of MJ Network observed in the median joining network (Fig. 2). Recent population expansion was also supported by highly significant negative values of both Tajima’s D (−2.2609, P < 0.01) and Fu’s Fst (−11.645, P < 0.01) tests indicating that the whole set of C. truncatum samples studied here does not fit to a simple model of neutral evolution.

Mismatch distribution of 98 rDNA sequences of C. truncatum from different countries representing the observed and expected pairwise differences under the sudden population expansion model
Discussion
Genus Colletotrichum is among the most important genera of plant pathogenic fungi worldwide and comprises of different species clades possessing species complex causing disease symptoms commonly known as anthracnose on a wide range of important crops, fruits and ornamental plants (O’Connell et al. 2012). The genetic variation in natural populations is the outcome of a balance between evolutionary and demographic processes provides tools for interpreting evolutionary processes determining the evolutionary potential of a species (Li et al. 2013). In this study, intraspecific genetic analysis of C. truncatum was carried out to understand the evolution and phylogeography of the species. Phylogenetic study suggests that C. truncatum is a single species with no distinct lineage as all the isolates originating from 23 countries clustered together in a single clade. The inference was also supported by the construction of median joining network and haplotypic analysis of the ITS data. Only 11 haplotypes were recognized in a group of 98 sequences with predominant haplotype H1 comprising of 81 individuals (89.01 % of total isolates) with diverse geographic origin and host specificities. These results are indicative of a high level of gene flow occurring among the geographically distinct populations of the pathogen.
Median joining network analysis of various C. truncatum isolates revealed that some of the haplotypes were population specific and unique whereas some of the populations suggests their haplotype specific evolution (H1 & H7)) and high gene flow among populations. The haplotypes H2-H4, H5-H6 and H8-H9, from North India, South India and Mexico seem to have originated from the predominant haplotype H1 as the same was also detected in C. truncatum populations of these regions. The haplotypes H10 and H11 are specific to the populations from Trinidad & Tobago and Bangladesh, respectively. These populations might have originated from haplotype H7 representing C. truncatum isolates from Nepal, South India, Thailand, Taiwan and the USA. These results suggest that haplotype H11 comprising of Bangladesh isolates most likely were introduced from South India, Nepal or Thailand than from Taiwan and USA due to geographic proximity of the these regions to Bangladesh. By contrast, the invasion of C. truncatum in Trinidad and Tobago seems to be more likely from USA than the other countries harboring H7 haplotype.
The overall low level of genetic differentiation between populations could also be due to recent history of the C. truncatum. Two neutrality (Tajimas’ D and Fu’s Fs) test statistics were used in this study to examine the sequences for evolutionary forces acting on the populations of C. truncatum. The significant and negative values of Tajimas’ D and Fu’s Fs values indicated that the C. truncatum populations do not fit a simple model of neutrality and reject the null hypothesis of constant population size. Whereas, mismatch distribution test was applied to determine the pattern of demographic expansion of C. truncatum and to test the null hypothesis of population growth. A typical unimodal mismatch distribution (Rogers and Harpending 1992) indicating a fit to the demographic expansion model test and further supported by a star like structure of median joining network. These results also suggest that the deviation from neutrality for constant population size (Tajima’s D and Fu’s Fs) was due to recent population expansion of the species. Introduction, domestication and co-cultivation of host plants of C. truncatum could be the reason for recent population expansion of the species as C. truncatum presently has pathogenic specialization on several wild and domesticated host plants. This kind of range expansion has also been observed earlier in different fungus and insects species (Agostini et al. 1993; Buchwaldt et al. 1996; Chen et al. 2007; Meyer et al. 2007; Prabhakar et al. 2012; Prabhakar et al. 2013; Wan et al. 2011).
In conclusion, with respect to metageographic population genetic study of C. truncatum, recent population expansion was detected in the species. Study also suggests that C. truncatum may have expanded its distribution in the past with the co-migration of the host plants as one of the haplotypes found predominant across the host and geographical regions. C. truncatum management strategies to target most predomint haplotypes for the development of durable resistance would prove to be the most practical.
Abbreviations
- rDNA:
-
Ribosomal deoxyribonucleic acid
- ITS:
-
Internal transcribed spacer
- MEGA:
-
Molecular evolutionary genetics analysis
- cTAB:
-
Cetyl trimethylammonium bromide
- DNA:
-
Deoxyribonucleic acid
References
Afanador-Kafuri L, Minz D, Maymon M, Freeman S (2003) Characterization of Colletotrichum isolates from Tamarillo, Passiflora and Mango in Colombia and identification of a unique species from the genus. Phytopathology 93:579–587
Agostini JP, Gottwald T, Timmer LW (1993) Temporal and spatial dynamics of postbloom fruit drop of citrus in Florida. Phytopathology 83(5):485–490
Bandelt H-J, Macaulay V, Richards M (2000) Median networks: speedy construction and greedy reduction, one simulation, and two case studies from human mtDNA. Mol Phylogenet Evol 16(1):8–28
Berbee ML, Taylor JW (2001) Fungal molecular evolution: gene trees and geologic time Systematics and Evolution. Springer, p 229–245
Bosland P, Votava EJ (2003) Peppers: vegetable and spice capsicums. Crop Production Science in Horticulture 12
Buchwaldt L, Morrall R, Chongo G, Bernier C (1996) Windborne dispersal of Colletotrichumtruncatum and survival in infested lentil debris. Phytopathology 86(11):1193–1198
Cai L et al (2009) A polyphasic approach for studying Colletotrichum. Fungal Divers 39(1):183–204
Chen P, Ye H, Mu Q (2007) Migration and dispersal of the oriental fruit fly, Bactrocera dorsalis, in regions of Nujiang River based on fluorescence mark. Acta Ecol Sin 6:2468–2476
Farr D, Rossman A, Palm M, McCray E (2007) Fungal databases. Systematic Botany & Mycology Laboratory, ARS, USDA http://nt.ars-grin.gov/sbmlweb/fungi/index.cfm
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791
Fu YX (1997) Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147:915–925
Ganley AR, Kobayashi T (2007) Highly efficient concerted evolution in the ribosomal DNA repeats: total rDNA repeat variation revealed by whole-genome shotgun sequence data. Genome Res 17(2):184–191
Garg R, Kumar S, Kumar R, Loganathan M, Saha S, Kumar S, Rai AW, Roy BK (2013) Novel source of resistance and differential reactions on chilli fruit infected by Colletotrichum capsici. Australas Plant Pathtol 42(2):227–233
Glass NL, Donaldson GC (1995) Development of primer sets designed for use with the PCR to amplify conserved genes from filamentous ascomycetes. Appl Environ Microbiol 61(4):1323–1330
Hadden J, Black L (1989) Anthracnose of pepper caused by Colletotrichum spp. In: Proceeding of the International Symposium on Integrated Management Practices: Tomato and Pepper Production in the Tropics Asian Vegetable Research and Development Centre, Taiwan, p 189–199
Hyde K et al (2009) Colletotrichum—names in current use. Fungal Divers 39:147
James TY et al (2006) Reconstructing the early evolution of Fungi using a six-gene phylogeny. Nature 443(7113):818–822
Karol KG, McCourt RM, Cimino MT, Delwiche CF (2001) The closest living relatives of land plants. Science 294(5550):2351–2353
Kimura M (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16(2):111–120
Li J, Ye Y, Wu C, Qi P, Guo B, Chen Y (2013) Genetic variation ofMytilus coruscusGould (Bivalvia: Mytilidae) populations in the East China Sea inferred from mtDNA COI gene Sequence. Biochem Syst Ecol 50:30–38
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25(11):1451–1452
McDermott JM, McDonald BA (1993) Gene flow in plant pathosystems. Ann Rev Phytopathol 31:353–373
McDonald BA (1997) The population genetics of fungi: tools and techniques. Phytopathology 87:448–453
McDonald BA, Linde C (2002) Pathogen population genetics, evolutionary potential and durable resistance. Ann Rev Phytopathol 40:349–379
Medina M, Collins AG, Silberman JD, Sogin ML (2001) Evaluating hypotheses of basal animal phylogeny using complete sequences of large and small subunit rRNA. Proc Natl Acad Sci 98(17):9707–9712
Meyer J, Taputuarai R, Killgore E (2007) Dissemination and impacts of the fungal pathogen, Colletotrichum gloeosporioides f. sp. miconiae, on the invasive alien tree, Miconia calvescens. In: Proceedings of the XIIth International Symposium on Biological Control of Weeds, La Grande Motte, France, p 22–27
Montri P, Taylor PWJ, Mongkolporn O (2009) Pathotypes of Colletotrichum capsici, the causal agent of chili anthracnose, in Thailand. Plant Dis 93:17–20
Murray M, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8(19):4321–4326
Nilsson RH, Ryberg M, Abarenkov K, Sjökvist E, Kristiansson E (2009) The ITS region as a target for characterization of fungal communities using emerging sequencing technologies. FEMS Microbiol Lett 296(1):97–101
O’Connell R, MLJ, Vaillancourt LJ et al (2012) Life-style transitions in plant pathogenic Colletotrichum fungi deciphered by genome and transcriptome analyses. Nat Genet 44:1060–1065
Park KS, Kim CH (1992) Identification, distribution and etiological characteristics of anthracnose fungi of red pepper in Korea. Plant Pathol J 8(1):61–69
Photita W, Taylor PW, Ford R, Hyde KD, Lumyong S (2005) Morphological and molecular characterization of Colletotrichum species from herbaceous plants in Thailand. Fungal Divers 18:117–133
Prabhakar CS, Mehta PK, Sood P, Singh SK, Sharma P, Sharma PN (2012) Population genetic structure of the melon fly, Bactrocera cucurbitae (Coquillett)(Diptera: Tephritidae) based on mitochondrial cytochrome oxidase (COI) gene sequences. Genetica 140(1–3):83–91
Prabhakar CS, Sood P, Mehta PK, Sharma PN (2013) Population genetic structure of the pumpkin fruit fly, Bactrocera tau (Walker) (Diptera: Tephritidae) in Himachal Pradesh, India. Biochem Syst Ecol 51:291–296
Rampersad SN (2013) Genetic Structure of Colletotrichum gloeosporioides sensu lato Isolates Infecting Papaya Inferred by Multilocus ISSR Markers. Phytopathology 103(2):182–189
Rampersad SN, Perez-Brito D, Torres-Calzada C, Tapia-Tussell R, Carrington CVF (2013) Genetic structure and demographic history of Colletotrichum gloeosporioides sensu lato and C. truncatum isolates from Trinidad and Mexico. BMC Evol Biol 13:130
Rogers AR (1995) Genetic evidence for a Pleistocene population explosion. Evolution 49:608–615
Rogers A, Harpending H (1992) Population growth makes waves in the distribution of pairwise differences. Mol Biol Evol 9:552–569
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4(4):406–425
Schoch CL et al (2012) Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc Natl Acad Sci U S A 109(16):6241–6246
Sharma PN, Kaur M, Sharma OP, Sharma P, Pathania A (2005) Morphological, pathological and molecular variability in colletotrichum capsici, the cause of fruit rot of chillies in the subtropical region of north‐western india. Phytopathol Z 153(4):232–237
Slatkin M, Hudson RR (1991) Pairwise comparisons of mitochondrial DNA sequences in stable and exponentially growing populations. Genetics 129(2):555–562
Soltis PS, Soltis DE (2000) The role of genetic and genomic attributes in the success of polyploids. Proc Natl Acad Sci U S A 97(13):7051–7057
Sreenivasaprasad S, Meehan B, Mills P, Brown A (1996) Phylogeny and systematics of 18 Colletotrichum species based on ribosomal DNA spacer sequences. Genome 39(3):499–512
Sutton BC (1992) Colletotrichum: biology, pathology and control. In: Bailey JA and Jeger MJ (eds), Colletotrichum: biology, pathology and control, CAB International, Wallingford, UK, pp 1–26
Sydow H (1913) Beitrage zur kenntnis der PilzHora des Siidlichen Ostindiens-I. Anv Myc, XI 329
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123(3):585–595
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28(10):2731–2739
Than P, Jeewon R, Hyde K, Pongsupasamit S, Mongkolporn O, Taylor P (2008) Characterization and pathogenicity of Colletotrichum species associated with anthracnose on chilli (Capsicum spp.) in Thailand. Plant Pathol 57(3):562–572
Torres-Calzada C, Tapia-Tussell R, Higuera-Ciapara I, Perez-Brito D (2013) Morphological, pathological and genetic diversity of Colletotrichum species responsible for anthracnose in papaya (Carica papaya L). Eur J Plant Pathol 135(1):67–79
Wan X, Nardi F, Zhang B, Liu Y (2011) The Oriental Fruit Fly, Bactrocera dorsalis, in China: origin and gradual inland range expansion associated with population growth. PLoS One 6(10), e25238
White TJ, Bruns T, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR protocols: a guide to methods and applications 18:315–322
Woese CR, Kandler O, Wheelis ML (1990) Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci U S A 87(12):4576–4579
Acknowledgments
The corresponding author is grateful to the Department of Science and Technology, Govt. of India, for financial assistance. We would like to thank the anonymous reviewers for their critical comments and suggestions for the improvement of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Katoch, A., Prabhakar, C.S. & Sharma, P.N. Metageographic population analysis of Colletotrichum truncatum associated with chili fruit rot and other hosts using ITS region nucleotide sequences. J. Plant Biochem. Biotechnol. 25, 64–72 (2016). https://doi.org/10.1007/s13562-015-0310-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13562-015-0310-1