
Abstract
Anthracnose caused by Colletotrichum spp. is one of main strawberry diseases worldwide. The disease recently arrived in Iran and become epidemic in the Kurdistan province. A combination of morphological, molecular and pathogen-host interaction approaches (referred to the consolidated species concept) were applied to isolates from symptomatic strawberry tissues collected in Iran. Multi-gene phylogenetic analysis based on ITS-rDNA, beta-tubulin (TUB2) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) identified the species as the S5 clade of Colletotrichum nymphaeae. No significant intraspecies variation was observed in terms of morphology and pathogenicity. No significant genetic variation was observed among the isolates using inter simple sequence repeat analysis (ISSR) and primer pair combination of ERIC1/BOX and ERIC2/BOX fingerprinting markers. Based on the results of this study, it can be postulated that the C. nymphaeae population in Iran established itself from a single origin due to a founder effect. More generally, molecular dating based on relaxed clocks indicates co-radiation of C. nymphaeae S5 and strawberry plants and suggests high reciprocal specificity between the host and pathogen.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Strawberry (Fragaria × ananassa Duch) is an important crop worldwide, representing a relatively small business in Iran, distributed all over the country, with Kurdistan being the leading strawberry-producing province (Tehranifar and Sarsaefi 2002). In 2013 strawberry anthracnose (Howard et al. 1992) was first reported in Iran (Ayoubi and Soleimani 2013) and the disease reached epidemic proportions in the Kurdistan province in 2014 and 2015.
The causal agents of strawberry anthracnose are commonly linked to three species of the Colletotrichum viz. genus (C. acutatum sensu lato, C. gloeosporioides sensu lato and C. fragariae) (Howard et al. 1992; Freeman and Katan 1997). Identification of these three species based on symptoms in the field and host specialisation is usually impracticable, because of poor host (Curry et al. 2002; Photita et al. 2005) and symptom specificity (Smith 2008). Furthermore, environmental factors may influence the stability of morphological traits and generate intermediate forms (Freeman and Katan 1997) and therefore morphological characteristics are often useless for discriminating between different species of the Colletotrichum genus. For example, C. acutatum and C. gloeosporioides can be distinguished based on the conidium shape and size on potato dextrose agar, but not on pea straw agar (Forster and Adaskaveg 1999). Colletotrichum acutatum has acute-ended rather than rounded conidia (Buddie et al. 1999), however this type of conidia has also frequently been recorded in other Colletotrichum spp. (Damm et al. 2012). In addition, isolates within C. acutatum sensu lato may also vary in terms of morphological traits (Sutton 1992; Johnston and Jones 1997). To help discriminate species of the Colletotrichum genus, besides morphology, various molecular and biological methods, such as benomyl sensitivity, vegetative compatibility, RAPD analysis, ITS and beta-tubulin gene phylogenies, have been suggested (Sreenivasaprasad et al. 1994; Lardner et al. 1999; Talhinhas et al. 2005; Sreenivasaprasad and Talhinhas 2005; Whitelaw-Weckert et al. 2007). Based on molecular phylogenetics, nine clades (A1 to A9) have previously been proposed for the C. acutatum complex; some of these clades were subsequently assigned to distinct species, namely C. lupini (A1), C. simmondsii (A2), C. fioriniae (A3), C. clavatum (A4) (Nirenberg et al. 2002; Shivas and Tan 2009; Faedda et al. 2011). Using a multi-gene approach including internal transcribed spacer region of ribosomal DNA (ITS-rDNA), chitin synthase 1 (CHS-1), histone3 (HIS3), actin (ACT), beta-tubulin (TUB2) andglyceraldehyde-3-phosphate dehydrogenase (GAPDH), various strains obtained from strawberry plants all over the world were assigned to six species including C. nymphaeae, C. simmondsii, C. fioriniae, C. acutatum sensu stricto, C. godetiae and C. salicis (Damm et al. 2012; Baroncelli et al. 2015); notably, the majority of strains isolated from strawberry were gathered in the C. nymphaeae clade. (Damm et al. 2012). The aforementioned examples highlight the advantage of the multi-gene approach in capturing the taxonomic diversity of C. acutatum sensu lato.
Accurate identification of plant pathogens is essential to decide on the appropriate plant protection strategies to be applied. For example, C. gloeosporioides isolates were found to be highly sensitive to benomyl, whereas C. acutatum isolates were more tolerant (Bernstein et al. 1995; Brown et al. 1996; Liyanage et al. 1992), indicating that correct species identification may lead to use of the most appropriate fungicides (Whitelaw-Weckert et al. 2007). Given the recent outbreak of anthracnose in strawberry fields in the Kurdistan province, this study aimed to characterise the genetic diversity of Colletotrichum in this region and to understand whether there was single or multiple introduction of the causal agent. We used a combination of morphological, molecular and ecological data, referred to as the consolidated species concept (Quaedvlieg et al. 2014). In order to clarify the evolution of Colletotrichum, initial molecular dating analysis of this genus was performed.
Materials and methods
Sampling and pathogen isolation
Strawberry fields from three different counties in the Kurdistan province (Sanandaj, Kamyaran and Sarvabad), where the disease appeared and become epidemic, were inspected from May to June 2014 (Table 1) to find symptomatic plants. Symptomatic petioles, leaves, blossoms, stems and fruits of strawberry plants with anthracnose symptoms were collected and immediately transferred to the laboratory. Isolation was carried out either by direct transfer of conidial ooze on fruits using a sterile needle and transfer to Petri plates on acidified potato dextrose agar (PDA; Merck, Germany), or by culturing infected plant tissues as follows: small segments were taken from the margin of the lesion and sterilised in sodium hypochlorite (1 %) for 30 s plus alcohol 70 % for 60 s and rinsed in sterile distilled water three times for 30 s. The segments were dried on sterile paper under a laminar hood and then placed on PDA in Petri plates. Inoculated Petri plates were incubated at 25 °C in darkness for 5 days. Monosporic cultures were established using a single spore technique (Arzanlou et al. 2015). Single-spore cultures were transferred onto potato carrot agar (20 g potato, 20 g carrot, 15 g agar, 1000 ml distilled water) in slants and stored in the Collection Culture of Tabriz University (CCTU) at 4 °C.
Pathogenicity of isolates on detached fruits
Pathogenicity of selected Colletotrichum isolates (Table 1) was evaluated on detached untreated fruits of Fragaria × ananassa cv. Camarosa. Fruits were surface-sterilised by dipping in 1 % sodium hypochlorite for 30 s and rinsed three times in sterile distilled water. Fruits were dried on sterile paper under a laminar hood before inoculation. For each fungal isolate, five fruits were inoculated with a drop (10 μl) of conidial water suspension with tween 20 (0.1 %; v/v) (1 × 106 conidia/ml) pipetted onto the fruit surface. Sterile distilled water plus 0.1 % Tween 20 was used instead of conidial suspension in untreated controls (Ureña-Padilla et al. 2002). After inoculation, fruits were placed in plastic containers with moistened filter papers at 20 °C to maintain high relative humidity (> 95 %). After seven days, anthracnose symptoms were recorded. To calculate disease severity, each strawberry fruit was imagined as having a conical shape (arbitrary index) and after calculating the total surface area approximately by measuring the lateral and base area, the following formula was applied: t/T × 100; where (t) and (T) are the infected and total areas respectively of each strawberry fruit. The percentage of infected fruits (incidence) out of the total was also calculated.
Morphological characterisation of isolates
The macroscopic morphological features of the isolates (shape, colour and growth rate) were described on colonies grown on PDA, malt extract agar (MEA; Sigma Aldrich, UK) and oat meal agar (OA; Sigma Aldrich, UK) for 7 days at 25 °C. Sporulation was induced by growth on pine needle agar (PNA) (Smith et al. 1996). Microscopic structures including conidiogenous cells and appressoria were studied using the slide culture technique (Arzanlou et al. 2015). The photographs were taken using a digital camera system (Canon, Japan) mounted on an optical microscope (Nikon ECLIPSE 80i, Japan) with high definition colour. Thirty measurements were made for each relevant microscopic feature, with derivation of 95th percentile intervals and the extremes in parentheses.
DNA extraction, amplification and sequencing
Axenic cultures of the selected isolates on PDA were used for DNA extraction using the protocol of Möller et al. (1992). DNA concentration was measured using a Nano Drop nd-1000 spectrophotometer (NanoDrop Technologies, Wilmington, USA) for each isolate, with three replications, and suitable dilutions were then prepared. In the polymerase chain reaction (PCR), the partial regions of three loci including ITS-rDNA, TUB2 and a part of 200 bp of the GDPAH gene were amplified using primers ITS1 + ITS4 (White et al. 1990), T1 + Bt2b (Glass and Donaldson 1995; O’Donnell and Cigelnik 1997) and GDF1 + GDR1 (Guerber et al. 2003) respectively. All PCR reactions were performed in a total volume of 25 μl, consisting of 50 ng genomic DNA, 1X Dream Taq Green PCR Mastermix (Fer-mentas, Lithuania) and 0.2 μM of each primer. Thermal cycling conditions were as described by Damm et al. (2012). Amplicons were visualised on a 1 % agarose gel stained with ethidium bromide and their size was determined in comparison to the GeneRuler molecular marker (GeneRuler™, #SM0243). For sequencing, PCR products were purified with Exo-Sap enzyme (Euroclone S.p.a., Italy) according to the manufacturer’s instructions. DNA amplicons were then sequenced using a BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, USA).
Phylogenetic analysis
Forward and reverse ABI raw trace files were used to create a consensus sequence using the Staden package program, version 2.0.0b9-src.tar.gz (Staden 1996). Consensus sequences were used as query to blast (Mega BLAST from the NCBIs) the GenBank nucleotide database. For phylogenetic analysis, subjects corresponding to ex-type strains obtained from strawberry belonging to the C. acutatum complex were downloaded from GenBank. All downloaded sequences, together with sequences generated in this study (Supplementary Table 1), were aligned individually (ITS, TUB2 and GADPH separately) using ClustalW multiple alignments implemented in BioEdit (Hall 1999) and subsequently manually checked. The three resulting alignments were concatenated using FASconCAT script (Kück and Meusemann 2010). The best evolutionary model for each of the alignments was obtained using MrModelTest software, v. 2.3 (Nylander 2004). Bayesian inference (BI) using MrBayes v. 3.2.1 (Ronquist and Huelsenbeck 2003) was used to build phylogenetic trees. We ran two distinct BIs, one using ITS and the other using the concatenated dataset (ITS, TUB2 and GADPH). For each of the two BIs, the heating parameter was set at 0.15 and four Markov Chain Monte Carlo (MCMC) chains were run, starting from random trees for 1 million generations, with trees sampled every 1000 generations. The first 25 % of saved trees were discarded as burn-in; consensus tree and posterior probabilities (PP) were determined from the remaining trees, and inspected and printed using Fig Tree v1.4.2. Trees were rooted using Monilochaetes infuscans (isolate CBS 869.96) and two strains of C. orchidophilum (CBS 128556 and CBS 632.80), respectively (Damm et al. 2012; Arzanlou et al. 2015). Sequences derived from this study were deposited at NCBI’s GenBank nucleotide database (http://www.ncbi.nlm.nih.gov; Supplementary Table 1).
Molecular clock analysis
Analysis using BEAST V.1.8 (Drummond and Rambaut 2007) was conducted on a restricted set of the ITS dataset; the subset included only species from the C. acutatum species complex, plus two out-groups and excluded species with identical sequences, in order to avoid the formation of spurious grouping. The ITS dataset was chosen because the only available prior rate in the literature was from a mixed ITS-rRNA dataset (Kasuga et al. 2002), which had the closest phylogenetic proximity to Sordariomycetes (the class containing Colletotrichum spp.). Kasuga et al. (2002) indicated high among-branch rate heterotachy and high variability of rate estimates when using different priors. As we were dating intra-generic species, the rate describing an intra-generic split (P. chrysogenum/P. crystaceum) was chosen by averaging six rates obtained using different priors, obtaining a rate of 0.00113 substitutions per site per million years (SSM). This rate was used as the mean of normal distribution with permissive standard deviation of 0.003, truncating the distribution to the highest and lowest of the six rates of Kasuga et al. 2002 (0.00976 and 0.00001 SSM respectively). The clock was modelled with uncorrelated relaxed lognormal distribution and the replacement model with a KYH model plus equal nucleotide frequencies, gamma distribution with four discrete categories plus invariable sites to match the SYM + G + I model, which was found by MrModeltest v. 2.3 to fit the ITS dataset better. A speciation Yule tree prior was used for all nodes in the tree and BEATS was allowed to infer the tree topology while estimating divergence time, starting from a random tree. Two independent chains were run for 10 million generations each, with sampling every 1000 generations. Satisfactory convergence of the various posteriors was checked with Tracer and a consensus divergence tree was calculated with TreeAnnotator v1.8.2 using burning-in of 1000 sampled trees.
The analysis was repeated, varying three key parameters: the calibration rate, the speciation tree prior and the clock priors. A broader replacement rate by averaging (and truncating accordingly) all the 36 rates of Kasuga et al. (2002) was used, therefore accounting for the higher diversity of rates within ascomycetes; with this approach normal distribution had a mean of 0.002266 SSM, a larger standard deviation of 0.005 and truncation at 0.00976 and 0.00001 SSM: the highest and lowest rates reported by Kasuga et al. (2002) for the ITS of fungi. BEAST analysis using a different speciation tree prior (birth-death process) and the strict clock was further repeated.
Repetitive-sequence-based PCR analysis
The fifteen selected isolates were subjected to polymorphic analysis with repetitive-sequence-based PCR (rep-PCR) genomic fingerprinting using primer pair combinations of ERIC2/BOXA1R and ERICF/BOXA1R (Versalovic et al. 1991, 1994). Amplifications were repeated twice to ensure that the results were reproducible. PCR reactions were prepared in 25 ml volumes, each containing 12.5 μl 2X Dream Taq Green PCR Mastermix (Fer-mentas, Lithuania) 9.5 μl sterile distilled water, 1 μl each of both primers (0.2 μmol/μl stock) and 1 μl of template DNA (50 ng). Distilled water (nuclease free) was added to the PCR reaction instead of DNA templates as a control. The PCR thermal profile consisted of initial denaturation at 96 °C for 2 min, followed by 35 cycles at 94 °C for 30 s, 52 °C for 1 min and 65 °C for 5 min. An aliquot of 5 μl PCR product of each isolate was loaded onto 1.5 % agarose gel 1 × Tris-Acetate-EDTA (TAE) buffer for 1.5 h and then stained with ethidium bromide. Amplicon size was determined in comparison to the GeneRuler molecular marker (#SM0333). Unambiguous bands were scored as “0” and “1” to show matrix data according to their presence or absence. UPGMA analysis was carried out using a simple matching coefficient with the MVSP multi-variate statistical package for each marker and finally a combined dendrogram was created using ERIC1/BOX and ERIC2/BOX patterns together.
Inter simple sequence repeat (ISSR) analysis
Simple sequence repeat analysis was performed using four ISSR primers including ISSR1, ISSR2, ISSR4 and ISSR5, as described by Armengol et al. (2010). Each PCR reaction (25 μl) consisted of 12.5 μl 2X Dream Taq Green PCR Mastermix, 1 μl each of primer (0.4 μmol/μl stock) and 1 μl of template DNA (50 ng), and finally the PCR reaction mixtures were adjusted to a final volume of 25 μl with water (nuclease free). Thermal cycles consisted of initial denaturation at 95 °C for 5 min, followed by 34 cycles at 95 °C for 1 min, four annealing temperatures containing 50, 41, 58 and 49 °C for 1 min for four primers of ISSR1, ISSR2, ISSR4 and ISSR5 respectively, 72 °C for 2 min and final extension of 72 °C for 10 min. Product viewing and gel image analysis were carried out as described above.
Statistical analysis
Experiments (including phylogenetic analysis) were carried out at least twice and one representative experiment is presented. Disease incidence and severity in the pathogenicity test were analysed with the SAS software package (SAS institute, Inc., USA, 2003) using one-way ANOVA. Duncan multiple-ranges test (DMRT) was used to compare the means.
Results
Isolates
In the field, typical symptoms associated with anthracnose were observed on the aerial parts of the strawberry plant, such as black, sunken lesions and conidial ooze on fruits (Supplementary Fig. 1c), sunken and girdled lesions on runner stems (Supplementary Fig. 1a), flower blight (Supplementary Fig. 1b) and the appearance of a reddish brown colour inside the crown tissue. Conidia on fruit were hyaline, unicellular, smooth-walled, cylindrical to fusiform, mostly with both ends acute, or rarely with one end acute and one end round, (11) 13–16 (19.5) × (3.5) 4–4.5 (5) μm in size (CCTUCch32 isolate). In total, ninety-five Colletotrichum isolates were recovered from different parts of symptomatic strawberry plants in sampled areas. Seventy-four isolates were obtained from fruit tissue and the remaining 21 isolates from other plant tissues including petiole (five isolates), stem (nine isolates), leaves (five isolates) and crown (two isolates). Fifteen representative isolates obtained from different plant parts of the plant and geographical locations were selected for the following investigations (Table 1).

Bayesian analysis of ITS sequence alignment using MrBayes v. 3.2.1. The arrows indicate isolates from Iran. The scale bar shows 0.2 expected changes per site. The tree was rooted to Monilochaetes infuscans (CBS 869.96)
Pathogenicity assay and morphological characterisation
After seven days, anthracnose symptoms developed on inoculated strawberries with all the fifteen isolates except on the untreated control. In terms of disease severity and incidence, no significant differences were observed between isolates (Supplementary Fig. 2).

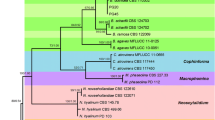
Bayesian inference phylogenetic tree of the Colletotrichum acutatum species complex. The tree was built using concatenated sequences of the ITS (internal transcribed spacer), GAPDH (glyceraldehyde 3-phosphate dehydrogenase) and TUB2 (beta-tubulin), with the GTR + G + I model. The tree was rooted using two strains of C. orchidophilum (CBS 632.80, CBS 119291). The scale bar shows 0.5 expected changes per site. Strawberry isolates are labelled with a strawberry and the isolates from Iran with an arrow
The growth rate of all of the isolates on the three media (PDA, OA and MEA) and the conidium sizes on PDA are presented in Supplementary Table 2. Macroscopic characterisation on PDA, MEA and OA including colony shape and colour, were in agreement with the study of Damm et al. (2012). (Supplementary Figs. 1d–f). Sexual form, sclerotia and chlamydospore were absent on all media. Since all the isolates were apparently morphologically identical, microscopic features are reported only for the CCTUCch32 isolate as a representative isolate. The mycelium was septate, hyaline, smooth, branched, thin-walled and (3) 4–5.5 (7) μm in size. Hyphal anastomosis was repeatedly visible between the vegetative mycelium (Supplementary Fig. 1t). Conidiogenous cells were hyaline, smooth-walled, cylindrical or elongate ampuliform, with periclinal thickening visible and 18.5–21 × 4–5 μm in size (Supplementary Figs. 1i and j). Conidia on PDA were unicellular, smooth-walled, cylindrical to fusiform, mostly with both ends acute or with one end acute and one end round, and rarely with both ends obtuse (Supplementary Fig. 1q). In the slide cultures, appressoria were terminally and intercalary formed on hyphae singly, in small groups or as pedunculate on conidia (Supplementary Fig. 1s), pale brown to brown and globose, subglobose, clavate, ellipsoid, ovoid and with other irregular shapes, entire or with undulated margins and (7) 8–17.5 (20) × (4) 4–9 (9) μm in size (Supplementary Fig. 1k to p). Setae formed on asexual structures on sterile pine leaves for some isolates, pale brown to brown, thick at the base and gradually tapered toward the end and 77–82 μm in length (Supplementary Fig. 1r and Supplementary Table 2).
Phylogenetic analysis
The final aligned ITS dataset contained 68 in-group taxa with a total of 546 characteristics containing 94 parsimonious informative sites. MrModeltest v. 2.3 found SYM + G + I and GTR + G + I to be the most fitting replacement models for ITS and the concatenated dataset, respectively. Bayesian analysis of the ITS dataset showed that all the Iranian isolates sequenced in this study belong to the C. acutatum complex (Fig. 1). The concatenated alignment of ITS, GAPDH and TUB2 contained 1268 characteristics (gene boundaries, ITS: 1–511, GAPDH: 512–776, TUB2: 777–1268), including 207 parsimonious informative sites (31, 86 and 90 for ITS, GAPDH and TUB2 respectively). Bayesian inference (Fig. 2) placed the Iranian isolates, as well as all strains from other countries obtained from strawberry, in six main clades including C. acutatum sensu stricto, C. nymphaeae, C. simmondsii, C. fioriniae, C. godetiae and C. salicis. The C. nymphaeae clade with high posterior probability contained six subclades. All the isolates generated in this study resided in clade S5. All strains located in the S5 clade were obtained from strawberry plants, except for one isolate, which was reported on Mahonia sp. from Italy. Other C. nymphaeae strains obtained from strawberry plants and other hosts or substrates containing Oenothera sp., Anemone sp., Phaselous sp., Protea sp., Nympheae sp., Nuphar sp., Caspicum sp., Pelargonium and litter were clustered in other subclades in the C. nymphaeae clade.
Repetitive-sequence-based PCR and inter simple sequence repeat analysis
Based on repetitive-sequence-based PCR analysis, the size of amplified bands ranged from 400 bp to 4500 bp and from 320 bp to 3000 bp for each primer pair of ERIC1/BOX and ERIC2/BOX respectively (Supplementary Fig. 5). Clustering analysis based on a simple matching coefficient using the data matrix combining both primer pairs, showed that the similarity of the two clades generated was 89.3 % (Supplementary Fig. 3). Selected isolates belonging to various regions and different parts of plant were clustered in three clades. Our isolates clustered in two clades, nine isolates were placed in clade 1 and the rest of the isolates in clade 2. Pilidium cancavum, which was used as an out-group, separated as a distinct group. The isolates obtained from various regions and different parts of the plant were distributed between these two clades (Supplementary Fig. 3).

Divergence estimates within the Colletotrichum acutatum species complex using a reduced version of the ITS dataset and BEAST. Black, green and red dots represent the diversification of C. acutatum species complex, C. nymphaeae and the S5 clade respectively
Following inter simple sequence repeat analysis, ISSR5 patterns were excluded due to inappropriate amplification. The next three primers of ISSR1, ISSR2 and ISSR4 showed no polymorphic variation for any of the isolates tested in this study. The size of the amplified bands for ISSR primers ISSR1, ISSR2 and ISSR4 ranged from 450 bp to 3500 bp, 750 bp to 3500 bp and 600 bp to 2000 bp respectively (Supplementary Fig. 5). The dendrogram revealed two clades including the P. cancavum clade as an out-group and isolates from this study clustered in a single clade (Supplementary Fig. 4).
Divergence estimates for the Colletotrichum acutatum species complex
Our molecular clock analysis (Fig. 3) indicated a relatively young age for the C. acutatum species complex (mean of 12.3 million years; my) and an extremely young age for the origin of the C. nymphaeae stem lineage (split from C. scovillei at 3.2 my). The divergence of C. nymphaeae (split between S5 and S3) was set to 2.2 my. The ITS dataset did not include enough substitutions to differentiate between other subclades (e.g. S1, S2) of C. nymphaeae. Repeating the analysis using different clock and tree priors returned similar estimates (Table 2), but employing a different rate prior had a major impact on the results (Table 2) with all the dates becoming extremely young. In all cases, the diversification of C. nymphaeae and the divergence of S5 followed diversification events within the strawberry genus Fragaria similar to the estimates of Njuguna et al. (2013). All the 95 % HPD intervals (height of the highest posterior densities, pink bars in Fig. 3, Table 2) were extremely high, indicating strong uncertainty regarding the mean estimates; this was probably due to a lack of fossil calibration and the use of permissive standard deviation in calibrating the rate.
Discussion
Because of the recent appearance of anthracnose in the country, the Kurdistan province offered an excellent case study, making it possible to understand whether introduction of the causal agent of strawberry anthracnose was the outcome of a single or multiple events. Indeed, several Colletotrichum spp. including C. acutatum sensu stricto, C. nymphaeae, C. simmondsii, C. fioriniae, C. godetiae and C. salicis have been reported on strawberry worldwide (Damm et al. 2012; Baroncelli et al. 2015). As some of these species cannot be easily identified based on morphological characteristics (Damm et al. 2012), we combined molecular phylogenetics with morphology and ecological essays. Multi-gene sequence data revealed that C. nymphaeae is the sole Colletotrichum species associated with anthracnose disease on strawberry in Iran.
Colletotrichum nymphaeae was initially described in detail on Nymphaea alba from The Netherlands (van der Aa 1978) and re-evaluated in specimens obtained from different hosts, including N. alba, Fragaria sp. and Mahonia aquifolium (Damm et al. 2012). In later studies, the conidial shape in association with the ex-type specimen was reported as ellipsoidal to cylindrical and cylindrical to cylindric-clavate respectively, while the conidia recovered from Fragaria sp. and M. aquifolium were mostly cylindrical to fusiform. Ayoubi and Soleimani (2013) reported that C. acutatum causing strawberry anthracnose in Iran produces cylindrical conidia. However, cylindrical to fusiform conidia were common among C. nymphaeae isolates examined in this study on strawberry (Supplementary Fig. 2Q), in agreement with the study of Damm et al. (2012). C. acutatum (in the broad sense) is primarily distinguished based on its fusiform conidia with acute ends (Simmonds 1965). This diagnostic criterion is however unreliable, because fusiform conidia with acute ends are prevalent in other taxa such as the C. dematium complex and because many strains within the C. acutatum complex exhibit a proportion of conidia with at least one acute end and cylindrical conidia (Damm et al. 2009; Damm et al. 2012). Nevertheless, the presence of fusiform conidia was used for initial screening of Colletotrichum isolates in this study. Nirenberg et al. (2002) noted that conidial shapes are discrepant among conidia formed in acervuli and on the aerial mycelium. The results of this study confirm the view that conidia morphology alone is not enough for diagnosis within the C. acutatum complex and that erroneous identification can be avoided when morphological studies are combined with other characteristics such as sequence data, biochemical and physiological characteristics and host range (Cai et al. 2009).
For this reason, we coupled morphological data with used sequence data from the ITS-rDNA region and two protein coding genes TUB2 and GADPH to ascertain phylogenetic placement of our isolates. ITS sequence data could only reveal the identity of our isolates at species complex level as C. acutatum (Fig. 1). Sequence data from the ITS-rDNA region has largely been used to identify phylogeny of Colletotrichum at species complex levels (Talhinhas et al. 2005; Arzanlou et al. 2015). Conversely, the addition of signals from TUB2 and GADPH genes to ITS (concatenated alignment) was successful in revealing more details of the affinities within the C. acutatum complex, which were in substantial agreement with other studies (Baroncelli et al. 2015; Damm et al. 2012). Our concatenated Bayesian tree clustered all of our specimens in the C. nymphaeae clade, with a high posterior probability (Fig. 2), and also revealed six highly supported subclades.
While the type strain of C. nymphaeae resided in clade S3, almost all specimens of clade S5 were from strawberry. This observation highlights a genetic discrepancy between strains obtained from strawberry and other strains obtained from other substrates. This confirms the view of Baroncelli et al. (2015) regarding host-preference for C. nymphaeae isolates obtained from the strawberry in comparison to other examined strains. Our phylogenetic data (Fig. 2) clearly indicated the S5 strawberry host preference. This view is reinforced by our molecular clock analysis (Fig. 3); the S5 clade diverged from S3 (green dot at 2.2 my) concomitantly with (or just after according to the alternative dates, Table 2) the diversification of the Fragaria genus (divergence of F. gracilis from F. chiloensis) at 2.1 my (Njuguna et al. 2013). Furthermore, the diversification within the S5 clade (red dot at 0.5 my, Fig. 3) happened after the origin of F. chiloensis, the parental species of the current hybrid. Although we suggest extreme care in interpreting our clock analysis - based on the use of suboptimal calibration rates from a different Ascomycota class - our S5 divergence estimates intriguingly match the diversification of strawberry plants and point toward a co-speciation event.
The results of pathogenicity assay supported high homogeneity among isolates in terms of disease incidence and severity rate (Supplementary Fig. 2). Complete uniformity in banding patterns was observed in a selected set of isolates based on ISSR analysis, performed with three primers (Supplementary Figs. 4 and 5), while these three primers have previously pinpointed a high rate of heterogeneity among different fungal populations, including Phaeoisariopsis griseola, Phytophthora cactorum and Rosellinia necatrix (Hantula et al. 1997; Mahuku et al. 2002; Armengol et al. 2010). On the other hand, the primer pair combinations of ERIC2/BOX and ERIC1/BOX showed two genetic groups (Supplementary Fig. 3) with a high level of uniformity (100 and 78.6 % respectively) for isolates. Continuing, the similarity level increased when the consensus dendrogram was created (89.3 %). However, the high level of homogeneity found among our isolates may be the result of recent epidemics in the sampled regions or the geographical vicinity of the sampled areas (Table 1). In previous studies a high rate of uniformity was recorded among a pathogenic population of C. acutatum from strawberry in a number of locations in the United States (Freeman and Katan 1997; Ureña-Padilla et al. 2002). However, no correlation was seen between these two clades in terms of genetic grouping and geographical origin. The lack of a clear relationship between genetic diversity and geographical area has been previously noted for Colletotrichum spp. on other hosts in some studies (Ratanacherdchai et al. 2010; Mahmodi et al. 2014). This phenomenon may imply the recent worldwide spread of pathogen populations from a single or few sources of origin (Denoyes-Rothan et al. 2003). For example, pathogen inoculum importation could occur through infected plant materials, including transplants in particular.
Identification of the C. nymohaeae in this study could provide useful information, making it possible to adopt a suitable management strategy for this disease, particularly in areas recently affected, such as Kurdistan (Iran). It has been shown that besides the strawberry, C. nymphaeae is able to infect multiple hosts, including important dicotyledonous plants such as the apple, pepper, olive and Protea spp. (Damm et al. 2012; Velho et al. 2014; Yamagishi et al. 2015; Nasehi et al. 2016). In general, Colletotrichum species have a cosmopolitan nature and it has been shown that several species can infect a single host, or a single species can infect several hosts (Cai et al. 2009; Hyde et al. 2009). The predominant carpospheric activity of C. acutatum sensu lato on strawberry plants (Freeman and Katan 1997) was confirmed in this study, as the disease symptoms were frequently observed on fruits over the fields and the number of isolates obtained from fruit was 77.89 % of all the isolates obtained.
To the best of our knowledge, C. nymphaeae is reported here for the first time as the causal agent of strawberry anthracnose in Iran. Given the genetic uniformity among the isolates tested in this study, it appears that the adoption of an appropriate quarantine programme could be helpful, in addition to chemical fungicides to prevent the advent of new pathotypes. Furthermore, it appears that further investigation with cross-infection assays, particularly on local weeds present in the area of cultivation, is needed to understand the full host range of C. nymphaeae.
References
Armengol, J., Vicent, A., Leo’n,M., Berbegal, M., Abad-Campos, P., & García-Jiménez, J. (2010). Analysis of population structure of Rosellinia necatrix on Cyperus esculentus by mycelial compatibility and inter-simple sequence repeats (ISSR). Plant Pathology, 59, 179–185.
Arzanlou, M., Bakhshi, M., Karimi, K., & Torbati, M. (2015). Multigene phylogeny reveals three new records of Colletotrichum spp. and several new host records for the mycobiota of Iran. Journal of Plant Protection Research, 55(2), 198–211.
Ayoubi, N., & Soleimani, J. (2013). Occurrence of strawberry anthracnose disease caused by Colletotrichum acutatum in Kurdistan province. 1th Iranian mycological congress. 3–5 September 2013. Rasht, Iran: University of Guilan.
Baroncelli, R., Zapparata, A., Sarrocco, S., Sukno, S. A., Lane, C. R., Thon, M. R., Vannacci, G., Holub, E., & Sreenivasaprasad, S. (2015). Molecular diversity of anthracnose pathogen populations associated with UK strawberry production suggests multiple introductions of three different Colletotrichum species. PloS One, 10(6), e0129140.
Bernstein, B., Zehr, E. I., Dean, R. A., & Shabi, E. (1995). Characteristics of Colletotrichum from peach, apple, pecan, and other hosts. Plant Disease, 79, 478–482.
Brown, A. E., Sreenivasaprasad, S., & Timmer, L. W. (1996). Molecular characterization of slow-growing orange and key lime anthracnose strains of Colletotrichum from citrus as C. acutatum. Phytopathology, 86, 523–527.
Buddie, A. G., Martínez-Culebras, P., Bridge, P. D., Garcia, M. D., Querol, A., Cannon, P. F., & Monte, E. (1999). Molecular characterization of Colletotrichum strains derived from strawberry. Mycological Research, 103(4), 385–394.
Cai, L., Hyde, K. D., Taylor, P. W. J., Weir, B. S., Waller, J., Abang, M. M., Zhang, J. Z., Yang, Y. L., Phoulivong, S., Liu, Z. Y., Prihastuti, H., Shivas, R. G., McKenzie, E. H. C., & Johnston, P. R. (2009). A polyphasic approach for studying Colletotrichum. Fungal Diversity, 39, 183–204.
Curry, K. J., Abril, M., Avant, J. B., & Smith, B. J. (2002). Strawberry anthracnose: Histopathology of Colletotrichum acutatum and C. fragariae. Phytopathology, 92, 1055–1063.
Damm, U., Woudenberg, J.H.C., Cannon, P.F., & Crous, P.W. (2009). Colletotrichum species with curved conidia from herbaceous hosts. Fungal Diversity, 39, 45–87.
Damm, U., Cannon, P. F., Woudenberg, J. H. C., & Crous, P. W. (2012). The Colletotrichum acutatum species complex. Studies in Mycology, 73, 37–114.
Denoyes-Rothan, B., Guérin, G., Délye, C., Smith, B., Minz, D., Maymon, M., & Freeman, S. (2003). Genetic diversity and pathogenic variability among isolates of Colletotrichum species from strawberry. Phytopathology, 93, 219–228.
Drummond, A. J., & Rambaut, A. (2007). BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evolutionary Biology, 7, 214.
Faedda, R., Agosteo, G. E., Schena, L., Mosca, S., Frisullo, S., Magnano, d. S. L. G., & Cacciola, S. O. (2011). Colletotrichum clavatum sp. nov. identified as the causal agent of olive anthracnose in Italy. Phytopathologia Mediterranea, 50, 283–302.
Forster, H., & Adaskaveg, J. E. (1999). Identification of subpopulations of Colletotrichum acutatum and epidemiology of almond anthracnose in California. Phytopathology, 89(11), 1056–1065.
Freeman, S., & Katan, T. (1997). Identification of Colletotrichum species responsible for anthracnose and root necrosis of strawberry in Israel. Phytopathology, 87, 516–521.
Glass, N. L., & Donaldson, G. (1995). Development of primer sets designed for use with PCR to amplify conserved genes from filamentous ascomycetes. Applied and Environmental Microbiology, 61, 1323–1330.
Guerber, J. C., Liu, B., Correll, J. C., & Johnston, P. R. (2003). Characterization of diversity in Colletotrichum acutatum sensu lato by sequence analysis of two gene introns, mtDNA and intron RFLPs, and mating compatibility. Mycologia, 95, 872–895.
Hall, T. A. (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symposium Series, 41, 95–98.
Hantula, J., Lilja, A., & Parikka, P. (1997). Genetic variation and host specificity of Phytophthora cactorum isolated in Europe. Mycological Research, 101, 565–572.
Howard, C. M., Maas, J. L., Chandler, C. K., & Albregts, E. E. (1992). Anthracnose of strawberry caused by the Colletotrichum complex in Florida. Plant Disease, 76, 976–981.
Hyde, K. D., Cai, L., Cannon, P. F., Crouch, J. A., Crous, P. W., Damm, U., et al. (2009). Colletotrichum: names in current use. Fungal Diversity, 39, 147–182.
Johnston, R. P., & Jones, D. (1997). Relationships among Colletotrichum isolates from fruit-rots assessed using rDNA sequences. Mycologia, 89(3), 420–430.
Kasuga, T., White, T. J., & Taylor, J. W. (2002). Estimation of nucleotide substitution rates in Eurotiomycete fungi. Molecular Biology and Evolution, 19(12), 2318–2324.
Kück, P., & Meusemann, K. (2010). FASconCAT: Convenient handling of data matrices. Molecular Phylogenetics and Evolution, 56, 1115–1118.
Lardner, R., Johnston, P. R., Plummer, K. M., & Pearson, M. N. (1999). Morphological and molecular analysis of Colletotrichum acutatum sensu lato. Mycological Research, 103, 275–285.
Liyanage, H. D., McMillan Jr., R. T., & Kistler, H. C. (1992). Two genetically distinct populations of Colletotrichum gloeosporioides from citrus. Phytopathology, 82, 1371–1376.
Mahmodi, F., Kadir, J. B., Puteh, A., Pourdad, S. S., Nasehi, A., & Soleimani, N. (2014). Genetic Diversity and Differentiation of Colletotrichum spp. Isolates Associated with Leguminosae Using Multigene Loci. RAPD and ISSR. Plant Pathology Journal, 30(1), 10–24.
Mahuku, G. S., Henrı’quez, M. A., Mun˜ oz, J., & Buruchara, R. (2002). Molecular markers dispute the existence of the Afro-Andean group of the bean angular leaf spot pathogen, Phaeoisariopsis griseola. Phytopathology, 92, 580–589.
Möller, E. M., Bahnweg, G., & Geiger, H. H. (1992). A simple and efficient protocol for isolation of high molecular weight DNA from filamentous fungi, fruit bodies, and infected plant tissues. Nucleic Acids Research, 20(22), 6115–6116.
Nasehi, A., Kadir, J., Rashid, T. S., Awla, H. K., Golkhandan, E., & Mahmodi, F. (2016). Occurrence of anthracnose fruit rot caused by Colletotrichum nymphaeae on pepper (Capsicum annuum) in Malaysia. Plant Disease. doi:10.1094/PDIS-08-15-0876-PDN.
Nirenberg, H. I., Feiler, U., & Hagedorn, G. (2002). Description of Colletotrichum lupini comb.nov. in modern terms. Mycologia, 94, 307–320.
Njuguna, W., Liston, A., Cronn, R., Ashman, T.-L., & Bassil, N. (2013). Insights into phylogeny, sex function and age of Fragaria based on whole chloroplast genome sequencing. Molecular Phylogenetics and Evolution, 66, 17–29.
Nylander, J. A. A. (2004). MrModeltest v2. Program distributed by the author. Evolutionary Biology Centre: Uppsala University.
O’Donnell, K., & Cigelnik, E. (1997). Two divergent intragenomic rDNA ITS2 types within a monophyletic lineage of the fungus Fusarium are non orthologous. Molecular Phylogenetics and Evolution, 7, 103–116.
Photita, W., Taylor, P. W. J., Ford, R., Hyde, K. D., & Lumyong, S. (2005). Morphological and molecular characterization of Colletotrichum species from herbaceous plants in Thailand. Fungal Diversity, 18, 117–133.
Quaedvlieg, W., Binder, M., Groenewald, J. Z., Summerell, B. A., Carnegie, A. J., Burgess, T. I., & Crous, P. W. (2014). Introducing the Consolidated Species Concept to resolve species in the Teratosphaeriaceae. Persoonia, 33, 1–40.
Ratanacherdchai, K., Wang, H. K., Lin, F. C., & Soytong, K. (2010). ISSR for comparison of cross-inoculation potential of Colletotrichum capsici causing chilli anthracnose. African Journal of Microbiology Research, 4, 76–83.
Ronquist, F., & Huelsenbeck, J. P. (2003). MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics, 19, 1572–1574.
Shivas, R. G., & Tan, Y. P. (2009). A taxonomic re-assessment of Colletotrichum acutatum, intro-ducing C. fioriniae comb. et stat. nov. and C. simmondsii sp. nov. Fungal Diversity, 39, 111–122.
Simmonds, J. H. (1965). A study of the species of Colletotrichum causing ripe fruit rots in Queensland. Queensland Journal of Agricultural and Animal Science, 22, 437–459.
Smith, B. J. (2008). Epidemiology and pathology of strawberry anthracnose: a north American perspective. Hortscience, 43(1), 69–73.
Smith, H., Wingfield, M. J., Crous, P. W., & Coutinho, T. A. (1996). Sphaeropsis sapinea and Botryosphaeria dothidea endophytic in Pinus spp. and Eucalyptus spp. in South Africa. South African Journal of Botany, 62, 86–88.
Sreenivasaprasad, S., & Talhinhas, P. (2005). Genotypic and phenotypic diversity in Colletotrichum acutatum, a cosmopolitan pathogen causing anthracnose on a wide range of hosts. Molecular Plant Pathology, 6, 361–378.
Sreenivasaprasad, S., Mills, P. R., & Brown, A. E. (1994). Nucleotide sequence of the rDNA spacer 1 enables identification of isolates of Colletotrichum as C. acutatum. Mycological Research, 98, 186–188.
Staden, R. (1996). The staden sequence analysis package. Molecular Biotechnology, 5(3), 233–241.
Sutton, B. C. (1992). The genus Glomerella and its anamorph Colletotrichum. In J. A. Bailey & M. J. Jeger (Eds.), Colletotrichum: biology, pathology and control (pp. 1–27). Wallingford: CAB International.
Talhinhas, P., Sreenivasaprasad, S., Neves-Martin, J., & Oliveira, H. (2005). Molecular and phenotypic analyses reveal association of diverse Colletotrichum acutatum groups and a low level of C. gloeosporioides with olive anthracnose. Applied and Environmental Microbiology, 71, 2987–2998.
Tehranifar, A., & Sarsaefi, M. (2002). Strawberry growing in Iran. Acta Horticulturae, 567, 547–549.
Ureña-Padilla, A. R., MacKenzie, S. J., Bowen, B. W., & Legard, D. E. (2002). Etiology and population genetics of Colletotrichum spp. causing crown and fruit rot of strawberry. Phytopathology, 92, 1245–1252.
van der Aa, H. A. (1978). A leaf spot of Nymphaea alba in the Netherlands. Netherlands Journal of Plant Pathology, 84, 109–115.
Velho, A. C., Stadnik, M. J., Casanova, L., Mondino, P., & Alaniz, S. (2014). First report of Colletotrichum nymphaeae causing apple bitter rot in southern Brazil. Plant Disease Note, 98(4), 56.
Versalovic, J., Koeuth, T., & Lupski, J. R. (1991). Distribution of repetitive DNA sequences in eubacteria and application to fingerprinting of bacterial genomes. Nucleic Acids Research, 19, 6823–6831.
Versalovic, J., Schneider, M., de Bruijn, F. J., & Lupski, J. R. (1994). Genomic fingerprinting of bacteria using repetitive sequence based polymerase chain reaction. Methods in Molecular and Cellular Biology, 5(1), 25–40.
White, T. J., Bruns, T., & Taylor, J. (1990). Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In M. A. Innis, D. H. Gelfand, J. J. Sninsky, & J.W. White (Eds.), A guide to molecular methods and applications (pp. 482). New York: Academic Press, USA.
Whitelaw-Weckert, M. A., Curtin, S. J., Huang, R., Steel, C. C., Blanchard, C. L., & Roffey, P. E. (2007). Phylogenetic relationships and pathogenicity of Colletotrichum acutatum isolates from grape in subtropical Australia. Plant Pathology, 56, 448–463.
Yamagishi, N., Ishiyama, Y., Ogiso, H., Sato, T., & Tosa, Y. (2015). Life cycle and control of Colletotrichum nymphaeae, the causal agent of celery stunt anthracnose. Journal of General Plant Pathology, 81(4), 279–286.
Acknowledgments
The authors would like to thank the research deputy of the University of Tabriz in Iran and the Fondazione Edmund Mach in Italy for financial support.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
ESM 1 Supplementary Fig. 1
(DOCX 749 kb)
ESM 2 Supplementary Fig. 2
(DOCX 49 kb)
ESM 3 Supplementary Fig. 3
(DOCX 112 kb)
ESM 4 Supplementary Fig. 4
(DOCX 120 kb)
ESM 5 Supplementary Fig. 5
(DOCX 200 kb)
ESM 6 Supplementary Table 1
(DOCX 24.4 kb)
ESM 7 Supplementary Table 2
(DOCX 14 kb)
Rights and permissions
About this article

Cite this article
Karimi, K., Babai Ahari, A., Arzanlou, M. et al. Application of the consolidated species concept to identify the causal agent of strawberry anthracnose in Iran and initial molecular dating of the Colletotrichum acutatum species complex. Eur J Plant Pathol 147, 375–387 (2017). https://doi.org/10.1007/s10658-016-1009-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10658-016-1009-4