
Abstract
The different coordination modes in carbon-gold(I) complexes are spotlighted with a focus on gold(I). The emergence of gold carbene complexes and their critical discussion in the community is presented, and an overview of their bonding fundamentals and synthetic procedures is given. Several intriguing and sometimes highly reactive gold-carbon coordination complexes that were recently synthesised and isolated are reviewed and a critical outlook into their potential applications is provided. This includes vinylidene and higher cumulenylidene complexes, free carbenes with gold substituents, carbodicarbene complexes, and non-Fischer/Schrock sesquicarbene complexes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Catalysis intermediates and new coordination complexes
Gold and its complexes have had to deal with the longstanding prejudice of being extraordinarily unreactive. We know now that the element has yet to be created for which the combined creativity of global chemistry and physics communities would fail to find an application. Rich chemical applications of homogeneous gold compounds have been found, especially in the catalytic activation of multiple bonds to give a manifold of products, including materials for organic electronics, natural products, and pharmaceuticals [1,2,3,4,5,6,7,8,9].
Even though gold(I) complexes are widely employed in highly selective C–C multiple bond activations even in the presence of air and moisture, it was still not until the advent of modern ligand systems that organogold intermediates of such activations were easily prepared. In early gold organometallics, mono-coordinated gold(I) complexes were found to react vigorously with water and other Lewis bases to form ate complexes with anions of [AuX2]− type [10]. LAu(I)X complexes are typically inert to oxygen and water if the X-type ligands are strongly coordinating, which is one of the reasons why such complexes were perceived to be not catalytically useful. But low reactivity of species is not always a crux. It is rather one of the main features that allowed the application of cationic gold complexes in the stabilisation of exotic species, and it also allows the use of air-stable catalysts and precursors for π-activation reactions. A manifold of catalytic intermediates such as carbene and higher cumulene complexes have been isolated, some of which even represent previously unknown coordination modes of carbon (Fig. 1).

Coordination modes in gold(I) complexes and their formal organic parent compounds
The discovery of gold’s potential in homogeneous catalysis
One of the reasons for the initial misconception of the reactivity of gold complexes is that the classification of transition metals in triads is misleading in group 11. Gold behaves differently to both silver and copper. This difference can be attributed to the onset of significant acceleration of electrons in the 1 s orbital of gold atoms. With 79 protons being close to the core electrons, Einstein’s relativity theory must be considered when describing gold’s electronic structure. Pyykkö described this in detail in 2004 and stressed that this effect even gives a powerful contraction of the outermost 6s orbital [11]. This effect propagates to the valence shell and leads to a contraction and lowered energy level of the valence 6 s orbital and a significant expansion and destabilisation of the 5d orbitals. Gold, thus, acts as a far stronger π-acid than one may have expected.
A consequence of this property is that gold is a surprisingly reactive catalyst for the activation of π-bonds. The oxidation states + I, + II, and + III of gold have all been shown to be useful in homogeneous catalysis. Gold(II) has been proposed as a transient intermediate in photocatalysis, and work is ongoing on discovering ways of employing and generating homogeneous gold(II) species. We still have limited access due to redox instability [12, 13]. However, in the case of gold(I) and gold(III), both species can well be stabilised by donor ligands such as phosphines, amines, and carbene ligands like N-heterocyclic carbenes, making them practical and often air-stable synthesis tools [14].
Researchers had found gold to be useful as π-acid already in the rearrangement of strained carbocycles in the 1970s [2]. Homogeneous gold complexes gained increasing attention when their usefulness was subsequently shown in asymmetric aldol reactions in 1986 by Ito and Hayashi [15, 16], in Teles’ finding of their high activity for the addition of nucleophiles to alkynes in 1998 [17, 18], and Hashmi’s discovery of catalytic C–C bond formation reactions in 2000 [2, 19, 20]. The investigation of the mechanisms of these reactions brought the field of gold-carbon coordination chemistry into the spotlight of the contemporary research. Simple gold(III) salts were often employed in early works, while more elaborate and more stable homogeneous complexes of gold(I) dominated the research landscape in the following years [4, 21, 22]. A typical example of a reaction cycle in gold(I) catalysis is given in Fig. 2. A cationic gold(I) complex 1 typically coordinated by a single phosphine or N-heterocyclic carbene ligand with an open coordination site is used to form a π-complex 3 with an alkyne or alkene 2. A nucleophile such as an amine then attacks the activated π-bond leading to a vinyl complex 4 by element-auration (e.g. aminoauration). The catalytic cycle is closed by electrophilic substitution of the gold moiety which recovers the cationic gold fragment (L)Au+ 1 and produces a 1,2-disubstituted alkene or alkane 5.

A schematic typical gold(I) catalysed π-activation
Gold(I) carbene complexes
Synthesis
The simple example shown before can satisfyingly be explained with simple π- and σ-coordinated gold fragments but it was observed that gold(I) fragments effectively function as electron-donating group (EDG) leading to initially unexpected behaviour of the intermediates generated by π-coordination. Gold carbene complexes soon appeared as peculiar mechanistic suggestion in such reactions [3, 23, 24]. These complexes have been discussed before that alongside the establishment of other carbene ligands that we nowadays consider commodity ligands in catalysis [25, 26]. The ensuing discussion focussed on carbocations bound to a gold centre carrying another neutral ligand which may be a phosphine ligand, an N-heterocyclic carbene ligand, or other related ligand systems.
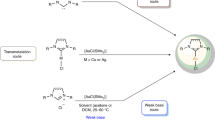
Convincing amounts of evidence have been gathered that gold carbene complexes are key intermediates in the activation of π-bonds and are crucial for explaining the observed distinct reactivity of gold with multiple bonds. As gold was expected to have a limited π-backbonding ability, this suggestion was viewed critically. It soon became clear that this concept can be used for broadening the scope of gold’s applications in homogeneous catalysis and several synthetic methods have been established for isolating gold carbene complexes (Fig. 3) [27,28,29,30]. Direct access can be furnished from diazo compounds 6 which can also be first captured with other metal centres to afford 8. The transfer of the resulting carbenes to cationic gold(I) complexes gives 7.

a–f Selected synthesis methods for gold carbenes
Bonding in gold(I) carbene complexes: carbene or carbenoid?
The nature of the chemical bonding in gold(I) carbenes has caused disputes of an extraordinary nature (Fig. 4a) [27]. They can be described by a σ-bond and a classical π-backbond to a cationic carbon’s empty p-orbital but the relevance of the backbond appeared to be unclear at the time (Fig. 4b). Several groups isolated formal gold carbene complexes with different aims. Some studies wanted to prove the critics wrong by showing the existence of “true” gold carbene complexes with a relevant double bond character. Others objected and showed the low contribution of π-backdonation in isolated complexes experimentally. Depending on the working hypothesis, both of these aims were fulfilled because both hypotheses were completely correct without contradicting each other. Several remarkable gold carbene complexes have been isolated, a selection of which is discussed later. Their publications have been used as a venue for discussing the relevance of the carbene resonance structure II.

Limiting structures of gold carbene complexes on the continuum ranging from σ-coordinated carbocations to carbon–metal double bonds (a). Binding orbital interactions in gold(I) carbene complexes (b)
One shall carefully absorb the following paragraphs as history lesson. While a background motif of the dispute regarding gold carbenes has been the relevance and strength of π-backbonds in gold carbenes, the predominating theme of this disaccord was purely semantic. The terms gold carbene and gold carbenoid were used to voice opinions on the relevance of the π-backbond. Gold carbene implies a two-electron donor with two orbitals involved in bonding [31]. They are usually classical Fischer carbene complexes with low oxidation state metals and π-donors on the carbene ligand. The lowest unoccupied molecular orbital (LUMO) constitutes the π-backbond to the carbene carbon which acts electrophilic and will be partially occupied depending on the electronic features of the complex. Typical gold carbenes contain weak π-backbonds, meaning that they are better described as metalated carbocations. While the backbonds in gold complexes are especially weak, one shall note that this is true for any Fischer carbene complex.
The term carbenoid was a poor way of describing gold carbene complexes. It was used to explicitly state the low relevance of the π-backbond. This name is broadly defined as “Complexed carbene-like entities that display the reactivity characteristics of carbenes, either directly or by acting as sources of carbenes.” in the IUPAC goldbook [32]. Any carbene complex can reasonably be viewed as carbenoid based on this definition which happened within this context, while the involved parties obviously meant that the complexes were like carbene complexes but not carbene complexes. It should be easy to see that this distinction is based on a subjectively selected discrete point on a continuous attribute. Now that the discussions seem settled, it can be summarised that the case of gold carbenes has once more brought to our attention the fragility of nomenclature definitions and Lewis representations of chemical structures.
As Echavarren stated more recently, it seems much more useful to restrict the term carbenoid to LAuCXR2 18 species that are related to Simmons-Smith carbenoids which may release a gold carbene complex 19 by elimination of X or it may release a carbene by α-elimination of LAuX (Scheme 1) [27]. We agree that this distinction makes the use of distinct terms relating to carbene reactivity more useful. Care is still advised both when reading and using the term “carbenoid” as it remains poorly defined. Highlighting the carbene reactivity from a carbenoid by this definition, Steinborn showed that [(PPh3)Au(CH2I)] decomposes quickly at room temperature to give ethylene and [(PPh3)AuI] [33]. Fürstner recently reported that such carbenoids can even be used for the transfer of formal difluorocarbenes [34]. The term “gold carbene complex” shall be considered gold coordinated by a carbene. Gold carbenes push the extremes towards the Fischer side of carbenes and are best described as metalated carbocations with most stabilisation originating from heteroatom or unsaturated substituents.

Gold carbenoids and gold carbene complexes
Isolated gold carbene complexes
Fürstner isolated the dianisole-stabilised gold carbene complex 22, calling it carbenoid at the time (Fig. 5) [35]. They employed an indirect synthesis method by first isolating a rhodium carbene complex generated from the diazonium precursor. Then, they transmetallated the carbene to gold. He would later with the isolation of another carbene complex note that the relevance of the carbene resonance structure II is low and that the discussion is of primarily semantic nature [36]. This dispute about the nature of gold carbene bonding has triggered the hunt for complexes with stronger and stronger carbene character to show the existence of true gold carbene complexes. Several non-heteroatom stabilised species were isolated in the following directly from diazonium precursors such as the tropylium-derived complex 20 by Widenhoefer [37], the bulky imidazole-protected dimesityl-stabilised carbene complex 21 by Straub [38], and Miqueu, Amgoune, and Bourissou’s carbene 23.[39]

Even though biscarbene complexes of gold(III) have been known since 1973, the landscape of gold(III) carbon/carbene complexes is underdeveloped compared to gold(I). Gold(III) carbene complexes with N-heterocyclic carbenes (NHC) of the type [(NHC)2AuX2] act as oxidants and react to the respective gold(I) complexes [(NHC)2Au]+X−.[40, 41] Only in 2017 Mézailles and co-workers synthesised reactive neutral gold(III) carbene complexes by adding a carbodianion to a dicationic gold(III) moiety and showed that these complexes are capable of carbene transfer reactions [42]. It was recently found again that gold(III) carbenes can efficiently give completely different products in catalyses with otherwise identical starting materials [43, 44]. Further elaboration of the carbon coordination chemistry of gold(III) and the in situ interconversion of gold(I) and gold(III) is expected to enhance the scope of homogeneous gold catalysis even further.
Trigonal planar gold(I) coordination allows the stabilisation of new complexes
Among all this progress, surprisingly little of what one would consider basic inorganic chemistry in transition metal coordination chemistry has yet been uncovered in well-defined molecular gold chemistry. Just over the last decade a manifold of π-complexes of gold(III) with C–C multiple bonds was reported [45, 46]. Bourissou has reported a π-allyl complex of gold(III) [47]. Much of this chemistry remains to be investigated in molecular gold(I) complexes. Significant strides forward have been reached, however, with the introduction of ligands that force gold(I) in trigonal planar geometry.
Bourissou’s carbene 23 already gave an outlook into this emerging field in gold’s coordination chemistry [34]. The employed o‐carborane diphosphine ligand forced gold(I) into an unusual trigonal planar geometry which was rare at the time. Trigonal planar gold(I) complexes were found to enable enhanced access to gold’s redox chemistry and even enabled oxidative addition reactions on gold(I) [48,49,50]. The first key step in both the activation of alkynes and alkenes is the π-coordination of the multiple bonds. Only limited structural information on such gold(I) complexes had been garnered [51]. Gold showed C–C hopping in π-complexes too rapidly for nuclear magnetic resonance analysis. Russel and later Hashmi and their co-workers have been able to isolate well-defined trigonal planar ethylene complexes 25 only in 2018 and 2019, respectively (Scheme 2) [49, 52]. Both found in these studies different modes to unlock oxidative addition/reductive elimination chemistry in these gold(I) complexes via the gold(III) complexes 26.[49, 52, 53] The hemilabile P/N-ligand MeDalPhos (di(1-adamantyl)-2-dimethylaminophenylphosphine) has been established in the meantime as gold standard for unlocking oxidative addition on gold(I) complexes, effectively merging the two rich fields of gold(I) and gold(III)-catalysed reactions [54,55,56].

Gold(I) ethylene complexes with bipyridine ligands unlock oxidative additions in gold(I) complexes
Higher cumulenes of gold(I) carbene complexes
Sometimes several exotic species need to be considered in a single mechanism. When two gold centres are employed, we can directly activate 1,5-diynes. The pre-catalysts that have proven most promising are the ones that are already pre-configured for the job, i.e. one gold atom is σ-coordinated (alkinyl complex) and one is π-coordinated (alkine complex) to propynyl. These are now called dual activation catalysts (DAC) [57]. This double activation is one of the methods to directly create gold carbenes in situ. A σ,π-complex such as 27 is formed when such catalysts are reacted with 1,5-diynes (Fig. 6a).

Access to vinylidenes by diyne-cyclisation (a) and to allenylidenes by alkylation of 3-oxoalkinylgold complexes (b)
The regioselectivity of such gold-catalysed cyclisation reactions follows the Zimmerman-Traxler model. This means that 5-endo-dig cyclisation is usually kinetically favoured over other options. This directly leads to a mixed organogold species containing a σ-bound gold centre and a gold vinylidene 28. CH-insertion can then be utilized for the annulation of further ring systems [58]. This selectivity can be overpowered by using a 5-membered ring as backbone for the diyne system. Two fused 5-membered rings are less stable than 5-membered rings fused to 6-membered rings which leads to the preference of a typically hindered 6-endo-dig cyclisation to give 29.[59]
The vinylidene intermediate 28 is a higher cumulene of a gold carbene: a gold carbene complex containing a double bond to the neighbouring carbon centre (Fig. 6b). Synthetic access was even found to allenylidenes containing two cumulated C–C double bonds and a gold carbene bond. This was enabled by electrophilic addition reactions to the oxygen atom in 3-oxoalkinylgold complex 30 which led to the rearrangement of the π-system to give the target allenylidene complexes 31.[60] Further analysis showed that the weak π-interaction with the gold centre leads to an electronic structure that is intermediary between a propargylic cation and an allenylidene.
Aurated carbenes
Whereas the previously discussed reactive species were mostly discovered and isolated in efforts of understanding mechanistic intricacies of known gold-mediated reactions, gold’s stabilising nature has been found useful in quests for novel carbon-based compounds as well. For example, vinylic gold(I) was calculated to have a strong stabilising effect similar to cyclopropyl and phenyl substituents on the ring strain energy (RSE) in cyclopropenes [61].

We discovered that the steric demand of gold centres, while decreasing the ring strain energy, leads to strongly distorted rings in cyclopropen-1-ylgold(I) complexes 33 (Scheme 3) [61]. Similar pre-activation was found in (NHC)(tricyclo[4.1.0.02,7]hept‐1‐yl)gold(I) complexes that contain an aurated bicyclobutane unit [62]. Both of these systems tend to undergo ring-opening reactions that lead to terminally bound gold moieties. This is both due to steric demand and due to gold stabilising the intermediates. Ring-opening of the cyclopropenyl complexes lead to an interesting arrangement: an aurated carbene wherein the carbon atom that is bound to gold(I) is a free carbene itself. This carbene intermediate was shown to be easily accessible at room temperature or with mildly elevated temperatures whereas non-aurated cyclopropenes would show the same reactivity only at temperatures of > 180 °C. The rearrangement to 1H‐inden‐1‐ylgold(I) complexes 34 succeeded with phenyl substituents installed in 3-position in the cyclopropenylgold(I) complexes.

Cyclopropenylgold(I) complexes derived from cyclopropenes and their rearrangement to indenylgold(I) complexes with an allylic gold centre
These indenylgold complexes 34 were the first isolated examples of allylgold(I) species [61]. Gold(I) had been proposed by computational efforts and based on indications from catalysis to show allylic substitution reactivity [63]. Indeed, the isolated indenyl complexes would favour the formation of indenes via an electrophilic SE’ mechanism under rearrangement even with an aggressive proton as electrophile from hydrogen chloride. We are still looking forward to the discovery of π-allylgold(I) complexes and further σ-allylgold(I) and their investigation regarding β-H-elimination and electrophilic addition mechanisms.
Bent allene and allenyl complexes
Schmidbaur has isolated and stabilised carbodiphosphoranes in the 1970s as digold complexes showing that allene-analogues III with strong mesomeric donor substituents (+ M) act as double ylides IV with formally dianionic central carbon atom that is capable of η1-σ-coordination (Fig. 7) [64,65,66,67]. (Me3P+)C2−(P+Me3) was successfully reacted with MeAu(PMe3) to give (Me3P+)2C(AuMe)2 35. This concept later saw a resurgence with upcoming investigations of carbodicarbenes in 2007 [68,69,70,71]. The donor atom X in carbodicarbenes is carbon which is substituted with π-donors and can be considered a carbene ligand for the central carbon atom of the allene. While not the focus of this article, it should be noted in the context of digold complexes that Schmidbaur also discovered aurophilic interactions in related work with the digold complexes of bis(diphenylphosphino)methane with AuCl, AuI, and AuMe [72,73,74,75]. This effect reasoned by relativistic effects in the d10-gold(I) complexes leads to changed reactivity and to the observation of shortened bond lengths of down to 2.5 Å compared to the ion’s van der Waals radius 3.8 Å [76]. (Me2N)2C = C = C(NMe2)2 by Fürstner’s group can be regarded as transient carbodicarbene. The molecule itself is linear along the allene carbon atoms, whereas η1-coordination with gold(I) leads to [(Me2N)2C]+2C−AuPPh3 with a bent allene (carbodicarbene) ligand. The Bertrand group extended the concept to a purely carbon-based carbodicyclopropeneylidene that was trapped with gallium, boron, and gold (36) [77]. Diauration was not found in these carbon-based examples. The discovery of these compounds was intimately intertwined with the coordination chemistry of gold and now they are an emerging class of ligands for their strong σ-donation and their interesting feature of potentially allowing double coordination at a single carbon centre [78,79,80].

Limiting structures of carbodicarbenes (a). Isolated gold carbodicarbenes complexes (b)
Related structures based on gold donors were synthesised from dilithiated propargylbenzene and phenylallene, respectively [81]. The lithiated reactants are easily accessible from the two triple bond isomers prop-1-yn-1-ylbenzene and prop-2-yn-1-ylbenzene and can be reacted with NHC gold(I) chloride precursors to give 37 and 38 (Fig. 8). Protodeauration succeeded selectively at C3 in both cases. The application of these promising entry points in further reactions remains to be investigated. 37 is a plausible access point toward a 3-metalated allenylidene by hydride abstraction. The resulting complex may also behave like an α-goldalkinyl gold carbene complex. 38 is a prototype carbodicarbene based on metallic π-backdonation rather than group 15 π-donation. 38 and analogues with a higher metalation grade or stronger π-donating metals are an interesting alternative to current generation carbodicarbenes and may tackle their stability issues and enable easier synthetic access.

Isolated allene isomeric alkynyl-propargyl digold complex and allen-1,3-diyl digold complex and plausible π-donation scenarios
1,1-Digoldallylium complexes
Just recently in 2020, Bertrand identified mono-coordinated monovalent carbanions as a feasible goal for future isolation [82]. A year prior to that, we reported that the known gold carbene complex synthesis method via ring-opening of cyclopropenes (Fig. 3f) can be used in conjunction with the cyclopropen-1-ylgold(I) complexes 32 to generate the digold(I) complexes 39 derived from said monovalent carbanions (Fig. 9a) [83]. Reacting 39 with dimethylsulfoxide (DMSO) quantitatively leads to the sulfoxonium salt 40 which was crystallised and appeared to be stable towards air and moisture for months. The optical properties indicate that comparably strong π-backbonding is present and a later computational study confirmed that both gold centres participate similarly strong in this (Fig. 9b). Each of the gold atoms donates back about as much as found in an average gold carbene complex which leads to a strong total backbonding and a first optical excitation in the near infrared spectrum [84]. It is interesting to note that this complex breaks the Fischer/Schrock carbene classification as the heteroatom-stabilisation is both weak and coming from a transition metal participating in carbene-bonding itself. The calculations furthermore showed that stabilisation by the allylic domain overpowers stabilisation by the second metal moiety by a factor of 3–4. This is an exotic addition to the portfolio of homodinuclear gold complexes [85], but more importantly a new coordination mode of carbon itself.

1,1-Digoldallylium complexes also termed sesquicarbene complexes (a). Limiting structures of gold sesquicarbene complexes and their average weights determined by intrinsic bond orbital calculations (b)
Future goals
The relatively young history of homogeneous gold catalysis has seen a spiralling rise based on initially identified applications in the first decade of our century. While the next decade brought increasingly elaborate applications, we have seen a particular interest in the mechanistic intricacies and in the isolation of unusual reactive species. Some fundamental blind spots on the map of gold(I)’s carbon coordination chemistry still exist, such as σ- and π-allylgold(I) complexes. We look forward to another golden decade of discovery that will utilize the toolbox that we have discussed:
-
Carbene complexes with comparably strong π-backbond
-
Ethylene complexes
-
Vinylidene and higher cumulenylidene complexes
-
Free carbenes with gold substituents
-
Carbodicarbene complexes with formally dianionic carbon ligand
-
Precursors for cumulenylidenes/carbodicarbenes chemistry based on auric π-backdonation
-
Non-Fischer/Schrock-type sesquicarbene complexes
It must be acknowledged that the by far most important contribution that gold-carbon coordination chemistry can currently make to the field of gold catalysis is the establishment of useful transformations based on organogold complexes (Scheme 4). An astonishing amount of established gold catalyses are terminated by a simple protodeauration rather than by the introduction of additional value from the resulting organogold species. It is highly desirable to establish simple standard protocols for avoiding the protodeauration of products such as 42 to introduce a non-proton electrophile E in 43. Direct (dissociative) substitution reactions are difficult due to the low polarity of C–Au bonds; however, C = Au and N = Au bonds can be utilized in carbene and nitrene transfer reactions under certain conditions [5]. There have been efforts directed towards circumventing this problem by transmetalation to other metal centres [86,87,88,89,90,91,92]. This would effectively unlock the cross-coupling chemistry or element-metal exchange chemistry of these other metal centres in lieu of protodeauration for the termination of gold catalyses [93,94,95]. Another promising way of achieving this is on the horizon with our increasing understanding of the interconversion of gold’s oxidation states. Gold(III) is typically tetracoordinate which allows for associative substitution or oxidative addition reactions which can release 43 by reductive elimination [9]. Further work is needed for the establishment of broadly applicable solutions.

Protodeauration versus electrophilic substitution with other reactants
One major advantage of gold(I) catalyses is that they often do not require any special procedures and can be performed in air which is not true for the discussed reactive species (yet). Their potential is currently severely limited due to partially elaborate synthetic methods and due to our limited understanding of their reactivity. The discussed portfolio of compounds extends our development platform for follow-up reactions of most conceivable catalysis products, but further research is needed to improve their synthesis and application. The primary goals for implementing these species are:
-
1)
Generate the organogold species in situ for enabling sustainable and procedurally convenient investigation and application
-
2)
Establish the complexes as reagents by developing useful and clean follow-up chemistry making use of the unique potential for multiple simultaneous bond formations
-
3)
Close catalytic cycles with the in situ generation of organogold species and useful follow-up chemistry
-
4)
Stabilise the carbon-gold(I) complexes against air and moisture either by electronic and steric tuning of the molecular structure or by establishing the complexes as short-lived intermediates in gold(I)-mediated or -catalysed reactions
Availability of data and material
Not applicable.
Code availability
Not applicable.
References
Hashmi ASK (2004) Homogeneous catalysis by gold. Gold Bull 37:51–65. https://doi.org/10.1007/bf03215517
Hashmi ASK (2003) Homogeneous gold catalysts and alkynes: a successful liaison. Gold Bull 36:3–9. https://doi.org/10.1007/bf03214859
Gorin DJ, Toste FD (2007) Relativistic effects in homogeneous gold catalysis. Nature 446:395–403. https://doi.org/10.1038/nature05592
Rudolph M, Hashmi ASK (2012) Gold catalysis in total synthesis—an update. Chem Soc Rev 41:2448–2462. https://doi.org/10.1039/C1CS15279C
Ye L-W, Zhu X-Q, Sahani RL, Xu Y, Qian, P-C, Liu R-S (2020) Nitrene transfer and carbene transfer in gold catalysis. Chem Rev. https://doi.org/10.1021/acs.chemrev.0c00348
Witzel S, Hashmi ASK, Xie J (2021) Light in gold catalysis. Chem Rev. https://doi.org/10.1021/acs.chemrev.0c00841
Hendrich CM, Bongartz LM, Hoffmann MT, Zschieschang U, Borchert JW, Sauter D, Krämer P, Rominger F, Mulks FF, Rudolph M, Dreuw A, Klauk H, Hashmi ASK (2021) Gold catalysis meets materials science – a new approach to π-extended indolocarbazoles. Adv Synth Catal 363:549–557. https://doi.org/10.1002/adsc.202001123
Cheng X, Zhang L (2020) Designed bifunctional ligands in cooperative homogeneous gold catalysis. CCS Chem 2:1989–2002. https://doi.org/10.31635/ccschem.020.202000454
Zheng Z, Ma Z, Cheng X, Zhao K, Gutman K, Li T, Zhang L (2021) Homogeneous gold-catalyzed oxidation reactions. Chem Rev. https://doi.org/10.1021/acs.chemrev.0c00774
Pope WJ, Gibson CS (1907) CCII.—the alkyl compounds of gold. J Chem Soc Trans 91:2061–2066. https://doi.org/10.1039/CT9079102061
Pyykkö P (2004) Theoretical chemistry of gold. Angew Chem Int Ed 43:4412–4456. https://doi.org/10.1002/anie.200300624
Mohamed AA, Abdou HE, Fackler JP Jr (2010) Coordination chemistry of gold(II) with amidinate, thiolate and ylide ligands. Coord Chem Rev 254:1253–1259. https://doi.org/10.1016/j.ccr.2009.10.017
Preiß S, Förster C, Otto S, Bauer M, Müller P, Hinderberger D, Hashemi Haeri H, Carella L, Heinze K (2017) Structure and reactivity of a mononuclear gold(II) complex. Nat Chem 9:1249–1255. https://doi.org/10.1038/nchem.2836
For further reading, I recommend the recent broader review of gold’s coordination chemistry: Herrera RP, Gimeno MC (2021) Main avenues in gold coordination chemistry. Chem Rev. https://doi.org/10.1021/acs.chemrev.0c00930
Ito Y, Sawamura M, Hayashi T (1986) Catalytic asymmetric aldol reaction: reaction of aldehydes with isocyanoacetate catalyzed by a chiral ferrocenylphosphine-gold(I) complex. J Am Chem Soc 108:6405–6406. https://doi.org/10.1021/ja00280a056
Sawamura M, Ito Y (1992) Catalytic asymmetric synthesis by means of secondary interaction between chiral ligands and substrates. Chem Rev 92:857–871. https://doi.org/10.1021/cr00013a005
Teles JH, Schulz M (1997) BASF AG Patent WO-A1 9721648 [Chem. Abstr. 127:121499]
Teles JH, Brode S, Chabanas M (1998) Cationic gold(I) complexes: highly efficient catalysts for the addition of alcohols to alkynes. Angew Chem Int Ed 37:1415–1418. https://doi.org/10.1002/(SICI)1521-3773(19980605)37:10%3c1415::AID-ANIE1415%3e3.0.CO;2-N
Hashmi ASK, Frost TM, Bats JW (2000) Highly selective gold-catalyzed arene synthesis. J Am Chem Soc 122:11553–11554. https://doi.org/10.1021/ja005570d
Dyker G (2000) An eldorado for homogeneous catalysis? Angew Chem 39:4237–4239. https://doi.org/10.1002/1521-3773(20001201)39:23%3c4237::AID-ANIE4237%3e3.0.CO;2-A
Jiménez-Núñez E, Echavarren AM (2008) Gold-catalyzed cycloisomerizations of enynes: a mechanistic perspective. Chem Rev 108:3326–3350. https://doi.org/10.1021/cr0684319
Fürstner A (2009) Gold and platinum catalysis—a convenient tool for generating molecular complexity. Chem Soc Rev 38:3208–3221. https://doi.org/10.1039/b816696j
Fürstner A, Stelzer F, Szillat H (2001) Platinum-catalyzed cycloisomerization reactions of enynes. J Am Chem Soc 123:11863–11869. https://doi.org/10.1021/ja0109343
Raubenheimer HG, Esterhuysen MW, Timoshkin A, Chen Y, Frenking G (2002) Electrophilic addition of Ph3PAu+ to anionic alkoxy Fischer-type carbene complexes: a novel approach to metal-stabilized bimetallic vinyl ether complexes. Organometallics 21:3173–3181. https://doi.org/10.1021/om020048g
Parks JE, Balch AL (1974) Gold carbene complexes: preparation, oxidation, and ligand displacement. J Organomet Chem 71:453–463. https://doi.org/10.1016/S0022-328X(00)95178-7
Schneider SK, Herrmann WA, Herdtweck E (2003) Synthesis of the first gold(I) carbene complex with a gold-oxygen bond - first catalytic application of gold(I) complexes bearing N-heterocyclic carbenes. Z anorg allg Chemie 629:2363–2370. https://doi.org/10.1002/zaac.200300247
A summary of established gold carbene complex syntheses can be found in the following publication and references therein: Wang Y, Muratore ME, Echavarren AM (2015) Gold carbene or carbenoid: is there a difference? Chem Eur J 21:7332–7339. https://doi.org/10.1002/chem.201406318
A discussion of 1,2- vs. 1,3-rearrangement relevant to route c can be found in: Wang S, Zhang G, Zhang L (2010) Gold-catalyzed reaction of propargylic carboxylates via an initial 3,3-rearrangement. Synlett 5:692–706. https://doi.org/10.1055/s-0029-1219527
The more recent α‐imino gold carbenes are discussed in this and the following reference: Tian X, Song L, Hashmi ASK (2020) α‐Imino gold carbene intermediates from readily accessible sulfilimines: intermolecular access to structural diversity. Chem Eur J 26:3197–3204. https://doi.org/10.1002/chem.201904869
Aguilar E, Santamaría J (2019) Gold-catalyzed heterocyclic syntheses through α-imino gold carbene complexes as intermediates. Org Chem Front 6:1513–1540. https://doi.org/10.1039/C9QO00243J
Salzer A (1999) Nomenclature of organometallic compounds of the transition elements (IUPAC Recommendations 1999). Pure Appl Chem 71:1557–1585. https://doi.org/10.1351/pac199971081557
Muller P (1994) Glossary of terms used in physical organic chemistry: (IUPAC Recommendations 1994). Pure Appl Chem 66:1077–1184. https://doi.org/10.1351/pac199466051077
Steinborn D, Becke S, Herzog R, Günther M, Kircheisen R, Stoeckli-Evans H, Bruhn C (1998) Heteroatomfunktionalisierte Methylgold-Komplexe: Synthese und Struktur von Chlormethyl(triphenylphosphin)- und Phenylthiomethyl(trimethylphosphin)gold. Z anorg allg Chemie 624:1303–1307. https://doi.org/10.1002/(SICI)1521-3749(199808)624:8%3c1303::AID-ZAAC1303%3e3.0.CO;2-R
Tskhovrebov AG, Lingnau JB, Fürstner A (2019) Gold difluorocarbenoid complexes: spectroscopic and chemical profiling. Angew Chem 131:8926–8930. https://doi.org/10.1002/ange.201903957
Seidel G, Fürstner A (2014) Structure of a reactive gold carbenoid. Angew Chem Int Ed 53:4807–4811. https://doi.org/10.1002/anie.201402080
Fürstner A, Morency L (2008) On the nature of the reactive intermediates in gold-catalyzed cycloisomerization reactions. Angew Chem Int Ed 47:5030–5033. https://doi.org/10.1002/anie.200800934
Harris RJ, Widenhoefer RA (2014) Synthesis, structure, and reactivity of a gold carbenoid complex that lacks heteroatom stabilization. Angew Chem Int Ed 53:9369–9371. https://doi.org/10.1002/anie.201404882
Hussong MW, Rominger F, Krämer P, Straub BF (2014) Isolation of a non-heteroatom-stabilized gold-carbene complex. Angew Chem Int Ed 53:9372–9375. https://doi.org/10.1002/anie.201404032
Joost M, Estévez L, Mallet-Ladeira S, Miqueu K, Amgoune A, Bourissou D (2014) Enhanced π-backdonation from gold(I): isolation of original carbonyl and carbene complexes. Angew Chem Int Ed 53:14512–14516. https://doi.org/10.1002/anie.201407684
Minghetti G, Bonati F (1973) Bis(carbene) complexes of gold(I) and gold (III). J Organomet Chem 54:C62–C63. https://doi.org/10.1016/S0022-328X(00)84984-0
Minghetti G, Bonati F, Banditelli G (1976) Carbene complexes of gold(III) and reactions of the coordinated ligand. Inorg Chem 15:1718–1720. https://doi.org/10.1021/ic50161a051
Pujol A, Lafage M, Rekhroukh F, Saffon-Merceron N, Amgoune A, Bourissou D, Nebra N, Fustier-Boutignon M, Mézailles N (2017) A nucleophilic gold(III) carbene complex. Angew Chem Int Ed 56:12264–12267. https://doi.org/10.1002/anie.201706197
Tian X, Song L, Farshadfar K, Rudolph M, Rominger F, Oeser T, Ariafard A, Hashmi ASK (2019) Acyl migration versus epoxidation in gold catalysis: facile, switchable, and atom-economic synthesis of acylindoles and quinoline derivatives. Angew Chem Int Ed 59:471–478. https://doi.org/10.1002/anie.201912334
Zhang C, Wang G, Zhan L, Yang X, Wang J, Wie Y, Xu S, Shi M, Zhang J (2020) Gold(I) or gold(III) as real intermediate species in gold-catalyzed cycloaddition reactions of enynal/enynone? ACS Catal 10:6682–6690. https://doi.org/10.1021/acscatal.0c00220
Savjani N, Roşca D-A, Schormann M, Bochmann M (2013) Gold(III)-olefin-komplexe. Angew Chem Int Ed 52:874–877. https://doi.org/10.1002/anie.201208356
Rekhroukh F, Blons C, Estévez L, Mallet-Ladeira S, Miqueu K, Amgoune A, Bourissou D (2017) Gold(III)-arene complexes by insertion of olefins into gold-aryl bonds. Chem Sci 8:4539–4545. https://doi.org/10.1039/c7sc00145b
Rodriguez J, Szalóki G, Sosa Carrizo ED, Saffon-Merceron N, Miqueu K, Bourissou D (2020) Gold(III) π-allyl complexes. Angew Chem Int Ed 59:1511–1515. https://doi.org/10.1002/anie.201912314
Yang Y, Antoni P, Zimmer M, Sekine K, Mulks FF, Hu L, Zhang L, Rudolph M, Rominger F, Hashmi ASK (2019) Dual gold/silver catalysis involving alkynylgold(III) intermediates formed by oxidative addition and silver-catalyzed C−H activation for the direct alkynylation of cyclopropenes. Angew Chem Int Ed 58:5129–5133. https://doi.org/10.1002/anie.201812577
Yang Y, Eberle L, Mulks FF, Wunsch JF, Zimmer M, Rominger F, Rudolph M, Hashmi ASK (2019) Trans influence of ligands on the oxidation of gold(I) complexes. J Am Chem Soc 141:17414–17420. https://doi.org/10.1021/jacs.9b09363
Gimeno MC, Laguna A (1997) Three- and four-coordinate gold(I) complexes. Chem Rev 97:511–522. https://doi.org/10.1021/cr960361q
Schmidbaur H, Schier A (2010) Gold η2-coordination to unsaturated and aromatic hydrocarbons: the key step in gold-catalyzed organic transformations. Organometallics 29:2–23. https://doi.org/10.1021/om900900u
Harper MJ, Arthur CJ, Crosby J, Emmett EJ, Falconer RL, Fensham-Smith AJ, Gates PJ, Leman T, McGrady JE, Bower JF, Russell CA (2018) Oxidative addition, transmetalation, and reductive elimination at a 2,2′-bipyridyl-ligated gold center. J Am Chem Soc 140:4440–4445. https://doi.org/10.1021/jacs.8b01411
Zhao X, Tian B, Yang Y, Si X, Mulks FF, Rudolph M, Rominger F, Hashmi ASK (2019) Gold-catalyzed stereoselective domino cyclization/alkynylation of N -propargylcarboxamides with benziodoxole reagents for the synthesis of alkynyloxazolines. Adv Synth Catal 361:3155–3162. https://doi.org/10.1002/adsc.201900264
Zeineddine A, Estévez L, Mallet-Ladeira S, Miqueu K, Amgoune A, Bourissou D (2017) Rational development of catalytic Au(I)/Au(III) arylation involving mild oxidative addition of aryl halides. Nat Commun 8:565. https://doi.org/10.1038/s41467-017-00672-8
Joost M, Zeineddine A, Estévez L, Mallet-Ladeira S, Miqueu K, Amgoune A, Bourissou D (2014) Facile oxidative addition of aryl iodides to gold(I) by ligand design: bending turns on reactivity. J Am Chem Soc 136:14654–14657. https://doi.org/10.1021/ja506978c
Rodriguez J, Zeineddine A, Sosa Carrizo ED, Miqueu K, Saffon-Merceron N, Amgoune A, Bourissou D (2019) Catalytic Au(i)/Au(iii) arylation with the hemilabile MeDalphos ligand: Unusual selectivity for electron-rich iodoarenes and efficient application to indoles. Chem Sci 10:7183–7192. https://doi.org/10.1039/c9sc01954e
Hashmi ASK (2014) Dual gold catalysis. Acc Chem Res 47:864–876. https://doi.org/10.1021/ar500015k
Echavarren AM, Muratore ME, López-Carrillo V, Escribano-Cuesta A, Huguet N, Obradors C (2017) Gold-catalyzed cyclizations of alkynes with alkenes and arenes. Organic reactions. Hoboken: Wiley, pp 1–288
Hansmann MM, Rudolph M, Rominger F, Hashmi ASK (2013) Mechanistic switch in dual gold catalysis of diynes: C(sp)-H activation through bifurcation-vinylidene versus carbene pathways. Angew Chem Int Ed 52:2593–2598. https://doi.org/10.1002/anie.201208777
Hansmann MM, Rominger F, Hashmi ASK (2013) Gold–allenylidenes – an experimental and theoretical study. Chem Sci 4:1552–1559. https://doi.org/10.1039/c3sc22227f
Mulks FF, Antoni PW, Rominger F, Hashmi ASK (2018) Cyclopropenylgold(I) complexes as aurated carbenoids or quasi-carbenes. Adv Synth Catal 360:1810–1821. https://doi.org/10.1002/adsc.201701526
Mulks FF, Faraji S, Rominger F, Dreuw A, Hashmi ASK (2018) Highly strained organogold complexes and their gold- or rhodium-catalyzed isomerizations. Chem Eur J 24:71–76. https://doi.org/10.1002/chem.201704652
Hashmi ASK, Schuster AM, Litters S, Rominger F, Pernpointner M (2011) Gold catalysis: 1,3-oxazines by cyclisation of allene amides. Chem Eur J 17:5661–5667. https://doi.org/10.1002/chem.201100132
Schmidbaur H, Gasser O (1976) Die ambidenten Ligandeigenschaften des Bis(trimethylphosphoranyliden)methans. Angew Chem 88:542–543. https://doi.org/10.1002/ange.19760881612
Schmidbaur H, Gasser O, Hussain MS (1977) Doppelylide, I. Synthese und Eigenschaften von Hexamethyl- und sym-Tetramethyldiphenylcarbodiphosphoran. Chem Ber 110:3501–3507. https://doi.org/10.1002/cber.19771101105
Gasser O, Schmidbaur H (1975) Bis(trimethylphosphoranylidene)methane, (CH3)3PCP(CH3)3. J Am Chem Soc 97:6281–6282. https://doi.org/10.1021/ja00854a077
Schmidbaur H (2006) Phosphor-Ylide in der Koordinationssphäre von übergangsmetallen: Eine Bestandsaufnahme. Angew Chem 95:980–1000. https://doi.org/10.1002/ange.19830951205
Tonner R, Frenking G (2007) C(NHC)2: zweibindige Kohlenstoff(0)-Verbindungen mit N-heterocyclischen Carbenliganden – theoretische Belege für eine Molekülklasse mit vielversprechenden Eigenschaften. Angew Chem 119:8850–8853. https://doi.org/10.1002/ange.200701632
Dyker CA, Lavallo V, Donnadieu B, Bertrand G (2008) Synthesis of an extremely bent acyclic allene (A “Carbodicarbene” ): a strong donor ligand. Angew Chem Int Ed 47:3206–3209. https://doi.org/10.1002/anie.200705620
Kaufhold O, Hahn FE (2008) Carbodicarbenes: divalent carbon(0) compounds. Angew Chem Int Ed 47:4057–4061. https://doi.org/10.1002/anie.200800846
Fürstner A, Alcarazo M, Goddard R, Lehmann CW (2008) Coordination chemistry of ene-1,1-diamines and a prototype “carbodicarbene.” Angew Chem Int Ed 47:3210–3214. https://doi.org/10.1002/anie.200705798
Schmidbaur H, Wohlleben A, Wagner F, Orama O, Huttner G (1977) Gold-Komplexe von Diphosphinomethanen, I. Synthese und Kristallstruktur zweikerniger Gold(I)-Verbindungen. Chem Ber 110:1748–1754. https://doi.org/10.1002/cber.19771100519
Schmidbaur H, Wohlleben A, Schubert U, Frank A, Huttner G (1977) Gold-Komplexe von Diphosphinomethanen, II. Synthese und Kristallstruktur achtgliedriger Ringverbindungen von Gold(I) mit Au–Au-Wechselwirkung. Chem Ber 110:2751–2757. https://doi.org/10.1002/cber.19771100810
Schmidbaur H, Wohlleben A, Wagner FE, Van de Vondel DF, Van der Kelen GP (1977) Gold-Komplexe von Diphosphinomethanen, III. Au -Verbindungen durch oxidative Addition von Halogen. Chem Ber 110:2758–2764. https://doi.org/10.1002/cber.19771100811
Schmidbaur H, Wagner FE, Wohlleben-Hammer A (1979) Gold-Komplexe von Diphosphinomethanen, IV. AuI-Verbindungen von Diphosphinoaminen und oxidative Addition von Chlor zu Au -und Au-Komplexen. Chem Ber 112:496–500. https://doi.org/10.1002/cber.19791120212
Wang W, Ji C-L, Liu K, Zhao C-G, Li W, Xie J (2021) Dinuclear gold catalysis. Chem Soc Rev 50:1874–1912. https://doi.org/10.1039/d0cs00254b
Pranckevicius C, Liu L, Bertrand G, Stephan DW (2016) Synthesis of a Carbodicyclopropenylidene: a carbodicarbene based solely on carbon. Angew Chem Int Ed 55:5536–5540. https://doi.org/10.1002/anie.201600765
Liu S, Shih W-C, Chen W-C, Ong T-G (2018) Carbodicarbenes and their captodative behavior in catalysis. Chem Cat Chem 10:1483–1498. https://doi.org/10.1002/cctc.201701577
Wang T-H, Chen W-C, Ong T-G (2017) Carbodicarbenes or bent allenes. J Chin Chem Soc 64:124–132. https://doi.org/10.1002/jccs.201600241
Munz D (2018) Pushing electrons - which carbene ligand for which application? Organometallics 37:275–289. https://doi.org/10.1021/acs.organomet.7b00720
Zargaran P, Mulks FF, Gall S, Rudolph M, Rominger F, Hashmi ASK (2019) Dinuclear NHC gold(I) allenyl and propargyl complexes: an experimental and theoretical study. Organometallics 38:1524–1533. https://doi.org/10.1021/acs.organomet.8b00943
Soleilhavoup M, Bertrand G (2020) Stable carbenes, nitrenes, phosphinidenes, and borylenes: past and future. Chem 6:1275–1282. https://doi.org/10.1016/j.chempr.2020.04.015
Mulks FF, Antoni PW, Gross JH, Graf J, Rominger F, Hashmi, ASK (2019) 1,1-Digoldallylium complexes: diaurated allylic carbocations indicate new prospects of the coordination chemistry of carbon. J Am Chem Soc 141:4687–4695. https://doi.org/10.1021/jacs.8b13395
Mulks FF, Hashmi ASK, Faraji S (2020) Sesquicarbene complexes: bonding at the interface between M-C single bonds and M═C double bonds. Organometallics 39:1814–1823. https://doi.org/10.1021/acs.organomet.0c00102
Bayrakdar TACA, Scattolin T, Ma X, Nolan SP (2020) Dinuclear gold(I) complexes: from bonding to applications. Chem Soc Rev 49:7044–7100. https://doi.org/10.1039/D0CS00438C
Hirner JJ, Shi Y, Blum SA (2011) Organogold reactivity with palladium, nickel, and rhodium: transmetalation, cross-coupling, and dual catalysis. Acc Chem Res 44:603–613. https://doi.org/10.1021/ar200055y
Hashmi ASK, Molinari L (2011) Effective transmetalation from gold to iron or ruthenium. Organometallics 30:3457–3460. https://doi.org/10.1021/om200360q
Shi Y, Blum SA (2011) Gold and rhodium transmetalation: mechanistic insights and dual-metal reactivity. Organometallics 30:1776–1779. https://doi.org/10.1021/om2001316
Al-Amin M, Roth KE, Blum SA (2014) Mechanistic studies of gold and palladium cooperative dual-catalytic cross-coupling systems. ACS Catal 4:622–629. https://doi.org/10.1021/cs400641k
Rangaraju SK, Gonela UM, Kavita A, Yadav JS, Mohapatra DK (2018) Synergistic gold and copper dual catalysis for intramolecular Glaser-Hay coupling: rapid total synthesis of ivorenolide B. Eur J Org Chem 2018:4376–4380. https://doi.org/10.1002/ejoc.201800708
Witzel S, Sekine K, Rudolph M, Hashmi ASK (2018) New transmetalation reagents for the gold-catalyzed visible light-enabled C(sp or sp2)–C(sp2) cross-coupling with aryldiazonium salts in the absence of a photosensitizer. Chem Commun 54:13802–13804. https://doi.org/10.1039/C8CC08227H
Long Y, Cao B, Xiong X, Chan ASC, Sun RW-Y, Zou T (2021) Bioorthogonal activation of dual catalytic and anti-cancer activities of organogold(I) complexes in living systems. Angew Chem 133:4179–4187. https://doi.org/10.1002/ange.202013366
Follow-up chemistry that could be leveraged by transmetallation reactions is discussed in this and the two references thereafter: De Meijere A, Bräse S, Oestreich M (eds.) (2013) Metal‐catalyzed cross‐coupling reactions and more, 1, 2 and 3. Weinheim: Wiley-VCH. https://doi.org/10.1002/9783527655588
Miyaura N (ed.) (2002) Cross-coupling reactions. Topics in current chemistry, vol 219. Berlin, Heidelberg: Springer. https://doi.org/10.1007/3-540-45313-X
Gentner TX, Mulvey RE (2021) Alkali-metal mediation: diversity of applications in main-group organometallic chemistry. Angew Chem Int Ed 60:9247–9262. https://doi.org/10.1002/anie.202010963
Acknowledgements
Florian F. Mulks is grateful to the Alexander von Humboldt-Foundation for a Feodor Lynen Research Fellowship. Proof-reading by Dr. Jan Wenz, Hanna Lee, Seok Yeol Yoo, Yerin Park, Dr. Yangyang Yang, and Patrick W. Antoni is acknowledged. Florian F. Mulks is grateful to the Institute for Basic Science (IBS-R10-A1) in Korea for financial support.
Funding
Not applicable
Author information
Authors and Affiliations
Contributions
Not applicable.
Corresponding author
Ethics declarations
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflict of interest
The author declares no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mulks, F.F. Gold carbene complexes and beyond: new avenues in gold(I)-carbon coordination chemistry. Gold Bull 55, 1–13 (2022). https://doi.org/10.1007/s13404-021-00298-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13404-021-00298-1