
Abstract
Purpose
Pancreatic ductal adenocarcinoma (PDAC) is a highly aggressive malignancy with a dismal prognosis which is, among others, due to a lack of suitable biomarkers and therapeutic targets. Previously, basic gene expression analysis methods have been used for their identification, but recently new algorithms have been developed allowing more comprehensive data analyses. Among them, weighted gene co-expression network analysis (WGCNA) has already been applied to several cancer types with promising results.
Methods
We applied WGCNA to miRNA expression data from PDAC patients. Specifically, we processed microarray-based expression data of 2555 miRNAs in serum from 100 PDAC patients and 150 healthy subjects. We identified network modules of co-expressed miRNAs in the healthy subject dataset and verified their preservation in the PDAC dataset. In the non-preserved modules, we selected key miRNAs and carried out functional enrichment analyses of their experimentally known target genes. Finally, we tested their prognostic significance using overall survival analyses.
Results
Through WGCNA we identified several miRNAs that discriminate healthy subjects from PDAC patients and that, therefore, may play critical roles in PDAC development. At a functional level, we found that they regulate p53, FoxO and ErbB associated cellular signalling pathways, as well as cell cycle progression and various genes known to be involved in PDAC development. Some miRNAs were also found to serve as novel prognostic biomarkers, whereas others have previously already been proposed as such, thereby validating the WGCNA approach. In addition, we found that these novel data may explain at least some of our previous PDAC gene expression analysis results.
Conclusions
We identified several miRNAs critical for PDAC development using WGCNA. These miRNAs may serve as biomarkers for PDAC diagnosis/prognosis and patient stratification, and as putative novel therapeutic targets.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Pancreatic ductal adenocarcinoma (PDAC) is an aggressive malignancy and the fourth most common cause of cancer death worldwide, with a five-year survival rate of approximately 3–6% and a median survival rate of 2–8 months [1]. These poor survival rates are mainly due to the asymptomatic and invasive nature of the disease at advanced stages, often with metastasis at the time of diagnosis, and to a chance of only 10% of being operable [2]. Therefore, suitable diagnostic and prognostic biomarkers and therapeutic targets are considered crucial, but they are currently scarce. Although several gene and protein biomarkers have been proposed [3], none of them is sensitive and/or specific enough. For example, the biomarkers carbohydrate antigen 19–9 (CA 19–9), carcinoembryonic antigen (CEA) [4] and LGR5 [5] have so far not met their initial expectations. Other biomarkers, such as KRAS, CDKN2A, TP53 and SMAD4, have been proposed based on their high mutational status and their involvement in important cellular pathways, such as those regulating apoptosis and cell cycle progression, DNA damage control, and Hedgehog, TGF-β, Wnt/Notch and KRAS signalling [2]. Also, microarray-based expression profiling studies have revealed hundreds of genes that are differentially expressed in PDAC tissues compared to normal pancreatic tissues that, as such, may serve as biomarkers [6, 7].
While the identification and validation of biomarkers are ongoing we should, however, reflect on their failures in order to guide future research. One major drawback of the interpretation of microarray-based expression data is that it addresses individual genes and does not consider correlations among genes. Certainly, the identification of differentially expressed genes may provide biologically and pathologically relevant information, but thanks to recent bioinformatic advancements, new and effective algorithms to interpret gene expression data may allow a more comprehensive identification of new biomarkers. As has extensively been reported [8–11], genes with similar expression patterns may code for proteins with similar biochemical and functional properties, that may form complexes or may act in similar pathways. Therefore, differential co-expression network analysis algorithms have been developed and applied to investigate gene and microRNA (miRNA) expression data [12–15]. As such, the weighted gene co-expression network analysis (WGCNA) algorithm [16] represents a novel systems biology approach that detects key genes related to a sample trait in a network of co-expressed genes. WGCNA is based on similarities in expression profiles among samples and detects, in the network, highly co-expressed genes that are closely connected. These sub-network regions are called modules that have been proven to be conserved across phylogenies, to be enriched in protein-protein interactions and often to consist of functionally related genes [8, 11, 17–19]. By constructing two gene networks, based on normal and tumour tissue expression data, it is possible to identify modules and genes that exhibit different network properties. These distinguishing elements may be involved in pathological processes and could serve as diagnostic and prognostic biomarkers or as potential therapeutic targets. Recently, researchers have been using WGCNA to obtain new and promising candidate biomarkers [20–23]. Previously, we also applied WGCNA to PDAC gene expression data [24] and identified new candidate biomarkers, besides others that were previously reported as potential PDAC biomarkers, thus confirming the reliability of this method.
In addition to genes and proteins, also miRNAs may serve as biomarkers since they can act as tumour promoters (“oncomiRs”) or suppressors, by silencing or promoting known cancer-related genes and pathways, respectively [25]. As for other cancers, aberrant expression of several miRNAs has been linked to PDAC development, and they have been proposed as diagnostic and prognostic biomarkers, and also as indicators of tumour aggression, metastasis, therapy resistance and response [25–27]. Furthermore, miRNAs have been found to play a role in epithelial-mesenchymal transition (EMT) [28], which has previously been suggested to be an essential process related to the progression of several cancers, including PDAC [29, 30]. Moreover, miRNA profiles can distinguish cancers from disorders with similar clinical presentations such as chronic pancreatitis and pancreatic intraepithelial neoplasia (PanIN) [31]. In recent years, miRNA expression profiles have extensively been generated from PDAC patient-derived blood samples, which has resulted in the identification of miRNAs for the diagnosis of pancreatic cancer [32, 33]. Although there are still some methodological challenges to be addressed for the correct quantification of these circulating miRNAs [34], they represent attractive alternatives to tissue-derived miRNAs, including abundance, non-invasive isolation and inexpensive amplification.
Here, we analysed microarray-based miRNA expression profiles from PDAC patient- and healthy individual-derived serum samples. In particular, we applied WGCNA to identify key miRNAs potentially involved in the pathogenesis of PDAC and validated them as diagnostic/prognostic biomarkers. These miRNAs may shed further light on the biological mechanisms underlying PDAC development. They may also serve as potential therapeutic targets.
2 Materials and methods
2.1 miRNA expression data and pre-processing
The recent microarray-based expression dataset GSE59856 [35] was downloaded from the NCBI Gene Expression Omnibus (GEO). It contains quantile normalized miRNA expression data of serum samples obtained from 150 healthy subjects and 100 PDAC patients. The dataset was produced using microarray platform 3D–Gene Human miRNA V20_1.0.0 and was based on miRNA sequences listed in a recent release (v20) of the miRBase database (www.mirbase.org) for probe design, which allows the expression assessment of 2555 miRNAs. As reported before [24], data analyses were performed using the R programming language (v3.3.0), Bioconductor packages (v3.0) and R functions in the WGCNA package [36]. The R codes for reproducing our analyses are provided in the Supplementary Materials.
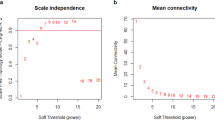
In order to detect outlier samples, we followed the standardized connectivity (Z. K) method as proposed before [37]. The Z. K score represents the overall strength of connections between a given node and all of the other nodes in a network. Samples with a Z. K score < −2 have to be considered as outliers [37]. To assess comparability between normal and PDAC samples, necessary for subsequent analyses, we used the softConnectivity function from package WGCNA. This function measures, across the two datasets, the correlation of the expression level of each miRNA and the correlation of the connectivity of each miRNA, i.e., its co-expression levels with other miRNAs, in the dataset. Datasets are comparable if these two correlations are positive and the p-values are significant. Moreover, higher correlation values (ranging from 0 to 1) indicate higher comparability between the normal and PDAC datasets. Generally, correlation values are better for expression than for connectivity parameters. We also applied the pickSoftThreshold function of the WGCNA package to assess whether the topology of the networks was scale-free, as is required for WGCNA analysis. This function provides a Scale-free Topology Fit Index that, for scale-free topology networks, reaches values above 0.9 for low powers (< 30) as defined in [16].
2.2 Weighted gene co-expression networks and their modules
We created two weighted gene co-expression networks based on the normal and PDAC miRNA expression data, using standard WGCNA procedures [16], including the creation of a matrix of adjacencies and its transformation into a topological overlap matrix (TOM) that was used as input for the hierarchical clustering analysis. In the resulting dendrograms, the function cutreeHybrid from the R package dynamicTreeCut was applied to identify network modules present in the normal dataset (used here as reference dataset). The preservation levels of normal network modules in the PDAC network were assessed by the function modulePreservation from the WGCNA package, that provides a preservation Z-score for each module. Modules with high Z-scores (> 10) are well preserved between normal and PDAC networks, whereas values <10 are indicative for a moderate to low preservation [38]. The grey and gold modules are special WGCNA modules that were not considered (for details see [24]).
2.3 Detection of hub miRNAs and their functional annotations
The WGCNA functions moduleEigengenes and signedKME were used to identify highly connected intra-modular miRNAs, also called hub miRNAs. In particular, the moduleEigengenes function calculates the eigengene of a module, that is a virtual gene representing the gene expression profile of the entire module. The signedKME function measures the distance of the expression profile of a gene to that of the module eigengene and, thus, quantifies how close a gene is to a module, i.e., it measures the module membership of a gene. Within a module, the genes with the highest module membership scores are considered hub genes of that module. In analogy to previous studies [24, 39], we here selected the 20 most connected hub miRNAs for each non-preserved module.
Next, we carried out functional enrichment analyses of known miRNA targets in order to facilitate the interpretation of the biological mechanisms related to these miRNAs. The most interesting miRNAs were used as input for the miRNet web tool [40] to identify the biological pathways, processes, molecular functions and cellular components statistically enriched for the corresponding miRNA target genes. In particular, this tool identifies the enriched KEGG and REACTOME pathways and Gene Ontology (GO) terms based on experimentally confirmed miRNA targets. Since this tool exploits validated miRNA targets, it ensures a higher reliability than tools based on predicted miRNA targets.
2.4 Survival analyses
We used the SurvMicro web tool [41] to carry out survival analyses in order to validate the identified hub miRNAs as prognostic PDAC biomarkers. Survival analyses were performed on an independent PDAC dataset present in The Cancer Genome Atlas (TCGA) (http://cancergenome.nih.gov/) containing miRNA expression and survival data derived from 54 PDAC patients (see Table 1 for the description of the GEO and TCGA cohorts). The SurvMicro tool uses the 2013 version of the TCGA collection and, therefore, allows the analysis of only 54 of 154 patients currently collected in TCGA. The SurvMicro tool divides samples into high-risk and low-risk groups through the median of the prognostic index obtained via a Cox regression model. After this, it generates the hazard ratios (HR) with relative confidence intervals (CI) and p-values.
3 Results
3.1 WGCNA prerequisites for normal and PDAC datasets: outlier samples, batch effects, comparability and scale-free topology
The GEO microarray expression dataset GSE59856 [35] contains miRNA expression data of serum samples obtained from 150 and 100 healthy subjects and PDAC patients, respectively. Since weighted gene co-expression network analysis (WGCNA) requires more than 15 samples to construct a network and to generate robust and refined results, our sample size was largely sufficient. The WGCNA approach is sensitive to the presence of outlier samples and batch effects [17], generally due to systematic and technical differences between different platforms and laboratories. The dataset we chose avoids batch effect issues since it is based on the same type of microarray platform and since the assays were carried out by the same operators. Based on the presence of outliers, we removed 6 and 5 samples from the normal and PDAC datasets, respectively. As required for WGCNA, we also verified whether the normal and PDAC datasets were comparable, i.e., sufficiently similar to highlight even slight differences. This assessment was carried out by the softConnectivity function, which provides positive correlation values when there is high comparability between two datasets. Our datasets were found to be comparable since the overall miRNA expression correlation was 0.85 (p < 1e-200) and the overall miRNA connectivity was 0.3 (p < 3.3e-51) (Supplementary Fig. 1). The connectivity parameter indicates how similar the co-expression level of a miRNA is compared to all other miRNAs in the network. An additional network requirement for WGCNA analysis is scale-free topology, a fundamental property of metabolic and signalling networks in which some nodes (here miRNAs) are more connected (“hub nodes” or “central nodes”) than others (“peripheral nodes”). We found that the normal and PDAC networks exhibited a scale-free topology since the Scale-free Topology Fit Index reached values above 0.9 for low powers (< 30), i.e., the powers were 7 and 5 for the normal and PDAC networks, respectively (Supplementary Fig. 2). This result also indicates that no batch-effects were present in the original microarray datasets.

Clustering dendrograms and modules identified by weighted gene co-expression network analysis (WGCNA). In (a) the clustering diagram and 12 modules for the normal dataset are shown. In (b) the clustering diagram of the PDAC dataset and the normal modules imposed on the PDAC network are shown

Meta-module identification. The module network dendrogram was constructed by clustering module eigengene distances. The horizontal line represents the threshold (0.3) used for defining the meta-modules. The Tan, Greenyellow, Purple, Magenta, Pink and Red modules clearly represent a meta-module (hereafter referred to as “M6” meta-module) in the PDAC network, while no meta-module was identified in the normal network (not shown)
3.2 Identification of modules able to distinguish PDAC from normal miRNA datasets
After constructing weighted gene co-expression networks derived from the normal and PDAC miRNA expression datasets, we set out to identify network modules. According to WGCNA, a module is a highly connected sub-graph, that is a group of strongly co-expressed genes. Generally, these modules are composed of genes with similar biochemical and functional properties or belong to similar pathways [8, 11, 17–19]. Through hierarchical clustering, we identified in the normal network 12 modules of different sizes in terms of miRNA number. Each module was labelled by a different colour, according to WGCNA package functions (Table 2). Considering normal as the reference network and PDAC as the test network, we assessed the preservation level across the two networks by evaluating whether the network properties of each reference module were maintained in the test network. This evaluation has previously been performed to e.g. identify sex- and species-associated differences in brain networks or to determine whether certain cellular pathways are perturbed in particular situations, including cancer [11, 20, 21, 38]. We found that, as expected, some modules exhibited similar characteristics in both networks while others were not preserved and, therefore, potentially related to the development of PDAC. By imposing modules belonging to the normal network onto the PDAC network, we found that many module-associated colour labels still clustered together (i.e., the turquoise, blue and black modules) indicating a good preservation between the two networks (Fig. 1). Instead, other modules (i.e., green and yellow) were found to be scattered throughout the PDAC network, indicating a limited preservation. In order to exactly quantify the module preservation levels, we used the modulePreservation function from the WGCNA package. For each module, a Z-score was calculated representing the preservation level (Table 2). The higher the Z-scores, the more the modules are preserved, whereas modules with Z-scores <10 are lowly preserved and, thus, may distinguish normal from pathological conditions. In particular, the greenyellow, purple, yellow, red, pink, green, magenta and tan modules were found to be not preserved.
We also ascertained whether there were relationships between modules within each network by verifying the presence of meta-modules [42]. By performing hierarchical clustering on the eigengenes of each module it is possible to determine how close, i.e., co-expressed, the modules are to each other. Therefore, a meta-module represents a cluster of co-expressed modules that can be considered as a higher order of organization of the miRNA profiles [42]. Within the PDAC network we identified a meta-module composed of the greenyellow, purple, red, pink, magenta and tan modules, hereafter referred to as the M6 meta-module (Fig. 2), that may be indicative of a common regulatory process encompassing the six modules.
3.3 Identification of hub miRNAs and functional enrichment analysis of their targets
Since the green and yellow modules and the M6 meta-module were found to be not preserved between the normal and PDAC networks, we set out to identify the central nodes (hub miRNAs) that represent them by selecting miRNAs with the highest module membership scores. These miRNAs may play important roles in the pathogenesis of PDAC. The top 20 hub miRNAs identified for each module in the PDAC network are listed in Table 3. In order to subsequently allow a biological and functional interpretation of these hub miRNAs, we performed a functional enrichment analysis limited to experimentally confirmed miRNA target genes. This choice was made to avoid false positive target genes due to prediction algorithms. The hub miRNA list of each module (or meta-module) was processed using the miRNet web tool to identify enriched KEGG and REACTOME pathways and Gene Ontology (GO) terms (Tables 4, 5 and 6). Genes targeted by the green module miRNAs were found to be significantly enriched in cancer pathways, including PDAC, and mainly related to gene expression regulation at both the transcription and translation level. Also the yellow module was found to be related to cancer pathways, including PDAC, and to be enriched in genes involved in cell cycle regulation. According to KEGG and REACTOME resources, the M6 meta-module was mainly related to cancer-associated pathways, apoptosis and transcription regulation.
Using the miRNet tool, miRNA-target interaction networks were created for each module to graphically highlight which miRNAs had a large number of interactions and to calculate the node degree and betweenness, both of which are indicators for node centrality (Fig. 3). We found that miR-302d-3p exhibited the highest node degree and betweenness in the green module, miR-6893-5p in the yellow module and miR-548aq-3p in the M6 meta-module. The interaction graph also allowed us to identify the genes (mRNAs) targeted by different miRNAs. By doing so, we found that the CREBRF, GATA6, NUFIP2 and YOD1 (green module), AGO2, CCNF, KREMEN1, LINC00598, RAB15, SAMD4B, SUMO1 and TPM3 (yellow module) and NUFIP2 (M6 meta-module) genes were regulated by at least five miRNAs.

Interaction network of hub miRNAs in the green (a) and yellow (b) modules and the M6 meta-module (c) with their experimentally validated target genes using the miRNet tool. This network graphically highlights which miRNAs exhibit a large number of interactions
3.4 miRNA candidate biomarkers allow the stratification of PDAC patients into high- and low-risk groups
In order to validate our candidate miRNAs as prognostic biomarkers, we assessed whether hub miRNAs in the green and yellow modules and the M6 meta-module were associated with the overall survival (OS) of PDAC patients [21, 24]. To this end, we performed both single-miRNA and multi-miRNA (top 20 hub miRNAs) survival analyses in each module by applying the SurvMicro tool to an independent miRNA expression dataset of PDAC patients. By using the top 20 hub miRNAs as input for the SurvMicro tool, we found that the miRNA profiles in all modules successfully stratified PDAC patients into high- and low-risk groups as revealed by Kaplan-Meier survival plots (Fig. 4). Specifically, we found that the OS times of the high-risk group patients were at least two-fold shorter than those of patients in the low-risk group (HR 2.79 [95% CI 1.46–5.33] p = 0.001939 for the green module, HR 2.05 [95% CI 1.08–3.89] p = 0.02841 for the yellow module and HR 2.44 [95% CI 1.28–4.65] p = 0.00645 for the M6 meta-module). In addition, we set out to identify miRNA expression signatures that were most significantly associated with PDAC survival outcomes by carrying out single-miRNA survival analyses. We found that an increased expression of each miRNA listed in Table 7, except miR-552-5p, was associated with a better OS.

Kaplan-Meier survival plots for overall survival related to the green (a) and yellow (b) modules and the M6 meta-module (c) using the top 20 hub miRNA signatures. The X and Y axes represent survival time (months) and percent of survival, respectively. Lower curves represent high-risk groups and upper curves represent low-risk groups
4 Discussion
Pancreatic ductal adenocarcinoma (PDAC) is a deadly tumour for which the identification of new diagnostic/prognostic biomarkers and therapeutic targets is of critical importance. Since circulating miRNAs may serve as such, we initiated the identification of miRNA biomarkers and targets through the analysis of microarray-based miRNA expression data obtained from serum of PDAC patients and healthy subjects. Through weighted gene co-expression network analysis (WGCNA) we compared networks in normal and PDAC samples and found that eight out of twelve network modules, i.e., network regions enriched in co-expressed miRNAs, in the normal network were not preserved in the PDAC network. These miRNA modules may play important roles in PDAC development and, therefore, the hub miRNAs within these modules may serve as new diagnostic/prognostic biomarkers or as therapeutic targets. We assessed the potential prognostic value of these miRNAs by validating them in an independent miRNA expression dataset. Through PDAC overall survival (OS) analyses we found that the miRNA expression profiles in all modules, and at least two hub miRNAs in each module, were able to effectively discriminate between two distinct prognosis groups.
Among the green module hub miRNAs, miR-302d-3p (previously known as miR-302d) has previously been found to be over-expressed in serum of both PDAC and chronic pancreatitis patients compared to healthy controls [43], whereas miR-942 has been found to be over-expressed in serum of PDAC patients compared to both patients with chronic pancreatitis [33] and healthy subjects [43]. In comparison with serum from healthy subjects, miR-513c-5p (also known as miR-513) has been found to be upregulated in serum of both PDAC and chronic pancreatitis patients [43]. Among the yellow module miRNAs, miR-135a-3p (previously known as miR-135a*) has been found to be 17-fold and 21-fold up-regulated in serum of patients with PDAC and pancreatitis, respectively, compared to serum of healthy subjects [43]. Also miR-575 was found to be higher expressed in serum of patients diagnosed with PDAC than in serum of healthy subjects, i.e., 2-fold [44] or 4-fold [45]. Conversely, miR-4497 was found to be down-regulated in serum of PDAC patients compared to healthy subjects [46]. Moreover, it has been reported that the serum levels of eight hub miRNAs in this module (miR-204-3p, miR-423-5p, miR-575, miR-4450, miR-4476, miR-4497, miR-4530 and miR-6893-5p) are individually able to discriminate PDAC patients from healthy individuals [35], as revealed by LIMMA R analysis. Among the hub miRNAs in the M6 meta-module, miR-196a-3p (also known as miR-196a*) has not been investigated in PDAC before, whereas the role of miR-196a-5p (also known as miR-196a), which originates from the same pre-miRNA precursor, has extensively been studied. miR-196a has repeatedly been found to be significantly over-expressed in serum of PDAC patients compared to both pancreatitis patients and healthy subjects [43, 47–49]. Moreover, it has been found that miR-196a levels may inversely correlate with patient survival [50]. At the functional level, miR-196a has been found to inhibit apoptosis and to promote the proliferation and invasion of PDAC cells by down-regulating the tumour suppressor gene ING5 (inhibitor of growth family member 5) [51] and the metastasis-related gene NFKBIA (NF-kappa-B inhibitor alpha) [52]. MiR-196a has also been identified in serum-derived exosomes of PDAC patients, although at very low levels [53]. Since the miR-196a levels are usually high in both PDAC cells and in serum of PDAC patients, its low level in exosomes is likely due to selective miRNA loading/sorting during exosome biogenesis. Specific sequence motifs are known to be involved in miRNA sorting into exosomes [54] following rules resembling those of the RNA export language [55].
Furthermore, we identified enriched KEGG and REACTOME pathways and GO terms through functional enrichment analysis of hub miRNA target genes. By doing so, we found that the obtained modules were enriched in PDAC-related pathways. Interestingly, also prostate and colon cancer-specific pathways emerged, and the miRNA profiles in prostate, colon and pancreatic cancers turned out to be more similar than those in lung and breast cancers [56]. In addition, we found that according to KEGG, REACTOME and GO, all modules were highly related to cell cycle and gene expression regulation.
We also identified genes targeted by the highest number of hub miRNAs, using only experimentally validated miRNA-target interactions. In the green module, the GATA6 transcription factor was found to be highly targeted. In the past, its role in PDAC has extensively been investigated. It has e.g. been reported that genomic rearrangements in PDACs frequently include amplification of the GATA6 gene which, accordingly, has been found to be over-expressed [57, 58]. Conversely, silencing of this gene has resulted in inhibition of pancreatic cancer cell growth [59]. A possible underlying mechanism may be a GATA6-mediated induction of the Wnt signalling pathway [60]. More recently, GATA6 has also been suggested to act as a tumour suppressor, since it can inhibit epithelial-mesenchymal transition (EMT), and has been associated with a better patient outcome [61]. Within the yellow module, we found that the tropomyosin 3 gene (TPM3), the small ubiquitin-like modifier 1 gene (SUMO1), the RISC catalytic component argonaute 2 gene (AGO2) and the cyclin F gene (CCNF) were highly targeted by hub miRNAs. Interestingly, by comparing the gene expression profiles of early-recurrent PDAC cases (within 12 months after surgery) with those of late-recurrent cases (over 12 months), it has been found that TPM3 can discriminate between these two groups [62]. The SUMO1, AGO2 and CCNF genes have respectively been found to be 42, 6 and 9-fold higher expressed in primary pancreatic tumours than in normal pancreatic tissues or normal duct epithelial cells [63–65]. Moreover, PDAC-associated missense mutations have been identified in the AGO2 (K709 M) [66] and SAMD4B (I668L) [67] genes, although their functional significance is still unclear [68–71]. Therefore, in addition to the suggested miRNAs, these genes deserve further investigation in relation to PDAC initiation and/or progression.
By comparing the experimentally validated targets of the key miRNAs identified in this work with the hub genes that we previously identified using the same method [24], a relationship emerges. In particular, we found that many key miRNAs target many hub genes (Table 8), which may explain the altered expression profiles of the hub genes that we previously highlighted. The BPNT1, CYCS, MANEAL and SEC23B hub genes are, for example, targeted by at least 2 key miRNAs. Possibly, also other relationships between our two studies may exist but, as yet, the experimentally validated miRNA targets are only partially known.
Taken together, we conclude that our work confirms the validity of some already suggested PDAC biomarkers and has revealed new candidate miRNAs that may be exploited as diagnostic or prognostic biomarkers or as therapeutic targets. Although there is overlap between known candidate biomarkers and the ones that we identified here, further studies are needed to evaluate their practical performance. In this respect, some factors should be carefully considered, including confounding factors such as age, gender and ethnicity of the patients [72–74], differences in tumour stages and treatment regimens, as well as technical factors, such as blood storage conditions and processing [75, 76]. Moreover, since miRNAs can be found in serum in a free form, associated to HDL or enclosed in exosomes or microvesicles, researchers have to pay special attention to the isolation of miRNAs from the desired fraction in order to avoid contamination [77]. Exosome sampling can e.g. also be performed using buccal swabs, thereby taking advantage of the non-invasiveness and higher yield than from blood. Moreover, it is possible to analyse PDAC-specific exosomal sub-populations based on the presence of the Glypican-1 antigen [78] and, by doing so, to validate candidate miRNA biomarkers for patient stratification or therapy response only in exosomes released from PDAC cells.
References
R.L. Siegel, K.D. Miller, A. Jemal, Cancer statistics, 2016. CA Cancer J Clin 66, 7–30 (2016)
M. Hidalgo, S. Cascinu, J. Kleeff, R. Labianca, J.M. Lohr, J. Neoptolemos, F.X. Real, J.L. Van Laethem, V. Heinemann, Addressing the challenges of pancreatic cancer: future directions for improving outcomes. Pancreatology 15, 8–18 (2015)
S. Majumder, S.T. Chari, D.A. Ahlquist, Molecular detection of pancreatic neoplasia: current status and future promise. World J Gastroenterol 21, 11387–11395 (2015)
D.P. Ryan, T.S. Hong, N. Bardeesy, Pancreatic adenocarcinoma. N Engl J Med 371, 1039–1049 (2014)
K. Andrikou, M. Santoni, F. Piva, A. Bittoni, A. Lanese, C. Pellei, A. Conti, C. Loretelli, A. Mandolesi, M. Giulietti, M. Scarpelli, G. Principato, M. Falconi, S. Cascinu, Lgr5 expression, cancer stem cells and pancreatic cancer: results from biological and computational analyses. Future Oncol 11, 1037–1045 (2015)
T.R. Donahue, L.M. Tran, R. Hill, Y. Li, A. Kovochich, J.H. Calvopina, S.G. Patel, N. Wu, A. Hindoyan, J.J. Farrell, X. Li, D.W. Dawson, H. Wu, Integrative survival-based molecular profiling of human pancreatic cancer. Clin Cancer Res 18, 1352–1363 (2012)
G. Zhang, A. Schetter, P. He, N. Funamizu, J. Gaedcke, B.M. Ghadimi, T. Ried, R. Hassan, H.G. Yfantis, D.H. Lee, C. Lacy, A. Maitra, N. Hanna, H.R. Alexander, S.P. Hussain, DPEP1 inhibits tumor cell invasiveness, enhances chemosensitivity and predicts clinical outcome in pancreatic ductal adenocarcinoma. PLoS One 7, e31507 (2012)
M.R. Carlson, B. Zhang, Z. Fang, P.S. Mischel, S. Horvath, S.F. Nelson, Gene connectivity, function, and sequence conservation: predictions from modular yeast co-expression networks. BMC Genomics 7, 40 (2006)
S.L. Carter, C.M. Brechbuhler, M. Griffin, A.T. Bond, Gene co-expression network topology provides a framework for molecular characterization of cellular state. Bioinformatics 20, 2242–2250 (2004)
J.M. Stuart, E. Segal, D. Koller, S.K. Kim, A gene-coexpression network for global discovery of conserved genetic modules. Science 302, 249–255 (2003)
J.A. Miller, S. Horvath, D.H. Geschwind, Divergence of human and mouse brain transcriptome highlights Alzheimer disease pathways. Proc Natl Acad Sci U S A 107, 12698–12703 (2010)
Y.X. Wang, H. Huang, Review on statistical methods for gene network reconstruction using expression data. J Theor Biol 362, 53–61 (2014)
C.F. Staehler, A. Keller, P. Leidinger, C. Backes, A. Chandran, J. Wischhusen, B. Meder, E. Meese, Whole miRNome-wide differential co-expression of microRNAs. Genomics Proteomics Bioinformatics 10, 285–294 (2012)
M. Bhattacharyya, S. Bandyopadhyay, Studying the differential co-expression of microRNAs reveals significant role of white matter in early Alzheimer's progression. Mol BioSyst 9, 457–466 (2013)
C.C. Lin, R. Mitra, F. Cheng, Z. Zhao, A cross-cancer differential co-expression network reveals microRNA-regulated oncogenic functional modules. Mol BioSyst 11, 3244–3252 (2015)
B. Zhang, S. Horvath, A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol 4, 17 (2005)
M.C. Oldham, G. Konopka, K. Iwamoto, P. Langfelder, T. Kato, S. Horvath, D.H. Geschwind, Functional organization of the transcriptome in human brain. Nat Neurosci 11, 1271–1282 (2008)
I. Voineagu, X. Wang, P. Johnston, J.K. Lowe, Y. Tian, S. Horvath, J. Mill, R.M. Cantor, B.J. Blencowe, D.H. Geschwind, Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474, 380–384 (2011)
P.S. Gargalovic, M. Imura, B. Zhang, N.M. Gharavi, M.J. Clark, J. Pagnon, W.P. Yang, A. He, A. Truong, S. Patel, S.F. Nelson, S. Horvath, J.A. Berliner, T.G. Kirchgessner, A.J. Lusis, Identification of inflammatory gene modules based on variations of human endothelial cell responses to oxidized lipids. Proc Natl Acad Sci U S A 103, 12741–12746 (2006)
R. Liu, C.X. Guo, H.H. Zhou, Network-based approach to identify prognostic biomarkers for estrogen receptor-positive breast cancer treatment with tamoxifen. Cancer Biol Ther 16, 317–324 (2015)
C. Clarke, S.F. Madden, P. Doolan, S.T. Aherne, H. Joyce, L. O'Driscoll, W.M. Gallagher, B.T. Hennessy, M. Moriarty, J. Crown, S. Kennedy, M. Clynes, Correlating transcriptional networks to breast cancer survival: a large-scale coexpression analysis. Carcinogenesis 34, 2300–2308 (2013)
Y.S. Lee, S.G. Hwang, J.K. Kim, T.H. Park, Y.R. Kim, H.S. Myeong, J.D. Choi, K. Kwon, C.S. Jang, Y.T. Ro, Y.H. Noh, S.Y. Kim, Identification of novel therapeutic target genes in acquired lapatinib-resistant breast cancer by integrative meta-analysis. Tumour Biol 37, 2285–2297 (2015)
Z. Mousavian, A. Nowzari-Dalini, R.W. Stam, Y. Rahmatallah, A. Masoudi-Nejad, Network-based expression analysis reveals key genes related to glucocorticoid resistance in infant acute lymphoblastic leukemia. Cell Oncol 40, 33–45 (2017)
M. Giulietti, G. Occhipinti, G. Principato, F. Piva, Weighted gene co-expression network analysis reveals key genes involved in pancreatic ductal adenocarcinoma development. Cell Oncol 39, 379–388 (2016)
M. Diab, I. Muqbil, R.M. Mohammad, A.S. Azmi, P.A. Philip, The role of microRNAs in the diagnosis and treatment of pancreatic adenocarcinoma. J Clin Med 5, E59 (2016)
V. Taucher, H. Mangge, J. Haybaeck, Non-coding RNAs in pancreatic cancer: challenges and opportunities for clinical application. Cell Oncol 39, 295–318 (2016)
C. Yu, M. Wang, Z. Li, J. Xiao, F. Peng, X. Guo, Y. Deng, J. Jiang, C. Sun, MicroRNA-138-5p regulates pancreatic cancer cell growth through targeting FOXC1. Cell Oncol 38, 173–181 (2015)
M.L. Abba, N. Patil, J.H. Leupold, H. Allgayer, MicroRNA regulation of epithelial to mesenchymal transition. J Clin Med 5, E8 (2016)
M. Beuran, I. Negoi, S. Paun, A.D. Ion, C. Bleotu, R.I. Negoi, S. Hostiuc, The epithelial to mesenchymal transition in pancreatic cancer: a systematic review. Pancreatology 15, 217–225 (2015)
F. Piva, M. Giulietti, M. Santoni, G. Occhipinti, M. Scarpelli, A. Lopez-Beltran, L. Cheng, G. Principato, R. Montironi, Epithelial to mesenchymal transition in renal cell carcinoma: implications for cancer therapy. Mol Diagn Ther 20, 111–117 (2016)
Y. Xue, A.N. Abou Tayoun, K.M. Abo, J.M. Pipas, S.R. Gordon, T.B. Gardner, R.J. Barth Jr., A.A. Suriawinata, G.J. Tsongalis, MicroRNAs as diagnostic markers for pancreatic ductal adenocarcinoma and its precursor, pancreatic intraepithelial neoplasm. Cancer Gene Ther 206, 217–221 (2013)
N.A. Schultz, C. Dehlendorff, B.V. Jensen, J.K. Bjerregaard, K.R. Nielsen, S.E. Bojesen, D. Calatayud, S.E. Nielsen, M. Yilmaz, N.H. Hollander, K.K. Andersen, J.S. Johansen, MicroRNA biomarkers in whole blood for detection of pancreatic cancer. JAMA 311, 392–404 (2014)
J. Xu, Z. Cao, W. Liu, L. You, L. Zhou, C. Wang, W. Lou, B. Sun, Y. Miao, X. Liu, T. Zhang, Y. Zhao, Plasma miRNAs effectively distinguish patients with pancreatic cancer from controls: a multicenter study. Ann Surg 263, 1173–1179 (2016)
L. Moldovan, K.E. Batte, J. Trgovcich, J. Wisler, C.B. Marsh, M. Piper, Methodological challenges in utilizing miRNAs as circulating biomarkers. J Cell Mol Med 18, 371–390 (2014)
M. Kojima, H. Sudo, J. Kawauchi, S. Takizawa, S. Kondou, H. Nobumasa, A. Ochiai, MicroRNA markers for the diagnosis of pancreatic and biliary-tract cancers. PLoS One 10, e0118220 (2015)
P. Langfelder, S. Horvath, WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008)
M.C. Oldham, P. Langfelder, S. Horvath, Network methods for describing sample relationships in genomic datasets: application to Huntington's disease. BMC Syst Biol 6, 63 (2012)
P. Langfelder, R. Luo, M.C. Oldham, S. Horvath, Is my network module preserved and reproducible? PLoS Comput Biol 7, e1001057 (2011)
E. Vauleon, A. Tony, A. Hamlat, A. Etcheverry, D.C. Chiforeanu, P. Menei, J. Mosser, V. Quillien, M. Aubry, Immune genes are associated with human glioblastoma pathology and patient survival. BMC Med Genet 5, 41 (2012)
Y. Fan, K. Siklenka, S.K. Arora, P. Ribeiro, S. Kimmins, J. Xia, miRNet - dissecting miRNA-target interactions and functional associations through network-based visual analysis. Nucleic Acids Res 44, W135-W141 (2016)
R. Aguirre-Gamboa, V. Trevino, SurvMicro: assessment of miRNA-based prognostic signatures for cancer clinical outcomes by multivariate survival analysis. Bioinformatics 30, 1630–1632 (2014)
P. Langfelder, S. Horvath, Eigengene networks for studying the relationships between co-expression modules. BMC Syst Biol 1, 54 (2007)
A.S. Bauer, A. Keller, E. Costello, W. Greenhalf, M. Bier, A. Borries, M. Beier, J. Neoptolemos, M. Buchler, J. Werner, N. Giese, J.D. Hoheisel, Diagnosis of pancreatic ductal adenocarcinoma and chronic pancreatitis by measurement of microRNA abundance in blood and tissue. PLoS One 7, e34151 (2012)
S. Ali, K. Almhanna, W. Chen, P.A. Philip, F.H. Sarkar, Differentially expressed miRNAs in the plasma may provide a molecular signature for aggressive pancreatic cancer. Am J Transl Res 3, 28–47 (2010)
M. Miyamae, S. Komatsu, D. Ichikawa, T. Kawaguchi, S. Hirajima, W. Okajima, T. Ohashi, T. Imamura, H. Konishi, A. Shiozaki, R. Morimura, H. Ikoma, T. Ochiai, K. Okamoto, H. Taniguchi, E. Otsuji, Plasma microRNA profiles: identification of miR-744 as a novel diagnostic and prognostic biomarker in pancreatic cancer. Br J Cancer 113, 1467–1476 (2015)
M.S. Lin, W.C. Chen, J.X. Huang, H.J. Gao, H.H. Sheng, Aberrant expression of microRNAs in serum may identify individuals with pancreatic cancer. Int J Clin Exp Med 7, 5226–5234 (2014)
J. Liu, J. Gao, Y. Du, Z. Li, Y. Ren, J. Gu, X. Wang, Y. Gong, W. Wang, X. Kong, Combination of plasma microRNAs with serum CA19-9 for early detection of pancreatic cancer. Int J Cancer 131, 683–691 (2012)
X. Kong, Y. Du, G. Wang, J. Gao, Y. Gong, L. Li, Z. Zhang, J. Zhu, Q. Jing, Y. Qin, Z. Li, Detection of differentially expressed microRNAs in serum of pancreatic ductal adenocarcinoma patients: miR-196a could be a potential marker for poor prognosis. Dig Dis Sci 56, 602–609 (2011)
J. Wang, J. Chen, P. Chang, A. LeBlanc, D. Li, J.L. Abbruzzesse, M.L. Frazier, A.M. Killary, S. Sen, MicroRNAs in plasma of pancreatic ductal adenocarcinoma patients as novel blood-based biomarkers of disease. Cancer Prev Res 2, 807–813 (2009)
M. Bloomston, W.L. Frankel, F. Petrocca, S. Volinia, H. Alder, J.P. Hagan, C.G. Liu, D. Bhatt, C. Taccioli, C.M. Croce, MicroRNA expression patterns to differentiate pancreatic adenocarcinoma from normal pancreas and chronic pancreatitis. JAMA 297, 1901–1908 (2007)
M. Liu, Y. Du, J. Gao, J. Liu, X. Kong, Y. Gong, Z. Li, H. Wu, H. Chen, Aberrant expression miR-196a is associated with abnormal apoptosis, invasion, and proliferation of pancreatic cancer cells. Pancreas 42, 1169–1181 (2013)
F. Huang, J. Tang, X. Zhuang, Y. Zhuang, W. Cheng, W. Chen, H. Yao, S. Zhang, MiR-196a promotes pancreatic cancer progression by targeting nuclear factor kappa-B-inhibitor alpha. PLoS One 9, e87897 (2014)
R. Que, G. Ding, J. Chen, L. Cao, Analysis of serum exosomal microRNAs and clinicopathologic features of patients with pancreatic adenocarcinoma. World J Surg Oncol 11, 219 (2013)
A.O. Batagov, V.A. Kuznetsov, I.V. Kurochkin, Identification of nucleotide patterns enriched in secreted RNAs as putative cis-acting elements targeting them to exosome nano-vesicles. BMC Genomics 12 Suppl 3, S18 (2011)
M. Giulietti, S.A. Milantoni, T. Armeni, G. Principato, F. Piva, ExportAid: database of RNA elements regulating nuclear RNA export in mammals. Bioinformatics 31, 246–251 (2015)
S. Volinia, G.A. Calin, C.G. Liu, S. Ambs, A. Cimmino, F. Petrocca, R. Visone, M. Iorio, C. Roldo, M. Ferracin, R.L. Prueitt, N. Yanaihara, G. Lanza, A. Scarpa, A. Vecchione, M. Negrini, C.C. Harris, C.M. Croce, A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A 103, 2257–2261 (2006)
B. Fu, M. Luo, S. Lakkur, R. Lucito, C.A. Iacobuzio-Donahue, Frequent genomic copy number gain and overexpression of GATA-6 in pancreatic carcinoma. Cancer Biol Ther 7, 1593–1601 (2008)
K.A. Kwei, M.D. Bashyam, J. Kao, R. Ratheesh, E.C. Reddy, Y.H. Kim, K. Montgomery, C.P. Giacomini, Y.L. Choi, S. Chatterjee, C.A. Karikari, K. Salari, P. Wang, T. Hernandez-Boussard, G. Swarnalata, M. van de Rijn, A. Maitra, J.R. Pollack, Genomic profiling identifies GATA6 as a candidate oncogene amplified in pancreatobiliary cancer. PLoS Genet 4, e1000081 (2008)
W.B. Chen, F.T. Huang, Y.Y. Zhuang, J. Tang, X.H. Zhuang, W.J. Cheng, Z.Q. Gu, S.N. Zhang, Silencing of GATA6 suppresses SW1990 pancreatic cancer cell growth in vitro and up-regulates reactive oxygen species. Dig Dis Sci 58, 2518–2527 (2013)
Y. Zhong, Z. Wang, B. Fu, F. Pan, S. Yachida, M. Dhara, E. Albesiano, L. Li, Y. Naito, F. Vilardell, C. Cummings, P. Martinelli, A. Li, R. Yonescu, Q. Ma, C.A. Griffin, F.X. Real, C.A. Iacobuzio-Donahue, GATA6 activates Wnt signaling in pancreatic cancer by negatively regulating the Wnt antagonist Dickkopf-1. PLoS One 6, e22129 (2011)
P. Martinelli, E. Carrillo-de Santa Pau, T. Cox, B. Sainz, Jr., N. Dusetti, W. Greenhalf, L. Rinaldi, E. Costello, P. Ghaneh, N. Malats, M. Buchler, M. Pajic, A.V. Biankin, J. Iovanna, J. Neoptolemos, F.X. Real, GATA6 regulates EMT and tumour dissemination, and is a marker of response to adjuvant chemotherapy in pancreatic cancer. Gut. (2016). doi:10.1136/gutjnl-2015-311256
T. Nakamura, Y. Furukawa, H. Nakagawa, T. Tsunoda, H. Ohigashi, K. Murata, O. Ishikawa, K. Ohgaki, N. Kashimura, M. Miyamoto, S. Hirano, S. Kondo, H. Katoh, Y. Nakamura, T. Katagiri, Genome-wide cDNA microarray analysis of gene expression profiles in pancreatic cancers using populations of tumor cells and normal ductal epithelial cells selected for purity by laser microdissection. Oncogene 23, 2385–2400 (2004)
C.A. Iacobuzio-Donahue, A. Maitra, G.L. Shen-Ong, T. van Heek, R. Ashfaq, R. Meyer, K. Walter, K. Berg, M.A. Hollingsworth, J.L. Cameron, C.J. Yeo, S.E. Kern, M. Goggins, R.H. Hruban, Discovery of novel tumor markers of pancreatic cancer using global gene expression technology. Am J Pathol 160, 1239–1249 (2002)
S. Jones, X. Zhang, D.W. Parsons, J.C. Lin, R.J. Leary, P. Angenendt, P. Mankoo, H. Carter, H. Kamiyama, A. Jimeno, S.M. Hong, B. Fu, M.T. Lin, E.S. Calhoun, M. Kamiyama, K. Walter, T. Nikolskaya, Y. Nikolsky, J. Hartigan, D.R. Smith, M. Hidalgo, S.D. Leach, A.P. Klein, E.M. Jaffee, M. Goggins, A. Maitra, C. Iacobuzio-Donahue, J.R. Eshleman, S.E. Kern, R.H. Hruban, R. Karchin, N. Papadopoulos, G. Parmigiani, B. Vogelstein, V.E. Velculescu, K.W. Kinzler, Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 321, 1801–1806 (2008)
A. Thakur, A. Bollig, J. Wu, D.J. Liao, Gene expression profiles in primary pancreatic tumors and metastatic lesions of Ela-c-myc transgenic mice. Mol Cancer 7, 11 (2008)
S.J. Murphy, S.N. Hart, J.F. Lima, B.R. Kipp, M. Klebig, J.L. Winters, C. Szabo, L. Zhang, B.W. Eckloff, G.M. Petersen, S.E. Scherer, R.A. Gibbs, R.R. McWilliams, G. Vasmatzis, F.J. Couch, Genetic alterations associated with progression from pancreatic intraepithelial neoplasia to invasive pancreatic tumor. Gastroenterology 145, 1098–1109 e1091 (2013)
A.V. Biankin, N. Waddell, K.S. Kassahn, M.C. Gingras, L.B. Muthuswamy, A.L. Johns, D.K. Miller, P.J. Wilson, A.M. Patch, J. Wu, D.K. Chang, M.J. Cowley, B.B. Gardiner, S. Song, I. Harliwong, S. Idrisoglu, C. Nourse, E. Nourbakhsh, S. Manning, S. Wani, M. Gongora, M. Pajic, C.J. Scarlett, A.J. Gill, A.V. Pinho, I. Rooman, M. Anderson, O. Holmes, C. Leonard, D. Taylor, S. Wood, Q. Xu, K. Nones, J.L. Fink, A. Christ, T. Bruxner, N. Cloonan, G. Kolle, F. Newell, M. Pinese, R.S. Mead, J.L. Humphris, W. Kaplan, M.D. Jones, E.K. Colvin, A.M. Nagrial, E.S. Humphrey, A. Chou, V.T. Chin, L.A. Chantrill, A. Mawson, J.S. Samra, J.G. Kench, J.A. Lovell, R.J. Daly, N.D. Merrett, C. Toon, K. Epari, N.Q. Nguyen, A. Barbour, N. Zeps, N. Kakkar, F. Zhao, Y.Q. Wu, M. Wang, D.M. Muzny, W.E. Fisher, F.C. Brunicardi, S.E. Hodges, J.G. Reid, J. Drummond, K. Chang, Y. Han, L.R. Lewis, H. Dinh, C.J. Buhay, T. Beck, L. Timms, M. Sam, K. Begley, A. Brown, D. Pai, A. Panchal, N. Buchner, R. De Borja, R.E. Denroche, C.K. Yung, S. Serra, N. Onetto, D. Mukhopadhyay, M.S. Tsao, P.A. Shaw, G.M. Petersen, S. Gallinger, R.H. Hruban, A. Maitra, C.A. Iacobuzio-Donahue, R.D. Schulick, C.L. Wolfgang, R.A. Morgan, R.T. Lawlor, P. Capelli, V. Corbo, M. Scardoni, G. Tortora, M.A. Tempero, K.M. Mann, N.A. Jenkins, P.A. Perez-Mancera, D.J. Adams, D.A. Largaespada, L.F. Wessels, A.G. Rust, L.D. Stein, D.A. Tuveson, N.G. Copeland, E.A. Musgrove, A. Scarpa, J.R. Eshleman, T.J. Hudson, R.L. Sutherland, D.A. Wheeler, J.V. Pearson, J.D. McPherson, R.A. Gibbs, S.M. Grimmond, Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 491, 399–405 (2012)
F. Piva, M. Giulietti, G. Occhipinti, M. Santoni, F. Massari, V. Sotte, R. Iacovelli, L. Burattini, D. Santini, R. Montironi, S. Cascinu, G. Principato, Computational analysis of the mutations in BAP1, PBRM1 and SETD2 genes reveals the impaired molecular processes in renal cell carcinoma. Oncotarget 6, 32161–32168 (2015)
F. Piva, M. Giulietti, L. Baldelli, B. Nardi, C. Bellantuono, T. Armeni, F. Saccucci, G. Principato, Bioinformatic analyses to select phenotype affecting polymorphisms in HTR2C gene. Humanist Psychol 26, 365–372 (2011)
F. Piva, M. Giulietti, B. Nardi, C. Bellantuono, G. Principato, An improved in silico selection of phenotype affecting polymorphisms in SLC6A4, HTR1A and HTR2A genes. Humanist Psychol 25, 153–161 (2010)
F. Bianchi, M. Raponi, F. Piva, A. Viel, I. Bearzi, E. Galizia, R. Bracci, L. Belvederesi, C. Loretelli, C. Brugiati, F. Corradini, D. Baralle, R. Cellerino, An intronic mutation in MLH1 associated with familial colon and breast cancer. Familial Cancer 10, 27–35 (2011)
B. Meder, C. Backes, J. Haas, P. Leidinger, C. Stahler, T. Grossmann, B. Vogel, K. Frese, E. Giannitsis, H.A. Katus, E. Meese, A. Keller, Influence of the confounding factors age and sex on microRNA profiles from peripheral blood. Clin Chem 60, 1200–1208 (2014)
H. Zhao, J. Shen, L. Medico, D. Wang, C.B. Ambrosone, S. Liu, A pilot study of circulating miRNAs as potential biomarkers of early stage breast cancer. PLoS One 5, e13735 (2010)
R. Duttagupta, R. Jiang, J. Gollub, R.C. Getts, K.W. Jones, Impact of cellular miRNAs on circulating miRNA biomarker signatures. PLoS One 6, e20769 (2011)
J.N. Boeckel, C.E. Thome, D. Leistner, A.M. Zeiher, S. Fichtlscherer, S. Dimmeler, Heparin selectively affects the quantification of microRNAs in human blood samples. Clin Chem 59, 1125–1127 (2013)
S. Grasedieck, N. Scholer, M. Bommer, J.H. Niess, H. Tumani, A. Rouhi, J. Bloehdorn, P. Liebisch, D. Mertens, H. Dohner, C. Buske, C. Langer, F. Kuchenbauer, Impact of serum storage conditions on microRNA stability. Leukemia 26, 2414–2416 (2012)
G. Occhipinti, M. Giulietti, G. Principato, F. Piva, The choice of endogenous controls in exosomal microRNA assessments from biofluids. Tumour Biol 37, 11657–11665 (2016)
S.A. Melo, L.B. Luecke, C. Kahlert, A.F. Fernandez, S.T. Gammon, J. Kaye, V.S. LeBleu, E.A. Mittendorf, J. Weitz, N. Rahbari, C. Reissfelder, C. Pilarsky, M.F. Fraga, D. Piwnica-Worms, R. Kalluri, Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature 523, 177–182 (2015)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
ᅟ
Conflict of interest
The authors declare that they have no conflict of interest.
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
ESM 1
(DOC 72 kb)
Rights and permissions
About this article

Cite this article
Giulietti, M., Occhipinti, G., Principato, G. et al. Identification of candidate miRNA biomarkers for pancreatic ductal adenocarcinoma by weighted gene co-expression network analysis. Cell Oncol. 40, 181–192 (2017). https://doi.org/10.1007/s13402-017-0315-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13402-017-0315-y