
Abstract
A multi-component starch-gelatin hybrid hydrogel was synthesized using eco-friendly approach. The starch-based hydrogel (without gelatin) was also synthesized to highlight the importance of gelatin in the hybrid hydrogel. The RSM-CCD (response surface methodology integrated central composite design) was used to optimize the synthesis of both the hydrogels to their maximum swelling capacity. The synthesis of hydrogel was confirmed using various characterization techniques including FTIR, SEM, XRD, thermal methods, compressive strength etc. The gelatin was found to enhance the compressive strength of the hybrid hydrogel. Further, silver nanoparticle-embedded starch and starch-gelatin hydrogels were synthesized. The hydrogel network itself reduced the silver ions to silver nanoparticles without the use of any external reductant and surfactant. The TEM images revealed the spherical AgNPs in the size range of 4–19 nm and quasi-spherical of 4–58 nm embedded in hybrid and starch hydrogels, respectively. Furthermore, the nanocomposite hydrid hydrogel was found to be better as stabilizing agent for stabilization of silver nanoparticles in comparison to nanocomposite starch hydrogel.The silver-embedded hybrid hydrogel exhibited good anti-bacterial activity for gram-negative (E. coli) along with gram-positive bacteria (S. aureus). Embraced with high mechanical strength, the various characteristics of silver-embedded starch-gelatin hydrogel such as good hydrophilicity, biocompatibility, biodegradability, non-cytotoxicity, and hemocompatibility reflect its utility as wound dressing material.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
The metal nanoparticles have been extensively investigated due to their distinctive physical and chemical properties along with comprehensive prospective applications. The important factors that decide the intrinsic properties of nanoparticles include their size, shape, crystallinity and structure [1]. Presently, silver nanoparticles (AgNPs) have gained importance in medical, health care, food, textile industry, catalysis, biosensing etc. [1,2,3]. The chemical reduction approach is quite common for synthesis of metal nanoparticles apart from the other approches like microorganism reduction, microwave-assisted method, photochemical reduction, laser ablation, γ irradiation etc. [4]. The different chemical reducing agents like sodium borohydride, hydrazine, hydroquinone, N, Nˈ-dimethylformamide, citrate, ascorbic acid etc. are required for the reduction of metal salts to synthesize nanoparticles [1, 2]. However, most of the reducing agents used are toxic in nature. Furthermore, synthesized metal nanoparticles are not stable in solution due to their small size and have tendency to lose their chemically active surface area through aggregation [5]. The non-toxic and biodegradable biological macromolecules such as polysaccharides and proteins can be useful for the synthesis as well as stabilization of metal nanoparticles [4]. The starch, carboxymethyl cellulose, sodium alginate, chitosan, gums, polypeptides, etc. could function as reducing and capping agents for the preparation of metal nanoparticles [6,7,8,9,10,11,12,13,14,15,16,17]. These cost-effective biomacromolecules are compatible for different biomedical applications [15]. Polysaccharides are frequently utilized for the synthesis of hydrogels due to their easy availability, biodegradability, high water swellability, and non-toxicity [18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34]. The chemical conversion of these polysaccharides to hydrogels results in three-dimensional cross-linked porous network structures. The controlled pore size distribution in hydrogels can easily manuplatethe overall size and shape of metal nanoparticles [2]. The metal nanoparticles get stabilized in the pores of hydrogel by interacting with the different functionalities present on the polymer backbone and results in the formation of nanoparticle-embedded hydrogel composites with improved physical characteristics.
The synthesis and growth of nanoparticles within the porous networks of hydrogel depend upon the chemical structure and magnitude of cross-linking in the polymeric backbone. Nowadays, the use of polysaccharide-based hydrogels for metal nanoparticles synthesis within a narrow size distribution and stabilization is a recognized approach [2, 35,36,37]. Further, porous network structure of hydrophilic hydrogels can regulate easy diffusion and sustained release of metal ions [5].
Moreover, to strengthen the thermal characteristics and compressive properties of hydrogels, they can be converted to multi-component networks including semi or interpenetrating polymer networks (semi-IPNs/IPNs) [38,39,40,41,42]. Such semi-IPNs and IPNs have a variety of applications in diversified fields [43, 44].
Starch contains two polysaccharide units: amylose and amylopectin having hydroxyl functional groups and is semi-crystalline in nature. Gelatin, on the other hand, is a linear polymer possessing ˗NH2 and -COOH functional groups. It is hydrophilic, non-toxic, biodegradable and biocompatible in nature. Gelatin improves the hydrogel network and enhances the hydrophilicity, functionality and mechanical strength of hydrogels [40, 45, 46]. The several food processing applications have been utilizing starch and gelatin in combination [45, 47]. However, the research work related to the cross-linking of starch and gelatin to produce a hybrid hydrogel network and further synthesis and growth of silver nanoparticles inside hydrogel’s matrix to produce nanocomposite hydrogel is limited. The resultant composite of nanaoparticles and hydrogel can be utilized for various applications. AgNPs are biocompatible, non-toxic and have antimicrobial activity. The AgNPs-embedded hybrid hydrogels with improved properties could be a good material of biomedical relevance and useful as wound dressing material, because of this reason AgNPs were used in present work.
Considering the importance of polysaccharides- and proteins-based hydrogels as green materials, the present work synthesized the novel semi-IPN hybrid starch-gelatin hydrogel in aqueous medium. For comparative studies; the starch-based hydrogel (without gelatin) was also synthesized. Further, the synthesized porous hydrogel acted as organic nano-reactor for generation and stabilization of AgNPs. The AgNPs-embedded hybrid hydrogel was studied for their antimicrobial activity, hydrophilicity, biocompatibility, biodegradability, non-cytotoxicity and hemocompatibility. The nanoparticle-embedded hybrid hydrogels with improved properties could be a good material of biomedical relevance and useful as wound dressing material.
2 Materials and methods
2.1 Materials
Soluble starch (S) was procured from Fisher Scientific, Mumbai, India. Gelatin (G) (gel strength-225 g Bloom, type B from bovine skin) was purchased from Sigma-Aldrich. N,N'–methylenebisacrylamide (MBA), acrylic acid (AA), silver nitrate, ammonium persulphate (APS) and were procured from Loba Chemie, Mumbai, India. E. coli and S. aureus strains are purchased from Institute of Microbial Technology, Chandigarh, India. The reagents used for the study was of analytical grade and the double distilled water was used for prepartion of different solutions.
2.2 The hybrid (starch and gelatin) hydrogel’s synthesis (RSM optimized synthesis as mentioned below)
Starch and gelatin (1:1) were kept in definite amount of distilled water (solvent) for 24 h to have a homogeneous mixture. The optimized amounts of APS (free radical initiator) and MBA (cross-linker) were mixed with stirring in the flask followed by the addition of AA (grafting monomer). The reaction mixture was kept at 70ºCfor 3 h. After 3 h, the synthesized hydrogel was taken out and kept in double-distilled water at ambient temperature so as to extract the synthesized graft copolymer. Then it was dried at 50ºC, ground and labeled as SG-g-poly(AA). A similar procedure was follwed for the synthesis of starch-based hydrogel without gelatin and labelled as S-g-poly(AA).
2.2.1 Sequential experimental design and statistical analysis
SG-g-poly(AA)
The effect of five independent variables including grafting agent, cross-linker, initiator, solvent amount and pH on swelling ratio were optimized using response surface methodology (RSM). Grafting agent (24–32 mmol), cross-linker (1–1.8 mmol), initiator (0.44–0.88 mmol), solvent (16–24 mL), and pH (8–12) were taken as independent variables during 23 randomized experiments.
S-g-Poly(AA)
Grafting agent (5–15 mmol), cross-linker (0.2–0.4 mmol), initiator (0.1–0.2 mmol), solvent (10–30 mL), and pH (8–12) were optimized using RSM to the maximum swelling of hydrogel.
In RSM, the synthesis is optimized by designing a sequence of experiments with variation of different operational parameters. The various reaction parameters are optimized simultaneously along with interaction between them. The experiments were performed by using central composite design (CCD) and optimized to maximum swelling of hydrogels. Design Expert 11 software was used for the experimental design and statistical analysis. The factorial model was confirmed by ANOVA and the non-significant terms were removed to obtain the reduced model. The coefficient of determination (R2) was used to evaluate the goodness of fit of the regression model.
2.3 Swelling studies
Swelling behavior of the hydrogel was studied by gravimetric method at 37 °C [34]. The dried hydrogels of known weight were kept in double distilled water at 37ºC. The samples were weighed after regular intervals till the attainment of constant weight. After that, the sample was taken out from water, wiped and weighed till the weight of sample became constant. The swelling ratio (SR) of the hybrid hydrogel was calculated using the following equation [34]:
where Wt and Wo is weight of the swelled and dry hydrogel, respectively.
2.4 Synthesis of AgNPs-embedded hybrid hydrogel
The dried hybrid hydrogel (0.5 g) was immersed in 5.0 mL of water. ThepH of the solution was maintained at 10 using 0.1 M NaOH. After that, the silver nitrate (0.2–0.8 mmol) solution was added drop wise and heated for 5 min over the flame. The color transformed to brownish yellow, confirms the formation of AgNPs. The AgNPs imbibed hybrid hydrogel was taken out from the solution. Further, it was washed and dried untill the attainment of constant weight [1, 3, 14, 26]. The in vitro release profiles of silver from silver nanoparticles loaded hydrogels were studied and hydrogels were, further, investigated for their antibacterial activity and other characteristics. A similar procedure was follwed for the synthesis of AgNPs imbibed starch-based hydrogel without gelatin. The silver nanoparticle-loaded hydrogels were named as SGH-AgNPs and SH-AgNPs for hybrid hydrogel and starch hydrogel, respectively.
2.5 Characterization
The Bruker Alpha FTIR spectrometer was used to record FTIR spectra (KBr pellet method). The SEM instrument (JEOL S150A) was used to study morphology of the synthesized hybrid hydrogels.The compression testing machine (100 Series Electromechanical Universal Test Machine, Testresources) was utilized for studying mechanical strength of the hydrogels (at a strain rate of 0.2 mm/min up to 50% strain). The thermal stability of hydrogels was studied using TGA/DTA 6300, SII EXSTAR 6000, INKARP instruments. The Rigaku Ultimia IV diffractometer was used for X-ray diffraction (XRD) measurements, operated at 40 kV and a current of 30 mA at a scan rate of 0.388 min−1 using parallel beam geometry, Cu-Kα radiation.The UV–visible absorption spectra were measured using double beam spectrophotometer (Shimadzu-2100, Japan). The JEOL-JEM-2100 transmission electron microscope (TEM) and energy-dispersive X-ray analysis (EDX) spectrum were used to investigate morphology and size of AgNPs. The dynamic light scattering (DLS) and zeta potential were utilized to study the average size of AgNPs.
2.6 Antibacterial activity
The disc diffusion method was used for antimicrobial activity of silver nanoparticle-embedded hydrogel with two bacterial strains, E. coli and S. aureus [48, 49].
2.7 In vitro silver release
0.5 g of dried hydrogel sample containing silver nanoparticles (SGH-AgNPs and SH-AgNPs) was dipped in 30 mL of PBS solution (pH 7.4) at 37 °C. The release behavior of silver from hydrogel was analyzed by measuring absorbanceof solution at 412 nmat different time periods using UV–visible spectrophotometer.
2.8 Water vapor transmission rate (WVTR)
At the mouth of a transmission bottle containing deionized water, the silver nanoparticle-embedded hydrogel samples with known dimensions were immobilized and stored at 37 °C. The WVTR values were calculated after 15 days according to the following equation.
Here, Wf and Wi are the final, and the initial weight of water remained in the bottle, respectively, t is the length of the experiment and A denotes the surface area of the hydrogels.
2.9 PBS absorption
The absorption of wound discharge by the hydrogel samples was assessed with the help of PBS absorption experiment. First, the silver nanoparticle-embedded hydrogel samples with known weights (Wi) were dipped in a bottle containing 20 mL of PBS (pH = 7.4) and incubated in vitro at 37 °C for a particular time. Later, after removing the excess PBS solution from the surface, the wet weight of the samples (Wf) was determined. The PBS absorption percentage was calculated using the following equation.
2.10 In vitro degradation
In vitro degradation of the silver nanoparticle-embedded hydrogel samples was carried out in a PBS solution at pH 7.4. The hydrogel samples were incubated in a PBS solution at 37 °C. Finally, the hydrogel samples after removing from PBS solution were washed and dried. The amount of degradation was examined looking into changes in morphology (scanning electron microscopy) for 4 weeks.
2.11 Moisture retention capability
The synthesized silver nanoparticle-embedded hydrogel samples were cut into pieces of uniform dimensions, weighted (Wi) and placed inside oven for 24 h at 50 °C. Afterwards, the samples were weighed again (Wf). The moisture retention capacity was determined by the following equation.
2.12 Gel fraction
The uniform pieces of silver nanoparticle-embedded hydrogel samples were dried in the oven to constant weight (Wi). The dried samples were put in de-ionized water for five days. Afterwards, the hydrogel samples were again dried in a vacuum oven to constant weight (Wf) is reached. The following equation was used to determine gel fraction.
2.13 Hydrogel porosity
The silver nanoparticle-embedded hydrogel samples were saturated in known volume of ethanol. Wi and Wf indicate the weight of samples before ethanol absorption and after ethanol absorption, respectively. V1 represents the volume of ethanol before absorption, V2 represents the volume of ethanol after absorption, and ρ is the density of alcohol at room temperature. The hydrogel porosity was determined using the following equation.
2.14 Swelling behavior
The swelling behavior represents the tendency of hydrogel to absorb liquid and swell in that medium. The swelling behavior of silver nanoparticle-embedded hydrogel samples was examined in water, 0.9% MgCl2, 0.9% NaCl and blood. The dried samples of the uniform dimensions were weighed (Wi) and immersed in the above-mentioned solutions for 24 h. The samples were removed from the different solutions and weighed (Wf) again after wiping extra solution from the hydrogel samples. The following equation was used to determine the swelling percentage.
2.15 In vitro cytotoxicity
TheWST-1 test was used to understand the cytotoxicity of hydrogel samples. The human skin fibroblast cells were cultured in 24 well culture plate at a cell density of 3 × 104 per well in Dulbecco’s modified Eagle medium (DMEM). The medium contained 10% fetal bovine serum and 1% penicillin. Afterwards in an atmosphere of 5% CO2, 5 mg of hydrogel sample was incubated for 48 h at 37 °C. The WST-1 colorimetric assay was used to determine the cell viability. Normal DMEM was used for the control sample [41].
3 Results and discussion
3.1 The proposed mechanism of hydrogel formation
The plausible mechanism for the synthesis of starch-gelatin hydrogel involves the free radical polymerization reaction in aqueous medium. The free radical mechanism involves three key phases: initiation, propagation and termination. During the initiation step, the free radical initiator (APS) undergoes homolytic cleavage to yield sulphate radicals along with other free radical species. These radical species abstract hydrogen from the -OH bonds in starch and -NH- bonds in gelatin and create radical sites over starch and gelatin for grafting of vinyl monomer. Alongside, the free radical produces radical sites over acrylic acid and MBA. During the propagation step, homopolymerization of vinyl monomers to form polyacrylic acid and grafting of polyacrylic acid onto starch and gelatin take place simultaneously. Further, the free radical sites on cross-linker (MBA) binds with the free radical sites over starch and gelatin and the three-dimensional network forms. Apart from linking through the cross-linker, the different physical interactions including Vander Waals and electrostatic interactions among and –OH, –COOH and–NH2 groups of starch and gelatin help to form a semi-IPN network (Scheme 1).

Schematic representation of cross-linked SG-g-Poly(AA)
3.2 RSM optimization of synthesis parameters
3.2.1 RSM optimization and model fitting of SG-g-poly(AA)
In order to attain the maximum swelling ratio (45 g/g), the optimized reaction parameters were AA (28 mmol), MBA (1.4 mmol), APS (0.66 mmol), solvent (20 mL) and pH (10). The regression coefficient in the form of coded equation
where A grafting agent, B cross-linker, C initiator, D solvent amount, E pH.
The final equation representing the actual factors
This equation can predict about the swelling ratio of the hydrogel; however, the relative impact of each factor should not be determined using it. The quadratic model is recommended by sequential model sum of square and lack of fit and further the Box-Cox plot (Fig. 1a) propose that no transformation is required in the data.

a–d RSM optimization of SG-g-poly(AA) (a) Box-Cox Plot; (b) correlation of the predicted and the experimental values of swelling; (c) response surface plot showing interaction between cross-linker and monomer concentration; and (d) response surface plot showing interaction between solvent volume and initiator concentration
3.2.2 Optimization of significant variables
The concentrations of AA, MBA, APS, and solvent volume are substantial parameters to affect the swelling ratio. The interaction of these parameters was analyzed using a central composite design (CCD). ANOVA, regression coefficients, R2 values, and lack of fit are summarized in Table 1. The model F-value of 55.93 implies the significance of quadratic model. That the model terms are significant is indicated by the P-values less than 0.0500. The non-significance of model terms is indicated by values greater than 0.1000. The Lack of Fit F-value of 4.69 suggests that Lack of Fit is not significant relative to the pure error. The non-significant lack of fit indicates the fitting of quadratic model. The negative predicted R2 (− 0.2793) indicates that the overall mean is a better predictor of the response (Fig. 1b). The optimal levels of independent variables on swelling percentage were determined by contour plots and 3-D response surface contour plots (Fig. 1c, d and Fig. S1a-d).
The equation in terms of unitless regression coefficients
The actual factors in terms of multiple regression equation
3.2.3 RSM optimization and model fitting of S-g-Poly(AA)
In order to attain the maximum swelling ratio (30 g/g), the reaction parameters optimized were AA (10 mmol), MBA (0.3 mmol), APS (0.15 mmol), solvent (20 mL), and pH (10). The regression coefficient in the form of coded equation
where A grafting agent, B cross-linker, C initiator, D solvent amount, E pH.
Final equation in terms of actual factors
This equation can be used to make predictions about the swelling. The quadratic model is recommended by sequential model sum of square and lack of fit and further the Box-Cox plot (Fig. 2a) suggest that power transformation is required in the data.

a–d RSM optimization of S-g-poly(AA)(a) Box-Cox Plot; (b) Predicted and experimental values of swelling; (c) response surface plot presenting interaction between cross-linker and monomer concentration; and (d) response surface plot describing interaction between solvent volume and pH
3.2.4 Optimization of significant variables
Here, AA concentration, MBA concentration, solvent volume, and pH are important terms to influence the swelling ratio. The interaction of the above-mentioned parameters was analyzed by a central composite design (CCD). ANOVA, regression coefficients, R2 values, and lack of fit are summarized in Table 2. The model F-value of 106.85 implies that quadratic model is significant. The significance of model terms was indicated by P-values less than 0.0500. The non-significance of Lack of Fit relative to the pure error was implied by the Lack of Fit F-value of 36.12. The fitting of quadratic model is indicated by the non-significant lack of fit. The predicted R2 of 0.2091 is not as nearer to the adjusted R2 of 0.9897; the dissimilarity is more than 0.2 (Fig. 2b). The optimal levels of independent variables on swelling percentage were determined by contour plots and 3-D response surface contour plots (Fig. 2c, d and Fig.S2 a-d).
The equation in terms of unitless regression coefficients
The multiple regression equation for actual factors
3.3 Swelling studies
The polymeric network of hydrogels expands in the presence of water and hydrates the various functional groups attached to various polymeric chains. Variation in swelling at different pH, help them utilization in biomedical sector. Higher swelling capacity means higher efficiency of drug loading. Higher swelling and retention capacity, make them biocompatible [50]. Figure 3(a) showed the effect of pH on water absorption tendency of SG-g-Poly(AA). In acidic condition, the carboxylic groups are protonated, which generate an additional cross-linking through hydrogen bonding and the network tends to shrink because of the restricted repulsions between the protonated carboxylate groups.It results in decreased swelling capacity of starch-gelatin semi-IPN at low pH [38]. However, at higher pH, the carboxylic groups are ionized to carboxylate ions, which results in increased repulsions between carboxylate groups and decreased hydrogen bonding. At higher pH, the expansion of the polymeric network increases the swelling capacity of hydrogel. The swelling studies indicate that SG-g-poly(AA) has good water absorption capacity and the maximum swelling of 4400% was achieved at pH 10 and the results are in good agreement with literature [45].

a–d Swelling kinetics of (a) SG-g-poly(AA) at different pH; (b) dynamic water uptake of SG-g-Poly(AA) at pH 10; (c) swelling kinetics of S-g-poly(AA) at different pH; and (d) dynamic water uptake of S-g-Poly(AA) at pH 10
Figure 3b represents the time dependent swelling of SG-g-poly(AA) hydrogel. The swelling capacity of hydrogel increased with time and the equilibrium swelling capacity was obtained in 140 min, approximately. Further, S-g-Poly(AA) hydrogel displayed the similar trend of growing swelling capacity with increased pH owing to the increased ionization of carboxylic groups as already mentioned above (Fig. 3c). The slight decrease in swelling capacity at pH 12 could be credited to the increased sodium ions concentration in the solution which interact and shield the carboxylate ions. It results in decreased ionic repulsions and reduction of the network expansion and consecutively, it decreases the swelling capacity of starch-based hydrogel. The equilibrium swelling of S-g-Poly(AA) hydrogel was obtained in about 210 min (Fig. 3d). The swelling capacity of starch-gelatin hydrogel was comparatively on higher side than the starch-based hydrogel. The presence of gelatin in the network structure adds rich hydrophilic functionalities to the three-dimensional network and results in better hydration.
3.4 Characterization of starch-gelatin and starch hydrogels
3.4.1 Fourier transform infrared spectroscopy (FTIR) of hydrogels
The FTIR spectra of starch, gelatin, starch-based hydrogel, and starch-gelatin hybrid hydrogel are displayed in Fig. 4(a–d). The characteristic peaks of starch (Fig. 4a) at 1020 cm−1, 2932 cm−1, and 3273 cm−1 are attributed to the C-O, C-H and O–H stretching vibrations, respectively. The peak at 1648 cm−1is resulted from the 1st overtone of OH bending and the stretching C–O–C vibrations. The frequency range between 920 and 1100 cm−1 is the typical profile of polysaccaharides as visible in the spectrum. Gelatin showed peaks at 3275 cm−1 (N–H stretching), 1629 cm−1 (amide-I), 1534 cm−1 (amide-II), 1239 cm−1 (amide-III), and 1448 cm−1 (symmetric stretching of –COO− group (Fig. 4b). The amide-I and amide-II bands represent the C = O stretching and the N–H in plane bending, respectively, in gelatin. The amide-III band represents the coupling of C-N stretching and N–H in plane bending of the bound amide.

a–d FTIR spectra of (a) starch; (b) gelatin; (c) S-g-Poly(AA), and (d) SG-g-Poly(AA)
The FTIR spectrum of S-g-Poly(AA) (Fig. 4c) showed a broad band with diminished intensity in the region of 3300–3600 cm−1 related to the stretching vibrations in-OH groups. The variation of band/peak intensity indicates the structural changes and the change in extent of hydrogen bonding. The peaks at 1700 cm−1 and 1200 cm−1 were recognized as the stretching vibrations of C = O and C-O of the grafted acrylic acid, respectively. The FTIR spectrum of SG-g-Poly(AA) showed amalgamation of characteristics bands of starch, gelatin and acrylic acid (Fig. 4d). There was shifting of gelatin peak from 1629 cm−1 to 1655 cm−1due to the formation of semi-IPN hydrogel owing to the various new molecular interactions or change in electronic distribution along the bond [40, 51]. The stretching band of C = O corresponding to acrylic acid was observed tobe overlapped with amide-I band of gelatin.
The hydrogels displayed broad absorption bands in the region of 3500–2500 cm−1 with change in intensity (Fig. 4c, d). The overlapping of O–H stretching band was seen with N–H stretching and C–H stretching peaks. The intermolecular hydrogen bonding of carbonyl groups (1710 cm−1) with hydroxyl groups of starch shift its stretching vibrational frequency. The presence of cross-linker in the hydrogel matrix was indicated by the presence of a peak at 1397 cm−1 with small intensity. Further, a peak at 1125 cm−1 was observed for C–O stretching vibration. The 920–1100 cm−1 region representing the typical profile of polysaccharides was found to be absent from the FTIR spectra of both the hydrogels. The intensities of different bands were different in SG-g-Poly(AA) and S-g-Poly(AA) hydrogels (Fig. 4c, d).
3.4.2 Scanning electron microscopy (SEM)
The morphological representation of starch, gelatin, starch-based hydrogel and starch-gelatin hydrogel surfaces is shown in Fig. 5(a–f). Figure 5(a) and (b) shows the spherically shaped starch particles and the gelatin in continuous phase, respectively. The morphology of S-g-Poly(AA) hydrogel was quite different from the starch (Fig. 5c, d). The original granular texture of starch was lost owing to formation of covalent crosslinks and the complex semi-IPN structure. Further, SG-g-Poly(AA) was found to possess a cross-linked and porous three-dimensional network structure with continuous morphology (Fig. 5e, f) The loss of unique morphologies of starch and gelatin with development of completely different morphology in hydrogel indicates the successful crosslinking of starch and gelatin [31]. Besides, the SEM images revealed the higher extent of crosslinking in the SG-g-Poly (AA) hydrogel as compared to S-g-Poly(AA).

a–f SEM images of (a) starch; (b) gelatin; (c-d) S-g-poly(AA), and (e–f) SG-g-Poly(AA)
3.4.3 Mechanical properties
Generally, the weak tensile strength of hydrogels made up from natural polymers limits their applications. The compressive modulus of SG-g-Poly(AA) was found to be superior to the hydrogel made from starch backbone without any gelatin (Table 1). The compressive modulus of the synthesized hybrid hydrogel synthesized in aq. medium was in the range as mentioned in [45], where the gelatin-starch hydrogel was synthesized in polar solvent [45]. The covalent cross-linking of starch and gelatin with cross-linker was responsible for improvement in mechanical strength of semi-IPN hydrogel. Apart from the covalent cross-linking, the different types of interactions including Vander Waals and electrostatic interactions among starch, gelatin and cross-linker might have further enhanced the mechanical strength of SG-g-Poly(AA).The various intermolecular interactions including physical binding and chemical bonding among the different components owing to the rich functionalities over starch-gelatin backbone helped to grow the three-dimensional structure and mechanical strength of SG-g-Poly(AA) (Table 3).
3.4.4 Thermal properties
Table 4 reveals the thermal behavior of starch, gelatin, S-g-Poly(AA) and SG-g-Poly(AA). The thermogravimetric analysis (TGA) of starch showed an initial weight loss (10%) at 194.1°Cwas mainly due to the exclusion of the absorbed moisture and volatile matter. The second stage degradation (with the weight loss of 32.9%) was observed between 194.1 and 293.0 °C and the third stage in the temperature range of 293.0–461.8 °C (with the weight loss of 70.8%) was mainly related to degradation of starch polymer. The decomposition of gelatin started with the removal of surface moisture with mass loss of 6.7% up to the temperature of 144.17 °C. Further, the mass loss was observed due to removal of bound water (8.74% loss) up to the temperature of 240.83 °C. Furthermore, the two stages decomposition process resulted in the mass loss of 61.67% and 99.152% up to the temperature of 446.58 °C and 625.81 °C, respectively. The initial and final decomposition temperatures of gelatin were 240.83 °C and 625.81 °C, respectively. The initial decomposition temperature (IDT) (247.2 °C) as well as the final decomposition temperature (FDT) (756.6 °C) in TGA were found to be higher for SG-g-Poly(AA) than that of starch and S-g-Poly(AA). The physical and chemical interactions of starch and gelatin via cross-linking and different functional groups enhanced interfacial adhesion and thus the thermal degradation temperature increased. DTA (differential thermal analysis) peaks were observed at higher temperature with less heat evolved (686 °C, 25.4 μV) in SG-g-Poly(AA) compared toS-g-Poly(AA) (627 °C, 57.6 μV). DTG (differential thermogravimetric analysis) studies showed that the rate of thermal decomposition was higher in S-g-Poly(AA) (0.475 mg/min) than in SG-g-Poly(AA) (0.466 mg/min). The grafting and crosslinking among different polymer chains resulted in enhanced thermal stability of the hydrogel.SG-g-Poly(AA) hydrogel was thermally more stable as compared to S-g-Poly(AA), in coherence with its higher mechanical strength (Table 4). The trends of increasing thermal stability of hydrogels as a result of grafting and cross-linking were in accordance with the previous results reporting the synthesis of polysaccharide hydrogels under similar conditions [31, 52].formation of silver nanoparticles (
3.4.5 X-ray diffraction (XRD) analysis
The crystallinity of starch originates from involvement of -OH groups in inter and intra molecular hydrogen bonding. It was observed from Fig. 6a that starch showed crystalline peaks at 2θ = 15.7°, 17.7°, and 22.7° as reported [53]. Gelatin showed a large broad amorphous peak at 22°at 2θ scale as reported [54]. Gelatin is amorphous in nature with a non-defined crystal structure, as clearly indicated by its XRD (Fig. 6b). In case of S-g-Poly(AA) hydrogel, the appearance of two broad peaks at 2θ values of 22° and 36° again points out its amorphous or non-crystalline nature (Fig. 6c). The peak intensity at 2θ of 22° was higher for starch hydrogel. Whereas, in SG-g-Poly(AA), the intensity of peak at 2θ of 22° was found to be comparatively lesser than starch hydrogel indicating its lesser crystallinity (Fig. 6d). The difference in peak intensities in starch and strach-gelatin hydrogels could be related to variation in their morphologies at the microscale level. The hybrid hydrogel displayed decreased crystallinity due to strong but random interactions among the various functional groups of starch and gelatin. Furthermore, the incorporation of poly(AA) chains in the matrix also results in decreased crystallinity [40].

a–d XRDs of (a) starch; (b) gelatin; (c) S-g-Poly(AA); and (d) SG-g-Poly(AA)
3.5 Characterization of the AgNPs-embedded hydrogels
3.5.1 UV–visible spectroscopic studies
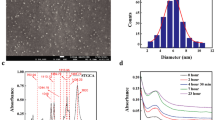
In the study, Ag(I) ions were loaded onto the hydrogels and were reduced to silver nanoparticles by the hydrogels. The synthesized silver nanoparticles were investigated through UV–Vis spectroscopy to get insight about the efficiencies of SG-g-Poly(AA) and S-g-Poly(AA) hydrogels as matrices for reduction and stabilization of silver nanoparticles (Fig. 7). The surface plasmon resonance (SPR) band of AgNPs was centered at 412 nm, depicting the presence of AgNPs [35]. The SPR is a result of collective oscillation of the electrons present in conduction band because of the resonant excitation by the incoming photonsand was found to be absent in both small silver nanoparticles (< 2 nm) and agglomerated one [14]. The vibrant yellowish color (Fig. 7e) supported the reduction and formation of Ag nanoparticles (AgNPs) in the hydrogel matrix.

a–e UV–Visible absorption spectra of AgNPs synthesized using (a & b) SG-g-poly(AA); (c & d) S-g-Poly(AA); and (e) color change due to synthesis of silver nanoparticles
The factors affecting the formation of silver nanoparticles in SG-g-Poly(AA) as well as S-g-Poly(AA) matrices were compared. It can be seen from Fig. 7(a & c) that the increasing the silver (I) salt, progressively increases the intensity of the SPR band due to increased concentration of silver nanoparticles. The absorption peak was found to be significantly less intense at AgNO3 concentration of 0.8 mmol. This might be due to the fact that AgNO3 concentration more than 0.6 mmol produces clusters or aggregates that in turn decreases the concentration of nanoparticles, thus results in decreased intensity of the SPR band [9].
Further, the influence of pH on the synthesis of AgNPs using SG-g-Poly(AA) hydrogel as well as starch hydrogel was investigated (Fig. 7b & d) in pH range of 10.0–13.0. The peak intensity of SPR band of the AgNPs increases with increase in the pH from 10.0 to 12.0, but the further increase in pH beyond 12 decreases the peak intensity. As the pH of solution increases, the functional groups (-COOH and -OH) present on the hydrogel matrices dissociate to form ions and provide electrons for the reduction of Ag (I)ions, which was true up to the pH 12.0. However, beyond pH 12, the silver ion gets precipitated as hydroxide, hence reduces the Ag (I) concentration in the solution [55].
Thus, the optimal condition for the AgNPs reduction and formation using hybrid hydrogel was observed at pH 12.0 with silver nitrate concentration of 0.6 mmol.
The synthesis of AgNPs was evaluated using both SG-g-Poly(AA) and S-g-Poly(AA) matrices. It was observed from the UV–Vis absorption spectra that silver nanoparticles with SG-g-Poly(AA) had higher absorption intensity in comparison to those with S-g-Poly(AA) hydrogel. Thus, the SG-g- Poly(AA) matrix could act as better reducing agent for the synthesis of AgNPs-embedded hydrogels compared to S-g-Poly(AA), because of the highly cross-linked network structure and the presence of additional functional groups.
3.5.2 XRD analysis
X-ray diffraction pattern of crystalline AgNPs synthesized using SG-g-Poly(AA) is given in Fig. 8(a). The presence of four sharp and well defined diffraction lines at 2θ = 38.28°, 44.58°, 64.59°, and 77.46° corresponding to (111), (200), (220), and (311) reflections of the face cubic centered (fcc) structure of silver, respectively. The results confirmed the crystalline structure of AgNPs [35]. The lattice parameters determined from XRD (a = b = c = 4.082975 A°) was in conformity with the literature (a = 4.086 Å) [35]. No other peaks of impurities or crystalline phases have been detected in the XRD pattern of silver nanoparticles.

a, b X-ray diffraction pattern of silver nanoparticles synthesized using (a) SG-g-poly(AA) and (b) SGH-AgNPs sample (silver nanoparticle-embedded starch-gelatin hydrogel)
The high intensity peak at (111) reflection indicates an orienting growth along the (111) direction and high degree of crystallinity of AgNPs. The broad diffraction peaks indicated small crystallite size of silver nanoparticles. The value of FWHH of AgNPs was determined to be 0.78 at 38.28°. The size of the Ag nanoparticles was 10.77 nm as calculated using the Debye–Scherrer equation [55].
Figure 8(b) represents the X-ray diffraction pattern of silver nanoparticle-embedded starch-gelatin hydrogel (SGH-AgNPs). The appearance of two broad peaks at 2θ values of 22° confirms the presence of amorphous starch-gelatin backbone. Further, the presence of four sharp and well-defined peaksat the 2θ values mentioned above indicated the presence of crystalline AgNPs in the hydrogel. The peak positions and peak intensities of the embedded silver nanoparticles (8b) were found to be different in comparison to the free silver nanoparticles in the solution (8a). The growth of the embedded silver nanoparticles might be oriented in different directions while inside the hydrogel matrix. The combination of sharp and broad peaks in the XRD confirmed the successful synthesis of silver nanoparticle-embedded starch-gelatin hydrogel.
3.5.3 Transmission electron microscopy (TEM) and selected-area electron diffraction (SAED) pattern
The EDX spectra of Ag nanoparticles synthesized in S-g-Poly(AA) and SG-g-Poly(AA) matrices, respectively, indicated the formation of silver nanoparticles (Supplementary data, Fig. S3a, b). The TEM images (Fig. 9) showed spherical and ellipsoidal morphology of silver nanoparticles in SGH-AgNPs and SH-AgNPs, respectively. The nanoparticles in SG-g-Poly(AA) were approximately 10 nm in diameter with predominantly spherical shape. The nanoparticles in S-g-Poly(AA) were spherical and elliptical with the large variation in diameter ranging from 4 to 58 nm. As visible from the TEM images, the nanoparticles were homogeneously distributed with particle size variation of 4–19 nm in SGH-AgNPs as compared to those in SH-AgNPs. The nanoparticle sizes determined by TEM and DLS were consistent (Table 5). Moreover, the nanoparticles in SGH-AgNPs were smaller in size as compared to those in SH-AgNPs. Undoubtedly the nucleation, growth and stabilization of nanoparticles takes place through different interactions in SG-g-Poly(AA) in comparison to S-g-Poly(AA).

a–f TEM images of (a, b) SGH-AgNPs; (d, e) SH-AgNPs; SAED pattern of (c) SGH-AgNPs and (f) SH-AgNPs
The SAED pattern showed diffraction rings and spots, which corresponded to the fcc phase of silver nanoparticles as inspected by XRD. The SAED spots depicted different crystallographic planes of fcc structure of elemental silver and are in agreement with the XRD results [56].
Zeta potential is used to study the charge stability of the nanoparticles in aqueous suspension. The higher zeta potential means stronger repulsion and higher stability of the nanoparticles.
The zeta potential values for silver nanoparticles in SGH-AgNPs were found to be higher as compared to silver nanoparticles in SH-AgNPs. This indicated the better stability of nanoparticles in SGH-AgNPs composite (Table 6). The positive zeta potential of silver containg hydrogels can better interact with bacterial cell wall and contribute to good antibacterial activity as discussed later [57]. The stability of nanoparticles in nanocomposites was again confirmed from the TEM images of silver nanoparticles containing hydrogels which were stored in closed bottles at room temperature for six months (Fig. 10a–d). The silver nanoparticles in SGH-AgNPs remained well dispersed without any significant change in particle size distribution in comparison to silver nanoparticles in SH-AgNPs which formed agglomerates. The UV–visible spectrum of the stored silver nanoparticles in SGH-AgNPs displayed the SPR at 411 nm without any major shift in absorption wavelength, indicating the insignificant change in particle size even after six months (Fig. 10e). Therefore, SG-g-Poly(AA) hydrogel with better cross-linking can act as better stabilizing matrix for nanoparticles as compared to S-g-Poly(AA) [58].

a–e TEM of SGH-AgNPs (a) at the time of synthesis and (b) after 6 months; TEM of SH-AgNPs; (c) at the time of synthesis and (d) after six months (e) UV–visible spectrum of SGH-AgNPs after 6 months
3.6 Antibacterial activity analysis of silver-embedded hydrogels
The contact of silver nanoparticles with bacteria changes their membrane structure and permeability. The interaction of AgNPs with the bacterial cell wall leads to the lysis of the cell wall. The magnitude of lysis of bacterial membrane increases with decrease in size of nanoparticles [49]. In this study, SGH-AgNPs exhibited good antibacterial activity against gram-negative (E. coli) as well as gram-positive bacteria (S. aureus) (Fig. 11). The higher positive surface charge on SGH-AgNPs as indicated from its zeta potential can better interact with the negatively charged bacterial cell wall [37, 58, 59]. There is difference in the sensitivity of both the bacteria (gram-positive and gram-negative) towards AgNPs because of differences in their cell wall structures [60]. As visible from the Fig. 11, starch as well as SG-g-Poly(AA) hydrogel did not show any kind of antibacterial activity. Whereas, SGH-AgNPs caused the inhibition of bacterial growth, both in E.coli and S.aureus.

Antibacterial activity of Starch (box1); SGH-AgNPs against E. coli (box 2); SG-g-poly(AA) hydrogel (box 3) and SGH-AgNPs against S. aureus (box 4)
3.7 In vitro release of silver from nanosilver hydrogels
Figure 12(a–d) represents the starch and hybrid hydrogels with and without silver nanoparticles. The liberation of AgNPs from the hydrogel matrix to the pathogenic environment is an important parameter to understand the antimicrobial activities of SGH-AgNPs and SH-AgNPs samples. The release of AgNPs from the hydrogel matrix was studied using UV–Visible spectroscopy.As shown in Fig. 12, the release of silver from SGH-AgNPs sample was fast in the begining as compared to SH-AgNPs. The total release of silver was also found to be higher in case of SGH-AgNPs. The hybrid hydrogel matrix is more hydrophilic owing topresence of more functional groups in comparison to starch hydrogel and favours the relase of silver from the SGH-AgNPs sample. Here, 0.5 g of SGH-AgNPs released 20.9 µg and 45.1 µg of silver during the first and second day, respectively and afterwards it became almost constant. The amount of silver released was found to be in clinically acceptable range and it indicates that SGH-AgNPs has potential to be used in wound healing applications [61].

a–e (a) SG-g-Poly(AA) hydrogel; (b) SGH-AgNPs hydrogel; (c) S-g-Poly(AA) hydrogel; (d) SGH-AgNPs hydrogel; and (e) in vitro release of silver from SGH-AgNPs and SH-AgNPs
3.8 Hydrophilicity of nanosilver hydrogel samples
The WVTR is an important characteristic of the wound dressing, indicating the rate of exchange of water vapor molecules through the wound dressing layer. The higher WVTR helps in quick drying of the wound and the lower WVTR decreases the process of healing and enhances the chance of bacterial infection. The ideal value of WVTR lies in the range of 8.33–10.42 mg/cm2hr for wound dressing [62]. For SGH-AgNPs and SH-AgNPs samples, the WVTR values were determined as 8.76 ± 0.78 mg/cm2hr and 8.99 ± 0.99 mg/cm2hr, respectively, indicates the ideal range for both the polymeric samples. The WVTR was slightly lower for SGH-AgNPs sample owing to the presence of more hydrophilic functional groups over the hydrogel backbone in comparison to SH-AgNPs.
The PBS absorption was observed to be 132% and 124% for SGH-AgNPs and SH-AgNPs samples, respectively. The hybrid silver nanoparticle-embedded hydrogel displayed better performance in PBS solution due to its better hydrophilic characteristics in comparison to silver nanoparticle-embedded starch hydrogel [62].
The moisture retention capability measures the tendency of a material to hold moisture and is inversely proportional to WVTR i.e. with higher tendency for loss of vapors, the moisture retention capability of the material decreases. The moisture retention capabilities of SGH-AgNPs and SH-AgNPs samples were found to be 94% and 89%, respectively. The large tendency of the material to retain moisture helps in better wound healing and tissue regeneration [63].
3.9 In vitro degradation of nanosilver hydrogels
The silver nanoparticle-embedded hydrogel samples primarily consist of natural polymers which have inherent tendency to degrade. The morphological analysis of SGH-AgNPs and SH-AgNPs hydrogels were done using SEM to confirm in vitro degradation of the samples in PBS solution at 37 °C for a month (Fig. 13a–d). Initially, the hydrogel surfaces were smoother and continuous. The hydrolysis and degradation of the material led to cleavage of cross-linkages and covalent bonds among various polymer chains and resulted in discontinuities and cracks whichare visible from Fig. 13(b, d). The degradation rate depends upon the chemical structure and hydrophilic nature of the material [64]. The better hydrophilicity of the material results in better hydrolysis and increased rate of degradation. The degradation of cross-linked polymer networks in a month’s period completely changed the surface morphologies of the hydrogel samples.

a–d (a) SGH-AgNPs hydrogel during the first week of degradation; (b) SGH-AgNPs hydrogel during the fourth week of degradation; (c) SH-AgNPs hydrogel during the first week of degradation; (d) SH-AgNPs hydrogel during the fourth week of degradation
3.10 Gel fraction of nanosilver hydrogels
The gel fraction determination helps in evaluating the cross-linker’s effectiveness in hydrogel synthesis [63]. The enhancement of cross-linking density increases the gel faction. Further, the strength and flexibility of the material is also related to gel fraction. The higher gel fraction helps the wound dressing material to sustain in cold/moist environment without disintegration [63]. The gel fraction of SGH-AgNPs and SH-AgNPs samples were found to be 93% and 87%, respectively. The higher value of gel fraction of SGH-AgNPs is attributed to good amount of cross-linking in the three dimensional network of hydrogel.
3.11 Porosity of nanosilver hydrogels
The porosity of hydrogel samples was measured using alcohol displacement method [63]. The wound dressing material should be porous to help in permeation of oxygen, water vapors, nutrients and further for migration of cells and protein molecules. The porous material accelerates the healing process and 30–40% porosity is ideal for wound dressing materials. The porosity of SGH-AgNPs and SH-AgNPs samples were found to be 43% and 37%, respectively.
3.12 Swelling behavior of nanosilver hydrogels
The capability of absorbing moisture and wound exudates by the wound healing materials is an important parameter to achieve the healing. The hydrophilic nature of the functional groups on the hydrogel’s polymeric chains attracts water molecules into its pores and results in swelling of the network. The swelling behavior of a material is affected by pH, temperature, chemical environment, degree of cross-linking etc. [63]. The swelling behavior of the prepared nanocomposite hydrogels were tested in water, MgCl2 solution, blood and NaCl solution as shown in Fig. 14.

Swelling behavior of nanosilver hydrogels in different media
All these mediums are available in the chemical environment inside the human body. The swelling of silver-embedded hydrogels was not affected much in different media. The swelling capacity was found to be quite comparable in all mediums including water, blood, MgCl2, and NaCl, making them suitable for wound dressing applications. The slight increase in swelling of hydrogels in blood could be attributed to its higher pH (7.45) in comparison to the other mediums where the pH was ≤ 7.
3.13 Hemocompatibility of nanosilver hydrogels
The compatibility of a biomaterial for blood is an important factor to be evaluated for its usability as wound dressing material. The hydrogel should be compatible with human blood and does not cause any adverse effects on red blood cells (RBCs). The hydrogel material in contact of blood might cause lysis of RBCs present in blood and releases hemoglobin into solution phase. Typically, the hemolytic ratio below 5% is requisite in order to consider biocompatibility of the material [41]. The hydrogel samples displayed low hemolysis ratio of 0.98 ± 0.23 and 0.86 ± 0.32 for SGH-AgNPs and SH-AgNPs samples. Both the nanocomposite hydrogel samples are haemocompatible for wound dressing applications. Figure 15a–c displayed the effect of positive control, negative control and hydrogel sample on RBCs. The human blood sample incubated with distilled water did cause the destruction of RBCs, whereas hydrogel and saline were safe and did not cause any destruction of RBCs.

a, b Hemolysis test on (a) distilled water (positive control) and (b) saline solution (negative control) (c) hydrogel extraction
3.14 In vitro cytotoxicity of nanosilver hydrogels
The cytotoxicity of hydrogel against human skin fibroblast cells was determined using WST-1 test [41]. The cytotoxicity analysis helps to access the potential of wound dressing hydrogels for providing healthy environment for cell growth. The fibroblast cells were cultured with gelatin, starch, and the nanosilver hydrogels. The percentage cell viability of starch, gelatin, SGH-AgNPs and SH-AgNPs was found to be 91%, 86%, 89%, and 87%, respectively. The hydrogels were non-cytotoxic due to biocompatibility of starch and gelatin and could be safely used for the wound dressing.
4 Conclusions
The semi-IPN of hybrid backbone containing starch and gelatin was synthesized and it was found to have good mechanical strength and thermal stability. For comparison, starch-based hydrogel without gelatin was also synthesized. Further, the silver nanoparticle–embedded semi-\PN hybrid- and starch-based hydrogels were also synthesized. The silver-embedded hydrogels contained well dispersed spherical shaped silver nanoparticles. The silver nanoparticles in hybrid hydrogel were smaller in size and more uniform (4–19 nm) as compared to those in starch hydrogel (4–58 nm). Moreover, the hydrid semi-IPN was found to act as better stabilizing agent for silver nanoparticles.The novel hybrid semi-IPN owing to its cross-linked network structure with large number of functional groups was hydrophilic, haemocompatible and biodegrdable. It exhibited good antibacterial activity and was suitable for wound healing applications. The synthesized nanocomposite hybrid hydrogel could be useful for biomedical applications owing to its improved thermal and mechanical characteristics and further, due to its biocompatible nature.
Data availability
The whole data of the present manuscript is available; it will be provided if asked.
References
Sharma VK, Yngard RA, Lin Y (2009) Silver nanoparticles: Green synthesis and their antimicrobial activities. Adv Colloid Interface Sci 145:83–96. https://doi.org/10.1016/j.cis.2008.09.002
Sahiner N (2013) Soft and flexible hydrogel templates of different sizes and various functionalities for metal nanoparticle preparation and their use in catalysis. ProgPolymSci 38:1329–1356. https://doi.org/10.1016/j.progpolymsci.2013.06.004
El-Nour KMMA, Eftaiha A, Al-Warthan A, Ammar RAA (2010) Synthesis and applications of silver nanoparticles. Arab J Chem 3:135–140. https://doi.org/10.1016/j.arabjc.2010.04.008
Khampieng T, Brikshavana P, Supaphol P (2014) Silver nanoparticle-embedded poly (vinyl pyrrolidone) hydrogel dressing: gamma-ray synthesis and biological evaluation. J BiomaterSciPolym Ed 25:826–842
Mittal H, Kumar V, Saruchi RSS (2016) Adsorption of methyl violet from aqueous solution using gum xanthan/Fe3O4 based nanocomposite hydrogel. Int J Biol Macromol 89:1–11
Saruchi KV, Mittal H, Alhassan SM (2019) Biodegradable hydrogels of tragacanth gum polysaccharide to improve water retention capacity of soil and environment-friendly controlled release of agrochemicals. Int J BiolMacromol 132:1252–1261
Saruchi KBS, Kumar V, Jindal R (2016) Biodegradation study of enzymatically catalyzed interpenetrating polymer network: evaluation of agrochemical release and impact on soil fertility. Biotechnol Rep 9:74–81
Kaith BS, Saruchi JR, Bhatti MS (2012) Screening and RSM optimization for synthesis of a gum tragacanth–acrylic acid based device for in situ controlled cetirizine dihydrochloride release. Soft Matter 8:2286–2293
Mittal H, Alili AA, Alhassan SM (2020) Solid polymer desiccants based on poly(acrylic acid-co-acrylamide) and laponite RD: Adsorption isotherm and kinetics studies. Colloids Surf A. 599:124813
Xie J, Zheng Y, Ying JY (2009) Protein-directed synthesis of highly fluorescent gold nanoclusters. J Am Chem Soc 131:888–889. https://doi.org/10.1021/ja806804u
Toroz D, Corni S (2011) Peptide synthesis of gold nanoparticles: the early steps of gold reduction investigated by density functional theory. Nano Lett 11:1313–1318. https://doi.org/10.1021/nl1043924
Cheviron P, Gouanvé F, Espuche E (2014) Green synthesis of colloid silver nanoparticles and resulting biodegradable starch/silver nanocomposites. Carbohydr Polym 108:291–298. https://doi.org/10.1016/j.carbpol.2014.02.059
Szabó T, Mihály J, Sajó I, Telegdi J, Nyikos L (2014) One-pot synthesis of gelatin-based, slow-release polymer microparticles containing silver Nanoparticles and their application in anti-fouling paint. Prog Org Coat 77:1226–1232. https://doi.org/10.1016/j.porgcoat.2014.02.007
Hebeish AA, El-Rafie MH, Abdel-Mohdy FA, Abdel-Halim ES, Emam HE (2010) Carboxymethyl cellulose for green synthesis and stabilization of silver nanoparticles. Carbohydr Polym 82:933–941. https://doi.org/10.1016/j.carbpol.2010.06.020
Saruchi KV (2019) Separation of crude oil from water using chitosan based hydrogel. Cellulose 26:6229–6239
Mittal H, Morajkar PP, Alili AA (2020) Alhassan SM In-Situ Synthesis of ZnO Nanoparticles using Gum Arabic Based Hydrogels as a Self-template for Efective Malachite Green Dye Adsorption. J Polym Environ 28:1637–1653
Saruchi KV, Kaith BS (2016) Synthesis of hybrid ion exchanger for rhodamine B dye removal: equilibrium, kinetic and thermodynamic studies. I & EC Research 55(39):10492–10499
Naushad M, Mittal A, Rathore M, Gupta V (2015) Ion-exchange kinetic studies for Cd(II), Co(II), Cu(II), and Pb(II) metal ions over a composite cation exchanger. Desalin Water Treat 54:2883–2890. https://doi.org/10.1080/19443994.2014.904823
Saruchi KV (2019) Adsorption kinetics and isotherms for the removal of rhodamine B dye and Pb+2 ions from aqueous solutions by a hybrid ion-exchanger. Arab J Chem 12:316–329
Mittal H, Kumar V, Alhassan SM, Ray SS (2018) Modification of gum ghatti via grafting with acrylamide and analysis of its flocculation, adsorption and biodegradation properties. Int J Biol Macromol 114:283–294
Naushad M, ALOthman ZA (2015) Separation of toxic Pb2+ metal from aqueous solution using strongly acidic cation-exchange resin: analytical applications for the removal of metal ions from pharmaceutical formulation. Desalin Water Treat 53:2158–2166. https://doi.org/10.1080/19443994.2013.862744
Mittal H, Alili AA, Alhassan SM (2020) High efficiency removal of methylene blue dye using jcarrageenan-poly(acrylamide-co-methacrylic acid)/ AQSOA-Z05 zeolite hydrogel composites. Cellulose 27:8269–8285
Zhou Y, Fu S, Zhang L, Zhan H (2013) Superabsorbent nanocomposite hydrogels made of carboxylated Cellulose nanofibrils and CMC-g-p (AA-co-AM). Carbohydr Polym 97:429–435. https://doi.org/10.1016/j.carbpol.2013.04.088
Saruchi, Thakur P., Kumar V., 2019. Kinetics and thermodynamic studies for removal of methylene blue dye by biosynthesize copper oxide nanoparticles and its antibacterial activity, J Environ Health Sci Eng 2019https://doi.org/10.1007/s40201-019-00354-1
Faisal AAH, Al-Wakel SFA, Assi HA, Naji LA, Naushad M (2020) Waterworks sludge-filter sand permeable reactive barrier for removal of toxic lead ions from contaminated groundwater. J Water Process Eng 33:101112. https://doi.org/10.1016/j.jwpe.2019.101112
Sethi S, Kaith BS, Saruchi KV (2019) Fabrication and characterization of microwave assisted carboxymethyl cellulose-gelatin silver nanoparticles imbibed hydrogel: Its evaluation as dye degradation. React Funct Polym 142:134–146
Naushad M (2014) Surfactant assisted nano-composite cation exchanger: Development, characterization and applications for the removal of toxic Pb2+ from aqueous medium. Chem Eng J 235:100–108. https://doi.org/10.1016/J.CEJ.2013.09.013
Saruchi, Verma, R., Kumar, V. et al. Comparison between removal of Ethidium bromide and eosin by synthesized manganese (II) doped zinc (II) sulphide nanoparticles: kinetic, isotherms and thermodynamic studies. J Environ Health Sci Engineer (2020). https://doi.org/10.1007/s40201-020-00536-2
Baysal K, Arogue AZ, Adiguel Z, Baysal BM (2013) Chitosan/alginate crosslinked hydrogels: Preparation, characterization and application for cell growth purposes. IntJBiolMacromol 59:342–348. https://doi.org/10.1016/j.ijbiomac.2013.04.073
Kaith BS, Sharma R, Kalia S, Bhatti MS (2014) Response surface methodology and optimized synthesis of guar gum-based hydrogels with enhanced swelling capacity. RSC Adv 4:40339–40344. https://doi.org/10.1039/C4RA05300A
Naushad M, Vasudevan S, Sharma G, Kumar A, Alothman ZA (2016) Adsorption kinetics, isotherms, and thermodynamic studies for Hg2+ adsorption from aqueous medium using alizarin red-S-loaded amberlite IRA-400 resin. Desalin Water Treat 57(39):18551–18559. https://doi.org/10.1080/19443994.2015.1090914
Saruchi BS, Kaith R, Jindal V, Kumar MS (2014) Bhatti, Optimal response surface design of Gum tragacanth-based poly[(acrylic acid)-co-acrylamide] IPN hydrogel for the controlled release of the antihypertensive drug losartan potassium. RSC Adv 4:39822–39829. https://doi.org/10.1039/C4RA02803A
Moura MJ, Faneca H, Lima MP, Gil MH, Figueiredo MM (2011) In situ forming chitosan hydrogels prepared via ionic/covalent Co-Cross-Linking. Biomacromol 12:3275–3284. https://doi.org/10.1021/bm200731x
Saruchi, Sharma M, Hatshan MR, Kumar V, Rana A (2020) Sequestration of Eosin Dye by Magnesium (II)-Doped Zinc Oxide Nanoparticles: Its Kinetic, Isotherm, and Thermodynamic Studies. J Chem Eng Data. https://doi.org/10.1021/acs.jced.0c00810
Vimala K, Sivudu KS, Mohan YM, Sreedhar B, Raju KM (2009) Controlled silver nanoparticles synthesis in semi-hydrogel networks of poly (acrylamide) and carbohydrates: A rational methodology for antibacterial application. CarbohydrPolym 75:463–471. https://doi.org/10.1016/j.carbpol.2008.08.009
Bajpai SK, Kumari M (2015) A green approach to prepare silver nanoparticles loaded gum acacia/poly (acrylate) hydrogels. IntJBiolMacromol 80:177–188. https://doi.org/10.1016/j.ijbiomac.2015.06.048
Xia B, Cui Q, He F, Li L (2012) Preparation of hybrid hydrogel containing Ag nanoparticles by a green in situ reduction method. Langmuir 28:11188–11194. https://doi.org/10.1021/la302011x
Sethi S, Kaith BS, Kaur M, Sharma N, Khullar S (2019) Study of a Cross-Linked Hydrogel of Karaya Gum and Starch as a Controlled Drug Delivery System. J Biomater Sci Polym Ed 30:1687–1708. https://doi.org/10.1080/09205063.2019.1659710
Stela E, Diana D, Apopei F (2013) Multiresponsivemacroporous semi-IPN composite hydrogels based on native or anionically modified potato starch. Carbohydr Polym 92:23–32. https://doi.org/10.1016/j.carbpol.2012.08.082
S. Sethi, B. S. Kaith, M. Kaur, N.Sharma& S. Khullar.A hydrogel based on dialdehyde carboxymethyl cellulose–gelatin and its utilization as a bio adsorbent. J Chem Sci 132 (2020). https://doi.org/10.1007/s12039-019-1700-z
Sapna S, Saruchi, Kaith BS, Kaur M, Sharma N, Kumar V (2020) Cross-linked xanthan gum-starch hydrogels as promising materials for controlled drug delivery. Cellulose 27:4565–4589. https://doi.org/10.1007/s10570-020-03082-0
Cui L, Jia J, Guo Y, Liu Y, Zhu P (2014) Preparation and characterization of IPN hydrogels composed of chitosan and gelatin cross-linked by genipin. Carbohydr Polym 99:31–38. https://doi.org/10.1016/j.carbpol.2013.08.048
Koul V, Mohamed R, Kuckling D, Adler H-JP, Choudhary V (2011) interpenetrating polymer network (IPN) nanogels based on gelatin and poly (acrylic acid) by inverse mini emulsion technique: Synthesis and characterization. Colloids Surf B Biointerfaces. 83:204–213. https://doi.org/10.1016/j.colsurfb.2010.11.007
Wang W-B, Huang D-J, Kang Y-R, Wang A-Q (2013) One-step in situ fabrication of a granular semi-IPN hydrogel based on chitosan and gelatin for fast and efficient adsorption of Cu2+ ion. Colloids Surf B Biointerfaces 106:51–59. https://doi.org/10.1016/j.colsurfb.2013.01.030
Chang C, Han K, Zhang L (2011) Structure and properties of cellulose/poly (N-isopropylacrylamide) hydrogels prepared by IPN strategy. PolymAdv Technol 22:1329–1334. https://doi.org/10.1002/pat.1616
Dragan ES (2014) Design and applications of interpenetrating polymer network hydrogels- A review. ChemEng J 243:572–590. https://doi.org/10.1016/j.cej.2014.01.065
Naseri N, Deepa B, Mathew AP, Oksman K, Girandon L (2016) Nanocellulose-based interpenetrating polymer network (IPN) hydrogels for cartilage applications. Biomacromol 17:3714–3723. https://doi.org/10.1021/acs.biomac.6b01243
Nieuwenhove IV, Salamon A, Petersb K, Graulusa G-J, Martins JC, Frankeld D, Kersemanse K, Vose FD, Vlierberghea SV, Dubruela P (2016) Gelatin- and starch-based hydrogels. Part A: Hydrogel Development, characterization and coating. Carbohydr Polym 152:129–139. https://doi.org/10.1016/j.carbpol.2016.06.098
Manna PJ, Mitra T, Pramanik N, Gnanamani A, Kundu PP, Kavitha V (2015) Potential use of curcumin loaded carboxymethylated guar gum grafted gelatin film for biomedical applications. Int J BiolMacromol 75:437–446. https://doi.org/10.1016/j.ijbiomac.2015.01.047
Burey P, Bhandari BR, Rutgers RPG, Halley PJ, Torley PJ (2009) Confectionery gels: a review on formulation, rheological and structural aspects. Int J Food Prop 12:176–210. https://doi.org/10.1080/10942910802223404
Maneerung T, Tokura S, Rujiravanit R (2008) Impregnation of silver nanoparticles into bacterial cellulose for antimicrobial wound dressing. Carbohydr Polym 72:43–51. https://doi.org/10.1016/j.carbpol.2007.07.025
Seo SY, Lee GH, Lee SG, Jung SY, Lim JO, Choi JH (2012) Alginate-based composite sponge containing silver nanoparticles synthesized in situ. CarbohydrPolym 90:109–115. https://doi.org/10.1016/j.carbpol.2012.05.002
Ismail H, Irani M, Ahmad Z (2013) Starch-based hydrogels: Present statusand applications. Int J Polym Mater 62:411–420. https://doi.org/10.1080/00914037.2012.719141
Tongdeesoontorn W, Mauer LJ, Wongruong S, Sriburi P, Rachtanapun P (2012) Mechanical and Physical Properties of Cassava Starch-Gelatin Composite Films. Int J Polym Mater. 61(2012):778–792. https://doi.org/10.1080/00914037.2011.610049
Sharma K, Kaith BS, Kumar V, Kumar V, Som S, Kalia S, Swart HC (2013) Synthesis and properties of poly (acrylamide-aniline)-grafted gum ghatti based nanospikes. RSCAdv 3:25830–25839. https://doi.org/10.1039/C3RA44809F
Holt KB, Bard AJ (2005) Interaction of silver (I) ions with the respiratory chain of Escherichia coli: an electrochemical and scanning electrochemical microscopy study of the antimicrobial mechanism of micromolar Ag. Biochemistry 44:13214–13223. https://doi.org/10.1021/bi0508542
Vasileva P, Donkova B, Karadjova I, Dushkin C (2011) Synthesis of starch-stabilized silver nanoparticles and their application as a surface plasmon resonance-based sensor of hydrogen peroxide. Colloids Surf A PhysicochemEng Asp 382:203–210. https://doi.org/10.1016/j.colsurfa.2010.11.060
Abdel-Halim ES, Al-Deyab SS (2011) Utilization of hydroxypropyl cellulose for green and efficient synthesis of silver nanoparticles. Carbohydr Polym 86:1615–1622
Sharma G, Pathania D, Naushad Mu, Kothiyal NC (2014) Fabrication, characterization and antimicrobial activity of polyaniline Th (IV) tungstomolybdophosphate nanocomposite material: efficient removal of toxic metal ions from water. Chem Eng J 251:413–421. https://doi.org/10.1016/j.cej.2014.04.074
Mohan YM, Premkumar T, Lee K, Geckeler KE (2006) Fabrication of silver nanoparticles in hydrogel networks. Macromol Rapid Commun 27:1346–1354. https://doi.org/10.1002/marc.200600297
Parang Z, Keshavarz A, Farahi S, Elahi SM, Ghoranneviss M, Parhoodeh S (2012) Fluorescence emission spectra of silver and silver/cobalt nanoparticles. Sci Iran F 19:943–947. https://doi.org/10.1016/j.scient.2012.02.026
Farajia S, Nowroozi N, Nouralishahi A, Shayeh JS (2020) Electrospun poly-caprolactone/graphene oxide/quercetin nanofibrous scaffold for wound dressing: Evaluation of biological and structural properties. Life Sci 257:118062. https://doi.org/10.1016/j.lfs.2020.118062
Altaf F, Niazi MBK, Jahan Z, Ahmad T, Akram MA, Safdar A, Butt MS, Noor T, Sher F (2021) Synthesis and Characterization of PVA/Starch Hydrogel Membranes Incorporating Essential Oils Aimed to be Used in Wound Dressing Applications. J Polym Environ 29:156–174. https://doi.org/10.1007/s10924-020-01866-w
Zou F, Sun X, Wang X (2019) Elastic, hydrophilic and biodegradable poly (1, 8-octanediol-co-citric acid)/polylactic acid nanofibrous membranes for potential wound dressing applications. Polym Degrad Stab 166:163–173. https://doi.org/10.1016/j.polymdegradstab.2019.05.024
Acknowledgements
The authors Sapna Sethi, Medha, Swati Thakur, and Neeraj Sharma are thankful to DAV University for providing infrastructure to carrying out this work. The authors thank the Researchers Supporting Project number (RSP-2021/169), King Saudi University, Riyadh, Saudi Arabia, for the financial support.
Author information
Authors and Affiliations
Contributions
Sapna Sethi, Medha, Swati Thakur, Balbir Singh Kaith, and Neeraj Sharma contributed in planning and synthesizing part. Sabah Ansar, Sadanand Pandey Saruchi, and Vaneet Kumar contributed in designing and finalizing this manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Sethi, S., Saruchi, Medha et al. Biopolymer starch-gelatin embedded with silver nanoparticle–based hydrogel composites for antibacterial application. Biomass Conv. Bioref. 12, 5363–5384 (2022). https://doi.org/10.1007/s13399-022-02437-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13399-022-02437-w