
Abstract
Our previous work in a mouse model of experimental rabies showed neuronal process (dendrites and axons) degeneration in association with severe clinical disease. Cultured adult rodent dorsal root ganglion (DRG) neurons infected with the challenge virus standard-11 (CVS) strain of rabies virus (RABV) showed axonal swellings and reduced axonal growth with evidence of oxidative stress. We have shown that CVS infection alters a variety of mitochondrial parameters and increases mitochondrial complex I activity and reactive oxygen species (ROS) production. Expression of a peptide from amino acid 139–172 of the CVS phosphoprotein (P) increased complex I activity and ROS generation similar to expression of the entire P. Site-directed mutational analyses illustrated the importance of the 145–151 and 157–169 regions of P and that serine residues at 162 and 166 are important single amino acid sites. Two CVS recombinant viruses with serine to alanine mutations at positions 162 (A162r) and 166 (A166r) did not increase complex I activity or ROS generation and also did not induce axonal swellings or inhibit axonal growth in DRG neurons. RABV infection is a mitochondrial disorder initiated by interaction of the RABV P and complex I; S162 and S166 are critical sites in the P for this interaction. The resulting mitochondrial dysfunction produces oxidative stress in neurons causing acute degenerative changes affecting neuronal processes resulting in a severe and fatal clinical disease. This information will be important for the future development of novel therapies for rabies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rabies is an acute viral infection of the nervous system affecting humans and animals that remains an important public health problem (Jackson 2013b). Rabies is virtually always fatal in humans and there is no effective therapy for the disease (Jackson 2013a, c). An incomplete understanding of rabies pathogenesis has served as an important barrier to the development of new approaches to therapy. We have made key observations that have led to new insights into basic mechanisms important in rabies pathogenesis.
The neuropathological changes in rabies include mild inflammatory changes with few degenerative neuronal changes in the central nervous system (Iwasaki and Tobita 2002; Rossiter and Jackson 2013). A lack of recognition of degenerative neuronal changes gave rise to the concept idea that rabies is caused by neuronal dysfunction (Fu and Jackson 2005). However, our studies in transgenic mice expressing the yellow fluorescent protein in a subpopulation of neurons that were inoculated peripherally with the challenge virus standard-11 (CVS) strain of fixed rabies virus (RABV) showed structural degenerative changes in neuronal processes, including both dendrites and axons, which explain the severity of disease (Scott et al. 2008). Cultures of adult rodent dorsal root ganglion neurons infected with CVS showed axonal swellings and reduced axonal growth and increased expression of 4-hydroxy-2-nonenal (4-HNE), which is associated with lipid peroxidation and serves as a marker of oxidative stress (Jackson et al. 2010). Hence, in RABV infection, injury to neuronal processes was associated with oxidative stress. Because mitochondrial dysfunction may be an important cause of oxidative stress, we evaluated a variety of mitochondrial functions in CVS infection in DRG neurons and other cell types. We found evidence of mitochondrial dysfunction, including increased activity of respiratory chain complex I, high mitochondrial membrane potential, high NADH/NAD+ ratio, and low ATP content (Alandijany et al. 2013). Multiple lines of evidence have implicated the RABV phosphoprotein (P) in this process, and interaction of the RABV P with complex I may explain the increased activity of complex I and generation of reactive oxygen species (ROS) (Kammouni et al. 2015), which play an important role in neuronal injury in oxidative stress. The region of the RABV P between amino acid positions 139–172 was found to be particularly important for RABV P–complex I interaction (Kammouni et al. 2015).
We have now further investigated the 139–172 region of the RABV P with a mutational approach in order to determine critical sites that are important for RABV P–complex I interaction and induction of oxidative stress. We have identified important amino acid mutations at positions 162 and 166 of the RABV P that affect complex I activity and generation of ROS. Furthermore, using recombinant RABVs, we have confirmed the importance of these sites in the induction of oxidative stress.
Materials and methods
Virus preparation and infection of continuous cell lines
The CVS-11 strain of fixed RABV (CVS), which was obtained from William H. Wunner (The Wistar Institute, Philadelphia, PA), was used in these studies. Viruses were grown in mouse neuroblastoma (MNA) cells that were obtained from Diagnostic Hybrids (Athens, OH) and cultivated in Eagle’s minimum essential medium (EMEM) (Lonza, Walkersville, MD) containing 1% sodium bicarbonate (Lonza), 40 mM l-glutamine (Sigma-Aldrich, St. Louis, MO), tryptose phosphate broth (Teknova, Hollister, CA) containing 0.14% tryptose, 0.014% glucose, 0.035% sodium chloride, 0.0175% sodium phosphate dibasic and 10% fetal calf serum at 37 °C in a 5% CO2 incubator. The virus titer in each sample was determined in triplicate by infecting MNA cells in 96-well plates with serial 10-fold dilutions of each sample. The plates were incubated at 37 °C for 48 h and then fixed with 80% acetone. The monolayers were stained with the Light Diagnostics Rabies DFA II FITC conjugate, containing a blend of two anti-nucleocapsid antibodies (EMD Millipore, Etobicoke, ON, Canada) and read using an Axiovert 40 CFL fluorescent microscope (Carl Zeiss, North York, ON, Canada).
Complex I activity and ROS levels were measured in monolayers of MNA cells infected with CVS or CVS recombinant (CVSr) or CVSr mutants (A159r, A162r, and A166r) at a multiplicity of infection (MOI) of 10 fluorescent foci forming units per cell. After 1 h of viral adsorption, fresh media was added. The human embryonic line (HEK-293T) was obtained from Thermo Fisher Scientific Open Biosystems and was cultivated in Iscove’s modified Dulbecco’s medium (IMDM) (Thermo Fisher Scientific, Waltham, MA) supplemented with 10% fetal bovine serum (Life Technologies, Grand Island, NY). MNA and HEK-293T cells were incubated at 37 °C in humidified air containing 5% CO2.
Site-directed mutagenesis
We obtained a plasmid from Zhen Fu (University of Georgia, Athens, GA) encoding the P gene of the CVS-B2c strain cloned into the pcDNA3 vector [GenBank accession number HQ891318] (Zhang et al. 2013). Recombinant plasmids expressing RABV P-GFP (full-length P of RABV strain B2c fused to GFP) as well as plasmids expressing amino acids 139–172 of P fused to GFP and GFP alone were generated as previously described (Kammouni et al. 2015). Saturation alanine mutagenesis was used to mutate overlapping triplicate adjacent sites within the region of amino acids 139–172 in the P of CVS. The following triplicate amino acids were mutated to alanine: RSS, SED, DKS, STQ, QTT, IGR, REL, LKK, KET, TTS, SAF, FSQ, QRE, ESQ, QPS, SKA, and KAR. Alanine was substituted for each of the three adjacent amino acids in each construct. The Q5 Site-Directed Mutagenesis kit (New England Biolabs) was used for site-directed mutagenesis of pAcGFP1 (Clontech), encoding the P gene of CVS. The primers used for each site-directed mutagenesis reaction are shown in Table 1. The serine residues at positions 159, 162, 166, and 169 were also mutated individually to both alanine (turn off or deactivated phenotype) and to aspartate (turn on or activated phenotype) by site-directed mutagenesis of pAcGFP1 encoding the P gene.
Plasmid transfection
HEK-293T cells were used for plasmid transfection due to the high transfection efficiency (~90%) of these cells. Cells were seeded at a density of 2.6 × 105 cells per well in a 24-well culture plate. The cells were transfected the following day using Lipofectamine 2000 reagent (Invitrogen, Burlington, ON, Canada) according to the manufacturer’s instructions and incubated for 72 h at 37 °C in 5% CO2.
Western blotting for P, neomycin, and vinculin expression
Mock and transfected HEK-293T cells (as described above) were incubated for 72 h at 37 °C in 5% CO2 and then lysed using ice-cold neurofilament stabilization buffer containing 0.1 M Pipes, 5 mM MgCl2, 5 mM EGTA, 0.5% Triton X-100, 20% glycerol, 10 mM NaF, 1 mM PMSF, and protease inhibitor cocktail. Protein amounts were determined by the DC protein assay. Equivalent amounts of the extracted proteins (20 μg/lane) were loaded onto a 10% SDS-PAGE gel and electrotransferred onto nitrocellulose membrane at 100 V for 1 h. The membrane was blocked with Odyssey blocking buffer (#927-50000, Li-Cor Biosciences, Lincoln, NE) and then incubated with specific primary antibodies overnight at 4 °C: living color mouse monoclonal anti-GFP (#632381, Clontech, CA) (1/1000), anti-vinculin (#18058, Abcam, Toronto, ON, Canada) (1/2000), and anti-neomycin antibody (#06747, Millipore, ON, Canada) (1/1000). Neomycin (NeoR) (expressed by transfected plasmids) was used as an internal control for the expression of RABV P and compared to vinculin, a cell lysate loading control. The gel was washed, probed with goat anti-mouse or goat anti-rabbit secondary antibodies (1:10,000) (#926-68020 and 926-32211, Li-Cor Biosciences), and washed again before imaging using the Odyssey imager (Li-Cor Biosciences). Data are expressed as the intensity of the protein scan values of GFP, neoR, or vinculin protein. Two biological replicates were performed.
Complex I activity
Complex I activity was measured as described previously (Alandijany et al. 2013), which was based on the ability of complex I to transfer electrons to complex III in the presence of NADH, resulting in the reduction of oxidized cytochrome c.
Determination of ROS levels
Transfected (HEK-293T) or infected (MNA) cells in 96-well plates were incubated for 72 h at 37 °C in 5% CO2. ROS production was then determined using the Amplex Red Hydrogen Peroxide/Peroxidase kit (Invitrogen, Burlington, ON, Canada). This kit is a one-step assay that detects hydrogen peroxide (H2O2) production from cells in the presence of horseradish peroxidase (HRP) activity. The media was removed from the cells, and 50 μL of reaction buffer containing 10 μM digitonin (to permeabilize plasma membranes) was added in the presence or absence of 10 mM succinate. The reaction was started by the addition of 50 μL of working solution containing 100 μM Amplex Red reagent and 0.2 U/mL HRP and incubated at 37 °C. The absorbance was measured at 560 nm after 2 h using a microplate reader as previously described (Alandijany et al. 2013).
CVS reverse genetics system
A reverse genetics system based on the CVS strain was established by cloning the full-length CVS genome by RT-PCR into the pCRII vector (Thermo Fisher Scientific, Burlington, ON, Canada) to generate pCRII-CVS. The CVS cDNA genome was cloned into pCRII under the control of a T7 RNA polymerase promoter. The sequence for the hepatitis delta virus ribozyme (HDVRz) was incorporated at the 3′ end of the viral genome to enhance viral replication. The genes encoding the nucleoprotein (N), phosphoprotein (P), and polymerase (L) of CVS and the T7 RNA polymerase were cloned into pCAGGS (Addgene, Cambridge, MA) under the control of a chicken β-actin promoter with a CMV enhancer for eukaryotic expression, to generate helper plasmids required for rescue of the recombinant CVS virus (CVSr). Mouse neuroblastoma (MNA) cells were co-transfected with 5 μg of pCRII-CVS, 5 μg of pCAGGS containing the T7 RNA polymerase, and 1.25 μg of each helper plasmid (pCAGGS-N, P, and L) using Fugene 6 (Promega, Madison, WI). The transfected cells were incubated for 6 days at 37 °C in 5% CO2. Infectious virus (CVSr) was isolated by passaging the supernatant onto fresh MNA cells which were incubated for a further 4 days 37 °C in 5% CO2. The supernatant was harvested from these cells and stored at −80 °C.
Three recombinant CVS viruses were generated using site-directed mutagenesis of pCRII-CVS, with each virus containing a single alanine substitution (in place of serine) at amino acid positions 159 (A159r), 162 (A162r), 166 (A166r) of the P. The Quick-Change II XL Site-Directed Mutagenesis kit (Agilent Technologies, Mississauga, ON, Canada) was used to introduce the mutations in the P gene by PCR. The primers used to generate the A162r and A166r mutants are shown in Table 2. Recombinant viruses were rescued by transfection of MNA cells as described above. The complete genome of each virus was sequenced and confirmed using the Mi-Seq system (Illumina, San Diego, CA) (data not shown).
Single-step and multi-step viral growth assays
Single-step and multi-step viral growth assays were performed using monolayers of MNA cells in T175 flasks infected with CVS, CVSr, A162r, or A166r at an MOI of 1 or 0.01, respectively. After 1 h of incubation at 37 °C, the inoculum was removed and the cells were washed twice with PBS. EMEM containing 10% FCS, 1% l-glutamine, and 7% tryptose phosphate broth was added to each flask followed by further incubation at 37 °C. Samples of culture supernatant were collected at 24, 48, 72, and 96 h after infection and the titer was determined as described previously.
Infection of DRG neuron cultures with CVSr, A162r, and A66r
DRG neurons were extracted from adult male 8- to 12-week-old Sprague Dawley rats (University of Manitoba, Winnipeg, Manitoba, Canada). The isolation method used has been described previously (Kammouni et al. 2012). Collected cells were resuspended in F-12 medium with 2 mM l-glutamine and Bottenstein’s N2 supplements without insulin: 0.1 mg/ml transferrin, 20 nM progesterone, 100 μM putrescine, 30 nM sodium selenite, and 0.1 mg/ml fatty-acid-free BSA. For immunohistochemistry, the cells were seeded on coverslips with a 12-mm diameter treated pre-treated with 0.5 mg/ml poly-dl-ornithine (P8638; Sigma-Aldrich) and coated subsequently with 2 μg/ml natural mouse laminin (23017-015; Invitrogen). The cells were incubated for 48 h in N2–F-12 medium at 37 °C in humidified air containing 5% CO2. After 2 days of incubation, the cultured DRG neurons was infected with CVSr, A162r, or A166r at a multiplicity of infection of 30 fluorescent focus-forming units per cell or cells were mock-infected, followed by the addition of fresh N2–F12 medium. The infected DRG neurons were incubated for 72 h at 37 °C.
Assessment of 4-HNE puncta and axonal outgrowth with recombinant viruses
Rat DRG neurons infected with the recombinant viruses (CVSr, A162r, and A166r) or mock-infected were incubated for 72 h, fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) (pH 7.4) for 15 min, and subsequently permeabilized with 0.3% Triton X-100 in PBS for 3 min. Coverslips were stained overnight using a mouse monoclonal antibody specific for β-tubulin isotype III (no. T8660; Sigma-Aldrich) (1:300) as a pan-neuronal marker to label cell bodies and neurites. Coverslips were also stained using a polyclonal rabbit anti-4-hydroxy-2-nonenal (4-HNE) antibody (no. ALX-210-767-R100; Alexis Biochemicals, San Diego, CA) (1:100) as was described previously (Jackson et al. 2010). Coverslips were mounted on slides with Vectashield mounting medium (H-1200, Vector Laboratories, Burlingam, CA) and examined using LSM700 laser scanning confocal microscope (Zeiss) equipped with fluorescein isothiocyanate (FITC) and cyanine (Cy3) filters with Zen 2009 software. Images of cells from a minimum of 10 random fields per well were taken, and the total number of neuronal cell bodies and the number of intersects of their neurites with a vertical grid were counted using a morphometric approach using ImageJ 1.42 software (available at http://rsbweb.nih.gov/ij/). The total number of intersects per neuron was taken as the parameter of total axon outgrowth as previously described (Zherebitskaya et al. 2009). The number of labeled puncta was analyzed relative to axonal length in micrometer.
Statistical analysis
Data were analyzed using GraphPad Prism version 4 software. Standard two-tailed unpaired Student’s t tests with Welch’s correction, which did not assume equal variances, were used for evaluating the significance of the difference between the means on assays comparing transfected or mock- and CVS-infected samples. A value of p < 0.05 was considered statistically significant.
Results
Site-directed alanine mutagenesis of the 139–172 region of the RABV P revealed important sites required by P for activation of complex I and ROS
We evaluated first the activity of complex I in HEK-293T cells transfected with plasmid expressing RABV P or plasmids expressing RABV P with overlapping triplicate adjacent alanine residues in the region 139–172 (Fig. 1a). In comparison to cells transfected with the vector coupled to RABV P (100%), cells expressing mutant P with alanine mutations at positions 145–151 and 157–169 significantly reduced complex I activity. These results provided evidence that those amino acids may play an important role in complex I activation in CVS infection.

Site-directed mutagenesis of the 139–172 region of the RABV P. Reduced activity (relative to plasmid P) of complex I and/or reduced generation of ROS were observed with site-directed mutagenesis for each of the three adjacent amino acids mutated to alanine at amino acid positions 145–151 and 157–169. Complex I activity was measured as rotenone-sensitive NADH-cytochrome c reductase activity, and determination of ROS levels was based on the ability of the Amplex Red reagent in the presence of horseradish peroxidase to detect the rate of H2O2 production in HEK-293T cells 72 h after transfection as we previously reported (Alandijany et al. 2013). *p < 0.05; **p < 0.01
Secondly, we determined ROS production for the phosphoproteins with the same alanine mutants (Fig. 1b). In comparison to RABV P protein in which ROS increased (100%), the recombinant proteins containing mutations in the regions 145–151 and 157–169 showed significantly reduced ROS production. On the basis of these results, we concluded that these regions were likely also important for ROS production.
High levels of expression of the RABV P-GFP with the alanine mutations, which was demonstrated by western blotting using anti-GFP antibody, were confirmed 72 h after transfection (data not shown). This indicates that reduced complex I activities and ROS levels associated with these mutations were not due to transfection inefficiencies or destabilization of the mutant phosphoproteins.
Complex I activities and ROS production were reduced with mutations of serine residues at positions 159, 162, 166, and 169 to alanine in the RABV P
The region 157 to 169 of P contains four serine residues at positions 159, 162, 166, and 169. Since phosphorylation at one or more of these serines could potentially be involved in the interaction of the RABV P and complex I, we separately mutated each of the serines to alanine (turned off phenotype) and to aspartate (turned on phenotype). We observed significantly reduced complex I activity in cells expressing the phosphoproteins containing each of the four alanine mutations versus the wild-type RABV P, and there was reduced activity for the phosphoprotein containing an aspartate mutation at 169, but not for aspartate mutations at the other positions (Fig. 2a). For all of the positions, we observed significantly reduced complex I activity for the phosphoproteins containing the alanine mutations versus the aspartate mutations. The reduction of activity of the phosphoprotein containing an aspartate mutation at 169 is of uncertain significance.

Complex I activity and ROS production with conversion of four individual serine residues to both aspartate and to alanine with site-directed mutagenesis. An in depth look at single amino acid mutations in the region 157 to 169. The four serine residues (which may be phosphorylated–e.g., position 162) were individually mutated to both alanine (deactivated phenotype) and to aspartate (activated phenotype). We observed a significant reduction in complex I activities (a) and lower ROS levels (b) with all of the phosphoproteins with serine to alanine mutations compared to RABV P in HEK-293T cells 72 h after transfection, but there was also a reduction for the serine to aspartate mutation at 169 (D169). *p < 0.05; **p < 0.01; ***p < 0.001
Evaluation of ROS production showed that there was also significant reduction of ROS production after transfection of plasmids expressing P with each of the four of alanine mutations separately, and there was also reduced activity for the P with the aspartate mutation at 169, but not at the other positions (Fig. 2b). For all of the positions, there was significantly reduced ROS levels for the alanine mutations versus the aspartate mutations. The reduced ROS levels detected in cells expressing the phosphoproteins with an aspartate mutation at 169 were consistent with the reduced complex I activity detected in the same cells. Because of the unexplained discordant results at position 169 for complex I activity and ROS levels, we decided to focus our further attention on the S159, S162, and S166 sites.
High expression of RABV P with serine to alanine mutations at positions 159, 162, 166, and 169 of P, evaluated 72 h p.i. by western blot with anti-GFP antibody, was demonstrated (Fig. 3). P expression was mildly reduced with the serine to aspartate mutation at position 162 (D162) and in the three double alanine mutants (A159 A162, A159 A166, and A162 A166) versus the unmutated P (Fig. 3), but these double mutants were not further evaluated. This indicates that reduced complex I activities and ROS levels could not be explained by reduced expression of the mutant proteins with the serine to alanine mutations.

Western immunoblot of RABV P, neomycin (NeoR) , and vinculin. Merged multiplex fluorescent near-infrared image in grayscale of total lysates assessed for the expression of RABV P-GFP , NeoR, and vinculin (loading control) in transfected and mock transfected cells after 72 h. Similar amounts of cell lysates were loaded for samples as reflected by the intensity of the vinculin marker. A good correlation was observed between the intensity of NeoR and GFP signals. In comparison to the expression of RABV P, high expression and similar levels of mutated RABV P expression was observed after transfection of plasmids expressing phosphoproteins with single mutations: A159, D159, A162, A166, D166, A169, and D169. The expression of mutant RABV P was slightly lower for the single mutant D162 and the three double mutants (A159A162, A159 A166, and A162 A166)
Recombinants viruses A159r, A162r, and A166r reduced complex I activity
CVSr with an identical genome sequence to CVS, which is neurovirulent in a mouse model, was generated by reverse genetics (Xue et al. 2014). Site-directed mutagenesis was used to change the serine residues to alanine at each of the 159, 162, and 166 positions of P. Rescued viruses retained their mutations as confirmed by RT-PCR and sequencing (data not shown). They were found to be replication competent (see below).
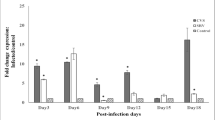
Complex I activity was assessed 72 h after infection of MNA cells with CVSr, A159r, A162r, and A166r. As previously demonstrated for CVS infection (Alandijany et al. 2013), CVSr strongly increased the activity of complex I (100%) (Fig. 4a). In comparison to the activity induced by CVSr, cells infected with all of the recombinant mutant viruses (A159r, A162r, and A166r) displayed significantly reduced activities of complex I (Fig. 4a).

Infection with recombinant A162r and A166r viruses did not increase complex I activity or ROS levels. A162r and A166r did not induce increased complex I activity (a) or ROS levels (b) 72 h post-infection of MNA cells vs. CVSr/CVS. ROS levels were increased with A159r, suggesting ROS generation via a mechanism other than through increased complex I activity. *p < 0.05; **p < 0.01; ***p < 0.001; NS not significant
Recombinant viruses A162r and A166r did not increase ROS generation
Evaluation of ROS production in MNA cells showed that there were high levels of ROS during infection with CVSr (Fig. 4b) similar to that demonstrated with CVS (Alandijany et al. 2013) and significantly lower ROS levels with A162r and A166r, but not with A159r virus (Fig. 4b). It appears that the alanine mutation at 159 was responsible for ROS generation by a mechanism independent of increased activity of complex I (unlike the A162 and A166 mutations). At this point, we decided to focus our attention on the mutations at S162 (A162) and at S166 (A166) for further evaluation.
Single-step and multiple-step viral growth curves for CVS, CVSr, A162r, and A166r
Recombinant viruses were prepared for the evaluation of the alanine mutations at positions 162 and 166 of the phosphoprotein, including CVSr, A162r, and A166r. Both single-step (Fig. 5a) and multiple-step (Fig. 5b) viral growth curves indicated that all three of our recombinant RABVs had similar profiles in comparison with CVS, which provides evidence that the A162 and A166 mutations did not interfere with viral replication. The recombinant viruses also grew to high viral titers in MNA cells (4.2 × 107 − 1.8 × 108 after four passages).

Viral growth curves of CVS, CVSr, A162r, and A166r in MNA cells. For single-step viral growth curves (a), MNA cells were infected with each virus at a MOI of 1, and for multiple-step viral growth curves (b) a MOI of 0.01 was used. Culture supernatants were harvested at 24, 48, 72, and 96 h after infection, and virus titers were determined in triplicate (p > 0.05). The values shown are the mean ± SEM
Infection with A162r and A166r did not induce axonal swellings or 4-HNE-labeled puncta in DRG neurons
DRG neurons were stained for β-tubulin at 72 h p.i., and oxidative stress was monitored by staining for 4-HNE. At 72 h post-infection, mock-infected DRG neurons exhibited normal axonal growth, whereas CVSr-infected DRG neurons displayed strong staining for 4-HNE with multiple axonal swellings (Fig. 6, arrowheads). DRG neurons infected with A162r and A166r showed markedly fewer axonal swellings and reduced 4-HNE staining at 72 h (Fig. 6).

A162r and A166r mutants do not induce oxidative stress. Seventy-two hours after infection, rat dorsal root ganglion neurons (MOI of 10) showed co-localization of immunofluorescent staining for tubulin (red) and 4-HNE (green) in axonal swellings (arrowheads) on the merged images (yellow), indicating induction of oxidative stress, after recombinant CVS (CVSr) infection, but not after infection with A162r or A166r or after mock infection
Infection of DRG neurons with A162r and A166r showed fewer axonal swellings positive for 4-HNE
Quantitative evaluation of the number of 4-HNE-immunostained puncta per 100 μm axon in mock-versus CVSr-infected cultures (Fig. 7a) showed a significantly (p < 0.01) increased numbers of labeled puncta in CVSr infection versus infections with A162r and A166r at 72 h p.i. We conclude that the serine residues at positions 162 and 166 of the RABV P are involved in CVSr-induced oxidative stress.

A162r and A166r mutants do not induce 4-HNE expression or inhibit axonal growth. There is quantitative evidence of oxidative stress with significantly more 4-HNE puncta (a) and significantly reduced axonal growth (b) in CVSr infection versus 72 h after infection with the A162r and A166r mutants (MOI 10) or in mock infection (*p < 0.05; **p < 0.01; ***p < 0.001)
Axonal growth of DRG neurons was not inhibited by infection with A162r and A166r
Total axonal outgrowth was assessed in DRG neurons 72 h after infection with the three recombinant viruses and after mock infection (Fig. 7b). After 72 h of infection, significantly reduced axonal growth was observed in CVSr-infected DRG neurons versus neurons infected with A162r and A166r, which were similar to the mock-infected neurons.
Discussion
Studies in a mouse model of experimental rabies showed that neuronal process degeneration occurs in association with the severe clinical disease (Scott et al. 2008). We have shown that oxidative stress is induced in RABV infection of neurons and that this is related to mitochondrial dysfunction (Alandijany et al. 2013). Furthermore, we showed that the region of the RABV P from amino acid positions 139 to 172 is important for increased activity of complex I and for generation of ROS (Kammouni et al. 2015), which plays an important role in oxidative stress. We have now focused our studies on this region of the P using a site-directed mutational analysis in an attempt to define critical sites that are important for RABV virus-induced oxidative stress.
Site-directed alanine mutagenesis was helpful in focusing attention on the 157–169 region of the P because of concordance of reduced complex I activities and reduced ROS levels with triplicate alanine mutations in this region (Fig. 1). Serine residues are potential phosphorylation sites, and there are four serine residues within this region at positions 159, 162, 166, and 169. Diverse classes of proteins kinases have been found to phosphorylate viral phosphoproteins (De and Banerjee 1997; De et al. 1997), and phosphorylation is thought to be involved in the function of the vesicular stomatitis virus (Pattnaik et al. 1997) and human parainfluenza virus type 3 phosphoproteins (HPIV3) (Huntley et al. 1995). In RABV infection, a unique protein kinase (packaged by the virion) and several isomers of PKC (in particular PKC ɤ) phosphorylate the P (Gupta et al. 2000). This phosphorylation could play an important role in mediating the interaction of RABV P and a subunit of complex I (Nishi et al. 2011) and intermediate host proteins that are known to bind to RABV P protein such as the signal transducer and activator of transcription 3 (STAT3) (Lieu et al. 2013) and the dynein light chain LC8 (Raux et al. 2000). Binding to STAT3 is of particular interest since STAT3 can modulate mitochondria function by decreasing the activity of complex I (Szczepanek et al. 2012). There is evidence that S162 of P is a RABV phosphorylation site (Gupta et al. 2000). We carefully evaluated each of these serine residue sites by mutating each of them to both aspartate and to alanine. Complex I activities and ROS levels were reduced for all, but there was disparity between the results at S169 and D169 and, hence, we focused on the other serine residues.
Recombinant RABVs were generated in order to better understand the importance of the serine residues during viral infection. We found that the A159r was associated with reduced complex I activity and high ROS levels, indicating ROS generation without activation of complex I activity. Because these results are inconsistent with our observations during CVS infection of DRG neurons (Jackson et al. 2010), we decided to focus on the A162r and A166r viruses for further studies.
We demonstrated that both A162r and A166r replicate well in culture (similar to CVSr and CVS) (Fig. 5) and they both failed to induce morphological changes of oxidative stress with reduced numbers of axonal swellings and less expression of 4-HNE in cultured DRG neurons. Hence, we have provided evidence that both S162 and S166 are critical sites in the RABV P for interaction with complex I, resulting in oxidative stress. The next step will be to further assess these mutations in the mouse model of rabies and determine their role in neurovirulence.
Therapy of rabies related to mitochondrial dysfunction will be an important therapeutic challenge for the future. Although we believe that increased complex I activity may play an important role in the generation of ROS in RABV-induced oxidative stress, there may be other mechanisms involved as well. Antioxidants are a potential therapy for rabies, but delivery in vivo may prove to be difficult. A mouse model of Parkinson’s disease is considered to be a mitochondrial complex I disorder with low complex I activity (Dawson et al. 2010). Kuan et al. (2012) provided therapy in a novel approach using a viral noncoding RNA that is expressed during human cytomegalovirus (HCMV) infection and functions to prevent mitochondrial-induced cell death by protecting complex I activity. The virus encodes an abundant 2.7-kb RNA transcript (β2.7) (Reeves et al. 2007) that functions as a noncoding RNA that interacts with genes associated with retinoid/interferon-induced mortality (GRIM)-19, which is a subunit of complex I that is essential for the assembly and electron transport activity of complex I (Huang et al. 2004). Interaction between complex I and GRIM-19 is essential for stabilizing the mitochondrial membrane potential and for continuing ATP production (Lu and Cao 2008). RNA interaction with complex I positively affects mitochondrial energy production reducing oxidative stress (Reeves et al. 2007). A similar approach may be taken for the therapy of rabies by addressing the clinical disease as a complex I disorder.
We now have an improved understanding of the mechanisms involved in CVS-induced mitochondrial dysfunction and oxidative stress. At the present time, we do not know if the same or similar mechanisms are involved in infection by street (wild-type) RABV infection in natural hosts. Infection of mice with street RABV variants may produce vastly different clinical and neuropathological features than in CVS infection or in natural hosts infected with street RABV variants (Jackson et al. 1989). Reduced activity of the mitochondrial electron transport system was reported in Mokola virus infection, which is a non-rabies virus lyssavirus. Studies using a yeast two-hybrid screening system indicated that the Mokola matrix protein interacts with subunit I of mitochondrial complex IV (cytochrome c oxidase) of the mitochondrial respiratory chain (Gholami et al. 2008). We have found that transfection with the Mokola virus P also results in significantly increased complex I activities and ROS levels (data not shown), suggesting that interaction of the RABV P with complex I may occur in different lyssaviruses, including wild-type lyssaviruses. We know that the RABV P plays an important role in CVS-induced mitochondrial dysfunction with oxidative stress and that S162 and S166 are critical single amino sites in this process, possibly related to phosphorylation of these amino acids. The next step will be to assess the role of these mutations in a mouse model with the goal of developing a novel therapeutic approach based on this improved understanding of rabies pathogenesis.
References
Alandijany T, Kammouni W, Roy Chowdhury SK, Fernyhough P, Jackson AC (2013) Mitochondrial dysfunction in rabies virus infection of neurons. J Neurovirol 19:537–549
Dawson TM, Ko HS, Dawson VL (2010) Genetic animal models of Parkinson’s disease. Neuron 66:646–661
De BP, Banerjee AK (1997) Role of host proteins in gene expression of nonsegmented negative strand RNA viruses. Adv Virus Res 48:169–204
De BP, Das T, Banerjee AK (1997) Role of cellular kinases in the gene expression of nonsegmented negative strand RNA viruses. Biol Chem 378:489–493
Fu ZF, Jackson AC (2005) Neuronal dysfunction and death in rabies virus infection. J Neurovirol 11:101–106
Gholami A, Kassis R, Real E, Delmas O, Guadagnini S, Larrous F, Obach D, Prevost MC, Jacob Y, Bourhy H (2008) Mitochondrial dysfunction in lyssavirus-induced apoptosis. J Virol 82:4774–4784
Gupta AK, Blondel D, Choudhary S, Banerjee AK (2000) The phosphoprotein of rabies virus is phosphorylated by a unique cellular protein kinase and specific isomers of protein kinase C. J Virol 74:91–98
Huang G, Lu H, Hao A, Ng DC, Ponniah S, Guo K, Lufei C, Zeng Q, Cao X (2004) GRIM-19, a cell death regulatory protein, is essential for assembly and function of mitochondrial complex I. Mol Cell Biol 24:8447–8456
Huntley CC, De BP, Murray NR, Fields AP, Banerjee AK (1995) Human parainfluenza virus type 3 phosphoprotein: identification of serine 333 as the major site for PKC zeta phosphorylation. Virology 211:561–567
Iwasaki Y, Tobita M (2002) Pathology. In: Jackson AC, Wunner WH (eds) Rabies. Academic Press, San Diego, pp. 283–306
Jackson AC (2013a) Current and future approaches to the therapy of human rabies. Antivir Res 99:61–67
Jackson AC (ed) (2013b) Rabies: scientific basis of the disease and its management, Third edn. Elsevier Academic, Oxford
Jackson AC (2013c) Therapy of human rabies. In: Jackson AC (ed) Rabies: scientific basis of the disease and its management, Third edn. Elsevier Academic, Oxford, pp. 573–587
Jackson AC, Kammouni W, Zherebitskaya E, Fernyhough P (2010) Role of oxidative stress in rabies virus infection of adult mouse dorsal root ganglion neurons. J Virol 84:4697–4705
Jackson AC, Reimer DL, Ludwin SK (1989) Spontaneous recovery from the encephalomyelitis in mice caused by street rabies virus. Neuropathol Appl Neurobiol 15:459–475
Kammouni W, Hasan L, Saleh A, Wood H, Fernyhough P, Jackson AC (2012). Role of nuclear factor-kB in oxidative stress associated with rabies virus infection of adult rat dorsal root ganglion neurons. J Virol 86:8139-8146.
Kammouni W, Wood H, Saleh A, Appolinario CM, Fernyhough P, Jackson AC (2015) Rabies virus phosphoprotein interacts with mitochondrial complex I and induces mitochondrial dysfunction and oxidative stress. J Neurovirol 21:370–382
Kuan WL, Poole E, Fletcher M, Karniely S, Tyers P, Wills M, Barker RA, Sinclair JH (2012) A novel neuroprotective therapy for Parkinson’s disease using a viral noncoding RNA that protects mitochondrial complex I activity. J Exp Med 209:1–10
Lieu KG, Brice A, Wiltzer L, Hirst B, Jans DA, Blondel D, Moseley GW (2013). The rabies virus interferon antagonist P protein interacts with activated STAT3 and inhibits Gp130 receptor signaling. J Virol 87:8261-8265.
Lu H, Cao X (2008) GRIM-19 is essential for maintenance of mitochondrial membrane potential. Mol Biol Cell 19:1893–1902
Nishi H, Hashimoto K, Panchenko AR (2011) Phosphorylation in protein-protein binding: effect on stability and function. Structure 19:1807–1815
Pattnaik AK, Hwang L, Li T, Englund N, Mathur M, Das T, Banerjee AK (1997) Phosphorylation within the amino-terminal acidic domain I of the phosphoprotein of vesicular stomatitis virus is required for transcription but not for replication. J Virol 71:8167–8175
Raux H, Flamand A, Blondel D (2000) Interaction of the rabies virus P protein with the LC8 dynein light chain. J Virol 74:10212–10216
Reeves MB, Davies AA, McSharry BP, Wilkinson GW, Sinclair JH (2007) Complex I binding by a virally encoded RNA regulates mitochondria-induced cell death. Science 316:1345–1348
Rossiter JP, Jackson AC (2013) Pathology. In: Jackson AC (ed) Rabies: scientific basis of the disease and its management, Third edn. Elsevier Academic, Oxford, pp. 351–386
Scott CA, Rossiter JP, Andrew RD, Jackson AC (2008) Structural abnormalities in neurons are sufficient to explain the clinical disease and fatal outcome in experimental rabies in yellow fluorescent protein-expressing transgenic mice. J Virol 82:513–521
Szczepanek K, Chen Q, Larner AC, Lesnefsky EJ (2012) Cytoprotection by the modulation of mitochondrial electron transport chain: the emerging role of mitochondrial STAT3. Mitochondrion 12:180–189
Xue X, Zheng X, Liang H, Feng N, Zhao Y, Gao Y, Wang H, Yang S, Xia X (2014) Generation of recombinant rabies virus CVS-11 expressing eGFP applied to the rapid virus neutralization test. Viruses 6:1578–1589
Zhang G, Wang H, Mahmood F, Fu ZF (2013) Rabies virus glycoprotein is an important determinant for the induction of innate immune responses and the pathogenic mechanisms. Vet Microbiol 162:601–613
Zherebitskaya E, Akude E, Smith DR, Fernyhough P (2009). Development of selective axonopathy in adult sensory neurons isolated from diabetic rats: role of glucose-induced oxidative stress. Diabetes 58:1356-1364.
Acknowledgements
We wish to acknowledge Michael Carpenter for assistance with the western blots, Stephanie Booth and Anna Majer for assistance with confocal microscopy, and Kym Antonation and Chantel Urfano for sequencing the RABV virus genomes (all at the Public Health Agency of Canada, Winnipeg, Manitoba, Canada).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding statement
This work was supported by a Research Manitoba Bridge Funding Award with the Department of Internal Medicine, University of Manitoba (to A.C. Jackson).
Conflict of interest
Wafa Kammouni, Heidi Wood, and Alan Jackson declare they have no conflicts of interest.
Rights and permissions
About this article

Cite this article
Kammouni, W., Wood, H. & Jackson, A.C. Serine residues at positions 162 and 166 of the rabies virus phosphoprotein are critical for the induction of oxidative stress in rabies virus infection. J. Neurovirol. 23, 358–368 (2017). https://doi.org/10.1007/s13365-016-0506-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13365-016-0506-8