
Abstract
Our previous studies in an experimental model of rabies showed neuronal process degeneration in association with severe clinical disease. Cultured adult rodent dorsal root ganglion neurons infected with challenge virus standard (CVS)-11 strain of rabies virus (RABV) showed axonal swellings and reduced axonal growth with evidence of oxidative stress. We have shown that CVS infection alters a variety of mitochondrial parameters and increases reactive oxygen species (ROS) production and mitochondrial Complex I activity vs. mock infection. We have hypothesized that a RABV protein targets mitochondria and triggers dysfunction. Mitochondrial extracts of mouse neuroblastoma cells were analyzed with a proteomics approach. We have identified peptides belonging to the RABV nucleocapsid protein (N), phosphoprotein (P), and glycoprotein (G), and our data indicate that the extract was most highly enriched with P. P was also detected by immunoblotting in RABV-infected purified mitochondrial extracts and also in Complex I immunoprecipitates from the extracts but not in mock-infected extracts. A plasmid expressing P in cells increased Complex I activity and increased ROS generation, whereas expression of other RABV proteins did not. We have analyzed recombinant plasmids encoding various P gene segments. Expression of a peptide from amino acid 139–172 increased Complex I activity and ROS generation similar to expression of the entire P protein, whereas peptides that did not contain this region did not increase Complex I activity or induce ROS generation. These results indicate that a region of the RABV P interacts with Complex I in mitochondria causing mitochondrial dysfunction, increased generation of ROS, and oxidative stress.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rabies is an acute viral infection of the nervous system in humans and animals that continues to be an important worldwide public health problem, particularly in Asia and Africa (Jackson 2013b). Human disease is virtually always fatal, and there have not been any recent important advances in therapy (Jackson 2013a, c). The development of new and effective approaches would benefit from an improved understanding of basic mechanisms that underlie the central nervous system disease in rabies. In an experimental model of rabies in mice with challenge virus standard (CVS) strain of rabies virus (RABV), our laboratory recognized that acute degeneration of neuronal processes (involving both dendrites and axons) explains the severe clinical disease in experimental rabies without major associated histopathological changes (Scott et al. 2008). Subsequent studies using cultured adult rodent dorsal root ganglion (DRG) neurons showed that axonal swellings and reduced axonal growth were associated with oxidative stress (Jackson et al. 2010). Oxidative stress has been recognized to play an important role in a variety of neurological diseases (Lin and Beal 2006; Reynolds et al. 2007), including diabetic neuropathy (Nishikawa et al. 2000; Obrosova et al. 2005, 2002; Russell et al. 2002).
Recently, it has been recognized that many viruses have important effects on mitochondrial function via a variety of different mechanisms (Anand and Tikoo 2013; Boya et al. 2004, 2003; Claus and Liebert 2014; Li et al. 2004, 2007; Lichty et al. 2006). For example, mitochondrial dysfunction is important in hepatitis C infection (Quarato et al. 2013; Wang and Weinman 2013), which is related, at least in part, to stimulation of the mitochondrial Ca2+ uniporter activity (Li et al. 2007). Virus-induced mitochondrial dysfunction may underlie many more diseases than have been recognized to date. We recently found evidence that mitochondrial dysfunction explains the occurrence of oxidative stress in rabies virus infection (Alandijany et al. 2013). Increased activity of mitochondrial Complex I was observed, which may, at least in part, explain the generation of reactive oxygen species (ROS) that induce neuronal injury affecting neuronal processes in the experimental animal model (Scott et al. 2008). In the present work, we have further investigated how RABV infection alters mitochondrial function resulting in oxidative stress. We assessed the presence of rabies virus proteins in purified mitochondrial extracts and the effects of expression of different RABV proteins on both Complex I activity and ROS generation. We have found evidence of a physical interaction of the RABV phosphoprotein (P) with mitochondrial Complex I in CVS-infected cultured cells and that a specific region of the RABV P plays an important role in increasing Complex I activity and ROS generation.
Materials and methods
Virus preparation and cell cultures
The fixed challenge virus standard (CVS) CVS-11 strain of rabies virus, which was obtained from Dr. William H. Wunner (The Wistar Institute, Philadelphia, PA, USA), was used in these studies. CVS was grown in baby hamster kidney (BHK) cells (C13 clone) in Dulbecco’s modified Eagle medium (DMEM) supplemented with 2 % newborn calf serum (NCS) (PAA Laboratories, Etobioke, ON, USA) at 37 °C in a 5 % CO2 incubator. Viral titration assays of stock virus were performed by counting fluorescent foci on BHK cell monolayers. Mouse neuroblastoma (MNA) cells were obtained from Diagnostic Hybrids (Athens, OH, USA) and were cultivated in minimum essential medium Eagle (Lonza, Walkersville, MD, USA) containing 1 % sodium bicarbonate (Lonza), 40 mM l-glutamine (Sigma-Aldrich, St. Louis, MO, USA), tryptose phosphate broth (Teknova, Hollister, CA, USA) containing 0.14 % tryptose, 0.014 % glucose, 0.035 % sodium chloride, 0.0175 % sodium phosphate dibasic, and 10 % fetal calf serum. MNA cells were infected with CVS at a multiplicity of infection (MOI) of 1 or 10 fluorescent foci forming unit(s) per cell. After 1 h of viral adsorption, fresh medium was added. The human embryonic kidney (HEK-293T) cell line was obtained from Thermo Scientific Open Biosystems and were cultivated in Iscove’s modified Dulbecco’s medium (IMDM) (Thermo Scientific, Waltham, MA, USA) supplemented with 10 % fetal calf serum. MNA and HEK-293T cells were kept at 37 °C in humidified air containing 5 % CO2.
Mitochondria extraction
Crude mitochondria extracts were obtained using the Cytoplasmic, Mitochondrial and Nuclear Protein Extraction kit from Zm Teck Scientific (Montreal, QC, Canada) and evaluated for Complex I interaction with the RABV P. Briefly, cells were centrifuged at 1600 rpm for 8 min and lysed in cytoplasmic lysis buffer, incubated on ice for 10 min, and then homogenized using Ten Broeck homogenizer (80 strokes). The cell extract was centrifuged twice at low speed (2500 rpm for 20 min and 3000 rpm for 8 min at 4 °C) in order to remove remaining cellular debris and nuclei. The supernatant was finally centrifuged at 14,000×g for 15 min at 4 °C to separate soluble cytoplasmic proteins (cytosol) and the mitochondrial pellet. Pellets were washed once with phosphate-buffered saline (PBS) and then resuspended and subjected to three freezing and thawing cycles before measuring total protein using the DC protein assay (BioRad, Mississauga, ON, Canada) prior to immunoprecipitation as described below.
Pure mitochondria extracts were obtained using the Q proteome mitochondria isolation kit (Qiagen, Toronto, ON, Canada). Briefly, the cells were washed with PBS and resuspended in lysis buffer, which disrupts the plasma membrane without solubilizing it, thereby releasing cytosolic proteins and organelles. After centrifugation at 1000 × g, the pellet was resuspended in disruption buffer and subjected to homogenization as described previously. The crude mitochondrial fraction was washed three times with PBS and resuspended in mitochondria purification buffer and then layered on top of a gradient consisting of mitochondria purification buffer above disruption buffer and then centrifuged at 14,000×g for 15 min. The pellet was then washed in mitochondria storage buffer as per the manufacturer’s instructions. The pure mitochondrial pellet was resuspended in Tris (10 mM, pH 7.4) supplemented with 0.1 % sodium dodecyl sulfate (SDS). Protein concentration was determined using the DC protein assay as described previously. The pure extracts were processed for proteomics analysis and Western blot analysis (see below).
Proteomics
MNA cells were cultured in T175 flasks at 37 ° Cin 5 % CO2. Confluent cells were either infected with CVS at a MOI of one or mock-infected and incubated for 72 h. Subcellular mitochondria fractionation was performed as described above with the Q proteome kit and subjected to nanoscale liquid chromatography coupled to tandem mass spectrometry (nano-LC-MS/MS) analysis. The mitochondrial pellet was resuspended in 20 μL of 8 M urea dissolved in 10 mM Tris buffer (pH 8.0). The samples were then digested using sequencing grade trypsin (Promega, Madison, WI, USA) (1 μg trypsin/20 μg protein). The digested peptides were desalted using C18 zip tip (Millipore, Billerica, MA, USA), and peptides were eluted from zip tip using 40 % acetonitrile. Peptides were then dried using a speed vacuum and suspended in water containing 0.1 % formic acid. Two microliters of the suspended peptides was used to determine the peptide concentration. Three micrograms of each sample was run on nano-reverse phase liquid chromatography-MS/MS using an A splitless Ultra 2D Plus (Eksigent, Dublin, CA, USA) system coupled to a high-speed Triple TOF™ 5600 mass spectrometer (AB SCIEX, Concord, ON, Canada) in duplicate. Peptides were injected via a PepMap100 trap column (0.3 × 5 mm, 5 μm, 100 Å, Dionex, Sunnyvale, CA, USA) (to capture the peptides and remove interfering substances), and a 100 μm × 150 mm analytical column packed with 5 μm Luna C18 (2) (to separate the peptides) was used prior to MS/MS analysis. Both eluents A (water) and B (98 % acetonitrile) contained 0.1 % formic acid as an ion-pairing modifier. The tryptic digest was analyzed by centrifugation for 170 min in a gradient of eluent B (0–35 % over 150 min, 35–80 % for 1 min, and finally kept at 80 % for 5 min) at a flow rate of 500 nL/min. A Thermo Scientific NanoDrop Spectrophotometer monitored the absorbance of the peptide bound at 205 nm UV wavelength and was used for quantitation measurements of peptides and proteins. Raw files (spectra) were run in ProteinPilot software version 4.2 and searched against rabies virus proteins (National Center for Biotechnology Information and Uniprot).
Western blotting for β-actin, α-tubulin, RABV P, Tim 23 VDAC, and NDUFS2 protein expression
CVS- and mock-infected MNA cells were incubated for 72 h at 37 °C in 5 % CO2 and then lysed using ice-cold neurofilament stabilization buffer containing 0.1 M pipes, 5 mM MgCl2, 5 mM EGTA, 0.5 % Triton X-100, 20 % glycerol, 10 mM NaF, 1 mM PMSF, and protease inhibitor cocktail (Fernyhough et al. 1999). Protein amounts were determined by the DC protein assay. Equivalent amounts of the extracted proteins (20 μg/lane) were loaded onto a 10 % SDS-PAGE gel and electrotransferred onto nitrocellulose membrane at 100 V for 1 h. The membrane was blocked with 5 % skim milk and 0.05 % Tween-20 in PBS for 1 h and then incubated with one of the specific primary antibodies: mouse monoclonal anti-P (M965) (Nadin-Davis et al. 2010) (obtained from C. Fehlner-Gardiner, Canadian Food Inspection Agency, Ottawa, ON, Canada) (1/1000); mouse monoclonal anti-Tim 23 (translocase of the inner membrane, which is an integral mitochondrial membrane protein) that was used as mitochondrial marker (BD Biosciences; Mississauga, ON, Canada) (1/2000), rabbit polyclonal anti-voltage-dependent anion channel (anti-VDAC) (VDAC is located in the outer mitochondrial membrane) that was also used as a loading control for mitochondrial extracts (Abcam, Toronto, ON, Canada) (1/700); goat polyclonal anti-NADH dehydrogenase [ubiquinone] iron-sulfur protein 2 (anti-NDUFS2; Pierce, Rockford, IL, USA) (1/1000) for detection of co-immunoprecipitated Complex I protein (NDUFS2 is a core subunit among about 45 different subunits that make up Complex I); or rabbit polyclonal anti-β-actin (Abcam) (1/2000) or mouse monoclonal anti-α-tubulin (Abcam) (1/1000) for detection of cytosolic proteins. All primary antibodies were incubated overnight in 5 % skim milk containing 0.05 % Tween-20 in PBS. The blots were then washed and incubated with horseradish peroxidase (HRP)-linked anti-rabbit or anti-mouse IgG (Jackson Immunoresearch Laboratories, West Grove, PA, USA) or VeriBlot anti-mouse or anti-goat IgG (Abcam) antibodies for 1 h. The proteins were detected using the Western Blotting Chemiluminescence Luminol Reagent (Santa Cruz Biotechnology) according to the manufacturer’s protocol. Membranes were stripped with Restore TM Western Blot Stripping Buffer (ThermoScientific) and reprobed with a different primary antibody. Quantity one software was used for densitometric scans of the blots. Data are expressed as ratio of protein scan value obtained divided by protein scan value of VDAC, NDUFS2, or β-actin. Two biological replicates were performed.
Immunocytochemistry for RABV P antigen and mitochondria localization
CVS- and mock-infected MNA cells were incubated for 72 h at 37 °C in 5 % CO2. The cells were fixed with MeOH for 5 min and permeabilized with 0.1 % Tween 20 in PBS for 10 min. The slides were blocked for 1 h at room temperature in blocking reagent (Roche, Mannheim, Germany), NCS, and PBS in a ratio of 1:1:3. Cultures were evaluated with fluorescent staining after incubation overnight at 4 °C with primary antibodies including mouse anti-P monoclonal antibody (M1034) (Nadin-Davis et al. 2010) (1/100) (obtained from C. Fehlner-Gardiner, Canadian Food Inspection Agency) and polyclonal rabbit anti-VDAC antibody (Abcam) (1/50). The secondary antibodies used were Alexa Fluor 488 conjugated donkey anti-mouse IgG (Life Technologies, Burlington, ON, Canada) (1/500) and Alexa Fluor 647-conjugated donkey anti-rabbit IgG (Life Technologies) (1/250), which were incubated in blocking buffer for 1 h at room temperature. Coverslips were mounted on slides with Prolong gold antifade reagent. The fluorescent signals were examined using confocal laser scanning microscopy LSM 710 (Carl Zeiss) and an Axio-Observer Z1 microscope.
Recombinant plasmid construction and site-directed mutagenesis
We obtained plasmids from Dr. Zhen Fu (University of Georgia, Athens, GA, USA) encoding four of the RABV proteins with M, G, N, and P genes cloned into the pcDNA3 vector from the CVS-B2c strain (GenBank accession number HQ891318; except M was derived from ERA strain) (Zhang et al. 2013). Transfection of the plasmids was evaluated in the human embryonic cell line, HEK-293T because of high transfection efficiency (e.g., about 90 %). Recombinant plasmids encoding various segments of the P gene of RABVstrain B2c (Fig. 1) were generated using a strategy similar to that used by Blondel et al. (2002). Briefly, the P gene of B2c was amplified by PCR using primers designed to anneal to the 5′ and 3′ ends of the intact P gene (see Table 1 for the specific oligonucleotide sequences); these primers also contained a specific 15 nucleotide sequence 5′ to the P gene sequence required for use with the In-Fusion Ready vector cloning protocol (Clontech, Mountain View, CA, USA). The pAcGFP1-N linearized vector (Clontech) contains ends that are complimentary to the specific 15 nucleotide sequences on both the sense and antisense primers, allowing directional insertion of the PCR product into the pAcGFP1-N vector. The FastStart PCR Master kit (Roche Applied Science) was used for all PCR reactions following the manufacturer’s instructions, using a plasmid encoding the P gene of rabies virus B2c as template. The PCR products were inserted into the linearized pAcGFP1-N, in-frame with the N terminus of the green fluorescent protein (GFP) to yield P-GFP with GFP located at the C terminus of the protein, using the In-Fusion Ready Vector cloning protocol, according to the manufacturer’s instructions. Deletion mutants were generated as follows: PΔN82-GFP, PΔN138-GFP, and PΔN172-GFP differed from P-GFP by 246, 414, and 516-bp deletions at the 5′ terminus of the P gene, respectively. PΔN52ΔC159-GFP differed from the P gene by a 156-bp deletion at the 5′ terminus and a 477-bp deletion at the 3′ terminus; PΔN172ΔC75-GFP differed from the P gene by a 516-bp deletion at the 5′ terminus and a 225-bp deletion at the 3′ terminus; and PΔN138ΔC125-GFP differed from the P gene by a 414-bp deletion at the 5′ terminus and a 372-bp deletion at the 3′ terminus. These deletions were created by PCR amplification of the wild-type B2c P gene using specific oligonucleotide primers designed to amplify the non-deleted regions (Table 1). The amplified PCR products were then inserted into pAcGFP1-N as described above using the In-Fusion Ready vector cloning protocol.

Generation of plasmids encoding the full-length phosphoprotein gene and deletion mutants of the P gene. Plasmids encoding the full-length phosphoprotein gene and deletion mutants lacking portions of the phosphoprotein gene were generated. The RABV P and deletion mutants of P were expressed as fusion proteins with GFP
Plasmid transfection
HEK-293T cells were seeded at a density of 4 × 105 cells per well in a 12-well culture plate. Transfection of HEK-293T cells was performed using lipofectamine 2000 reagent (Invitrogen, Burlington, ON, Canada) according to the manufacturer’s instructions. MNA cells were seeded at a density of 2 × 104 cells per well in a 96-well cell culture plate and transfected using Fugene (Promega, Madison, MI, USA) according to the manufacturer’s instructions (transfection efficiency about 73 %). Transfected cells were incubated for 72 h at 37 °C in 5 % CO2.
Complex I activity
Transfected HEK-293T cells were incubated for 72 h at 37 °C in 5 % CO2 and then harvested in KPi buffer (20 mM, pH7.4). The cells were subjected to 3 cycles of freezing and thawing to measure Complex I activity. This activity was assessed as rotenone-sensitive NADH-cytochrome c reductase activity as described previously (Roy Chowdhury et al. 2010). Addition of rotenone, a specific inhibitor of Complex I, results in blockade of Complex I rotenone sensitive activity, which is determined by subtraction of Complex I kinetic curve slopes before and after addition of rotenone. The kinetic reaction was performed in a 1000-μL cuvette with a reaction buffer containing 20 mM potassium phosphate buffer (pH 7.4), 100 μM NADH, 1 mM KCN (Sigma-Aldrich), and 20 μg of sample. The mixture was preincubated for 3 min, followed by the addition of 100 μM oxidized cytochrome c (Sigma-Aldrich). The reaction was followed at 550 nm for 4 min and subsequently for another 4 min after addition of 25 μM rotenone.
Immunoprecipitation of Complex I
Complex I was immunoprecipitated using 10 μg of anti-Complex I immunocapture antibody (Abcam). Nonspecific binding of the antibody was evaluated using 10 μg of control IgG (mouse IgG2b kappa monoclonal) (Abcam). Briefly, mitochondria pellets from CVS- and mock-infected MNA cells were resuspended in PBS. A 20-μL aliquot of the suspension was subjected to three freezing and thawing cycles, and the protein content was measured as described previously. A total of 250 μg of mitochondria suspension in a volume of 180 μL was treated with 20 μL of 10 % n-dodecyl β-d-maltoside (Sigma) and supplemented with a protease/phosphatase cocktail inhibitor (ZmTechScientific, Montreal, QC, Canada). The extracts were centrifuged at 14,000×g for 30 min. The supernatants were incubated with 10 μL solid beads (Pierce, Rockford, IL, USA) overnight with mixing. The beads were collected at 500×g for 10 s and washed three times with PBS supplemented with lauryl maltoside 0.05 % (w/v). Complex I was eluted with 1 % SDS, and the supernatant was mixed with 5× loading buffer. Electrophoresis and Western blotting were performed as described previously to detect P, VDAC, and NDUFS2.
Assessment of hydrogen peroxide production
ROS generation was assessed in MNA cells using the Amplex Red Hydrogen Peroxide/Peroxidase kit (Invitrogen, Burlington, ON, USA), a one-step assay that detects hydrogen peroxide (H2O2) or peroxidase activity. Cells were seeded overnight as indicated previously at 2 × 104 cells per well in a 96-well plate and were transfected the following day with each plasmid construct. The transfected cells were incubated for 72 h at 37 °C in 5 % CO2. The medium was then removed and 50 μL of reaction buffer with 10 μM digitonin was added in the presence or absence of 10 mM succinate. Digitonin was added in order to permeabilize plasma membranes of the cells to permit access of substrates to mitochondrial membranes. The reaction was started by the addition of 50 μL of working solution containing 100 μM Amplex Red reagent and 0.2 U/mL HRP and incubated at 37 °C, and the absorbance was measured at 560 nm at 2 h using a microplate reader. The kinetics over 2 h had been previously documented in MNA cells (Alandijany et al. 2013).
Statistical analysis
Data were analyzed using GraphPad Prism version 4 software. Standard two-tailed unpaired Student’s t tests with Welch’s correction, which did not assume equal variances, were used for evaluating the significance of the difference between the means on assays comparing transfected or mock- and CVS-infected samples. A value of p < 0.05 was considered statistically significant.
Results
Identification of RABV P as the most abundant RABV protein in CVS-infected and enriched mitochondria extracts
We used a proteomic approach in order to make an assessment of the presence of CVS proteins in purified mitochondria. Using nano-LC-MS/MS, we identified peptides belonging to P, G, and N with 25.9 % protein coverage at 95 % confidence for RABV P versus 5.7 % for the RABV nucleocapsid protein (N) and 4.4 % for the RABV glycoprotein (G) (Table 2). We found mitochondria most highly enriched with the RABV P and less enriched with the two larger viral proteins, N and G. The enrichment for P in the mitochondrial extracts is even more significant in light of the smaller size of P (297 aa) in comparison with N (450 aa) and G (524 aa). We did not detect the RABV matrix (M) protein in the mitochondrial extracts. The pattern of the tryptic digest of the RABV P indicated that tryptic peptides were identified throughout a large portion of the P sequence (data not shown). Hence, the presence of P in mitochondria indicates that it could potentially interact with one or more mitochondrial components, resulting in mitochondrial dysfunction and increased activity of mitochondrial Complex I, which we have previously reported (Alandijany et al. 2013).
CVS infection induced RABV P expression in mitochondrial extracts of infected MNA cells
In order to confirm the expression of P in mitochondrial extracts of CVS-infected MNA cells, we performed Western blots and probed the extracts with an anti-P monoclonal antibody (Fig. 2). We also evaluated the expression of P relative to Tim 23 and VDAC as indicators of mitochondria loading by probing the Western blots with anti-Tim 23 and anti-VDAC antibodies. The expression of P in CVS- versus mock-infected mitochondrial extracts probed with anti-P antibody showed that there was induction of expression of P in CVS-infected cytosolic extracts and a smaller induction of P expression in CVS-infected mitochondrial extracts (Fig. 2). The purity of the mitochondrial extracts was evaluated by probing the extracts with markers specific for mitochondria (Tim 23 and VDAC) and others specific for the cytosol (β-actin and α-tubulin), in the CVS- and mock-infected cultures at 72 h p.i. (Fig. 2). In the mitochondrial fractions, evaluation of the expression of VDAC in CVS- and mock-infected MNA cells by Western blots showed markedly higher levels of expression of Tim 23 and VDAC in both the CVS- and mock-infected mitochondrial samples and, similarly, markedly higher levels of expression of β-actin and α-tubulin in the cytosolic fractions of CVS- and mock-infected cells (Fig. 2). These assays confirmed purity of the mitochondrial extractions.

Western immunoblots of markers for mitochondrial and cytosolic subfractions. Purified mitochondrial extracts were assessed for the expression of RABV P with p965 antibody in CVS- and mock-infected MNA cultures 72 h p.i. In CVS infection, P was expressed in the cytosolic fraction and to a much lesser extent in the mitochondrial fraction. Representative Western blots of markers of mitochondrial (Tim 23 and VDAC) and cytosolic (β-actin and α-tubulin) subfractions were evaluated to assess the purity of mitochondrial extracts from CVS- and mock-infected cultures 72 h p.i. High expression of the mitochondrial markers Tim 23 and VDAC (and undetectable signals for β-actin and α-tubulin) was observed in the mitochondrial fractions, whereas high expression of the cytosolic markers β-actin and α-tubulin (and undetectable signals for Tim 23 and VDAC) was observed in the cytosolic fractions
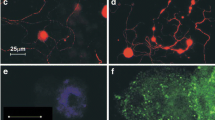
RABV P co-localized with mitochondria in MNA cells
We have shown that P was expressed in CVS-infected MNA mitochondrial extracts 72 h p.i. by proteomics and Western blot analysis. We also evaluated co-localization of the expression of RABV P and mitochondrial VDAC in cells by immunofluorescent staining. Staining of the nucleus with DAPI (Fig. 3a), RABV P (Fig. 3b), and VDAC (Fig. 3c) are shown in CVS-infected MNA cells 72 h p.i. CVS-infected MNA cells showed strong staining for P in the cytoplasm and also in mitochondria (Fig. 3b). The merged images showed co-localization of signals for RABV P and mitochondria (Fig. 3d). Background staining of mock-infected cells for RABV P was low with omission of primary antibodies on CVS-infected cells (data not shown).

Colocalization of the RABV P and mitochondrial markers in infected cells. CVS-infected MNA cells at 72 h p.i. were stained for nuclei with DAPI (a) and immunostained for RABV P with anti-P monoclonal antibody M1034 (green) (b) and for mitochondrial marker VDAC with an anti-VDAC antibody (red) (c). The merged image shows colocalization of RABV P staining in mitochondria at higher (yellow) and lower (orange) levels (d)
A plasmid encoded with the RABV P increased Complex I activity
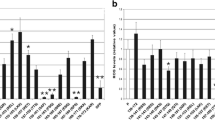
We have previously reported alterations in mitochondria parameters and, in particular, the activity of respiratory chain complexes mainly affecting Complexes I and IV (Alandijany et al. 2013). Complex I is an important site of ROS production and was found to better correlate with the susceptibility to infection of cell types. We next wanted to determine if Complex I activity is directly affected by expression of the P. Therefore, we measured Complex I activity after transfection of plasmids without any insert (empty pcDNA3 vector) and with plasmids expressing each of four rabies virus proteins, N, G, P, and M, in the human embryonic line HEK-293T (Fig. 4). Complex I activity was increased significantly after transfection of the plasmid encoding the P, but not with transfection of the plasmids encoding G or N, whereas M protein significantly reduced Complex I activity. Hence, the P was the only rabies virus protein that we found that induced increased Complex I activity.

Complex I activity after transfection with plasmids expressing the RABV proteins. We evaluated the effects of the expression of viral proteins on the activity of Complex I after transfecting HEK-293T cells with empty plasmid (pcDNA3) or plasmids expressing M, G, N, and P 72 h after transfection. Only P significantly increased the activity of Complex I (194.5 % ± 12.6, *p = 0.023) unlike G (99.0 % ± 8.5, p = 0.952, ns) and N (131.1 % ± 4.9, p = 0.0623, ns), whereas the M protein significantly reduced the activity of Complex I (46.6 % ± 2.2, *p = 0.0048). Data are representative of three independent experiments with triplicate samples expressed as mean ± SEM, and significance is indicated by single asterisk (p < 0.05)
RABV Pco-immunoprecipitated with Complex I
We have shown that the P induces increased activity of Complex I and also co-localizes in the mitochondria. The next step was to determine whether P produces its effects through a direct physical interaction with Complex I. Therefore, we purified Complex I from mitochondrial extracts from CVS-infected MNA cells by immunoprecipitation and assessed for the presence of P in the immunocomplex. In order to verify that the immunoprecipitation of Complex I was efficient, we probed for NDUFS2 (core subunit of the mitochondrial membrane respiratory chain complex I) (Fig. 5a). NDUFS2 was present in CVS- as well as in mock-infected samples after immunoprecipitation with anti-Complex I antibody, but not with an IgG control. After normalization of scan values of mitochondrial protein expression with VDAC, NDUFS2 showed no significant difference in expression in CVS- vs. mock-infected samples (Fig. 5b). However, P was detected in the purified mitochondrial extracts and co-immunoprecipitated with Complex I in CVS-infected samples, but was not found in mock-infected samples (Fig. 5a, d). On the contrary, P was not immunoprecipitated with the IgG control in CVS- and mock-infected samples (Fig. 5a, d), confirming the specificity of the anti-Complex I antibody.

Detection of the RABV P after immunoprecipitation of Complex I from mitochondrial extracts. CVS- and mock-infected MNA cells were cultured for 72 h p.i. and then subjected to mitochondrial subfractionation. Extracts were treated with specific antibody to Complex I or antibody to IgG 2b kappa as a nonspecific control. Complex I was immunoprecipitated uniquely from extracts treated with antibody specific for Complex I (a). Western blots were performed on purified mitochondria and probed for VDAC and, after immunoprecipitation of Complex I, were first probed for RABV P and then stripped and reprobed for NDUFS2. Scan values of duplicate samples indicated that there was an absence of NDUFS2 protein in mitochondrial lysates from CVS- and mock-infected cells treated with the nonspecific control antibody, whereas NDUFS2 was present in CVS- and mock-infected extracts treated with anti-Complex I antibody (2.59-fold ± 0.56 vs. 1.71-fold ± 0.05 versus input VDAC, respectively, p = 0.365) (b). The same extracts were probed for P and showed that P was immunoprecipitated in CVS-infected extracts treated with the specific antibody and not in mock-infected cells treated with either the specific antibody or the nonspecific antibody control with normalization to input VDAC (c) and NDUFS2 (d). Data are representative of three independent experiments expressed as mean ± SEM
P-GFP and deletion mutants of P-GFP containing sequence 139–172 of RABV P increased activity of Complex I
Plasmids encoding the RABV P gene and deletion mutants of the RABV P gene were generated. These plasmids expressed P or deletion mutants of P as fusion proteins linked to GFP. Complex I activity was measured after transfection of these plasmids into HEK-293T cells (Fig. 6). Complex I activity was significantly increased 72 h after transfection of P. In comparison to the activity of Complex I obtained when full-length P was expressed in cells, the activity of Complex I was reduced significantly when deletion mutants of P that did not contain the amino sequence from 139 to 172 (P∆N172-GFP, P∆N172∆C75-GFP, P∆N52∆C159-GFP, and GFP), were expressed, whereas deletion mutants containing the 139–172 region (P-GFP, P∆N82-GFP, P∆N138-GFP, and P∆N138∆C125) all increased Complex I activity. We conclude that the 139–172 region is important for the interaction with Complex I resulting in its increased activity. Fluorescence-activated cell sorting (FACS) analysis confirmed that reduction or loss of activity of Complex I observed for some constructs was not due to inefficiency in transfection (data not shown).

Complex I activity after transfection with plasmids containing deletions in the RABV P gene. Plasmids expressing the full-length P and a variety of P deletion mutants fused to GFP were transfected into HEK-293T cells in order to determine the effects on the activity of Complex I. Transfection of cells with the plasmid encoding the full-length P induced Complex I activity by 7.9-fold vs. expression of GFP alone (empty vector). In comparison with P-GFP, there was a significant reduction of Complex I activity after transfection of HEK-293T cells with plasmids expressing P∆N52∆C159 (48.8 % ± 11.1, *p = 0.018), P∆N172 (47.8 % ± 10.3, *p = 0.015), P∆N172 P∆C75 (19.8 % ± 0.4, **p = 0.001), and (for comparison purposes) GFP alone (12.6 %, ±0.04 p = 0.002). In comparison to cells transfected with the plasmid expressing full-length P, no significant difference in activity was observed in extracts from cells transfected with plasmids expressing P∆N82 (95.9 % ± 15.3, p = 0.826), P∆N138 (87.3 ± 18.3, p = 0.256), and P∆138F∆C125 (90 % ± 13, p = 0.527). Hence, the region associated with increased Complex I activity can be localized to amino acids sequence 139–172. Data are representative of three independent experiments with triplicate samples expressed as mean ± SEM and significance is indicated by single asterisk (p < 0.05) and double asterisk (p < 0.01)
RABV P increased ROS production
Increased activity of Complex I induced by P likely plays a central role in ROS production resulting in oxidative stress (Alandijany et al. 2013; Jackson et al. 2010). In order to confirm that the increased activity of Complex I was associated with ROS generation, we evaluated the production of ROS in MNA cells transfected with plasmids expressing P as well as N, M, and G (Fig. 7). In comparison to cells transfected with the empty vector, pcDNA3, cells expressing P induced a significant increase in ROS generation, but cells expressing N, M, and G did not, providing further evidence that the P plays a central role in the induction of mitochondrial dysfunction in RABV infection.

Generation of ROS after transfection with a plasmid expressing full-length RABV P, but not plasmids expressing other RABV proteins. ROS levels were measured in MNA cells in the presence of succinate (10 mM) after transfection with plasmids. Optical density is shown at 120 min and significance level is indicated by single asterisk (p < 0.05). In comparison to pcDNA3 (21.8 % ± 10.9), there were a significant increases of ROS by P (100 % ± 28.1, *p = 0.033), but not by M (33.6 % ± 9.0, p = 0.492), N (31.1 % ± 7.4), p = 0.505), or G (11.3 % ± 4.0, p = 0.298). Data are representative of two independent experiments with three replicates
P-GFP and deletion mutants containing amino acid sequence 139–172 induced ROS production
We have also observed that the P and deletion mutants containing the amino acid sequence from 139 to 172 induced an increase in Complex I activity. Thus, we also determined if this activation correlated with an increased amount of ROS generation. In comparison to the full-length P-GFP, only deletion mutants containing the amino acid sequence from 139 to 172 induced a significant increase in ROS generation (P-GFP, P∆N82-GFP, P∆N138-GFP, and P∆N138∆C125), whereas deletion mutants without this region (P∆N172-GFP, P∆N172∆C75-GFP, P∆N52∆C159-GFP, and GFP alone) did not (Fig. 8).

Generation of ROS after transfection with plasmids expressing full-length P and deletion mutants of P. Plasmids encoding deletion mutants of P were transfected into MNA cells in order to measure ROS production. In comparison to cells transfected with plasmid expressing full-length P, no significant difference in activity was observed in extracts from cells transfected with plasmids expressing P∆N82 (93.5 % ± 5.8, p = 0.519), P∆N138 (124.8 % ± 7.8, p = 0.077), and P∆138∆C125 (117.9 % ± 23.0, p = 0.451). ROS production was reduced significantly after transfection of plasmids expressing deletion mutants of P without the amino acid sequence 139–172: P∆N52∆C159 (51.4 % ± 8.8, **p = 0.003), P∆N172 (47.1 % ± 13.2, **p = 0.0055), P∆N172∆C75 (49.5 % ± 4.5, **p = 0.009), and, for comparison purposes, GFP alone (47.4 %, ±3.6, **p = 0.004). Data are representative of three independent experiments expressed as mean ± SEM and significance is indicated by double asterisk (p < 0.01)
Discussion
The basic mechanisms by which rabies virus infection produce neurological disease in an animal host are not yet well understood (Jackson and Fu 2013). CVS infection in mice inoculated experimentally by the peripheral route was shown to induce acute degeneration of neuronal processes that explained the severe and fatal encephalomyelitis (Scott et al. 2008). Further studies in CVS-infected cultured rodent DRG neurons have shown evidence of oxidative stress resulting in axonal degeneration and impaired axonal growth (Jackson et al. 2010). Investigations in different CVS-infected cell types have shown mitochondrial dysfunction that is associated with increased activity of mitochondrial Complex I and increased generation of ROS, which induce neuronal injury (Alandijany et al. 2013). Although we also found increased activity of Complex IV, increases in Complex I activity, but not in Complex IV activity, correlated with susceptibility of different cell types to CVS infection (Alandijany et al. 2013). Hence, we concluded that increased Complex I activity was of greater importance. How does rabies virus infection induce mitochondrial dysfunction? We hypothesized that one or more rabies virus proteins induce mitochondrial dysfunction by interacting with and altering the activity or function of one or more mitochondrial components leading to oxidative stress. We initially took a proteomic approach in order to determine the rabies virus proteins that could be detected in purified mitochondrial extracts and found the greatest enrichment for the RABV P. Western blots of mitochondria extracts (Fig. 2) and confocal microscopy studies (Fig. 3) also strongly supported the presence of P in mitochondria. These findings raised the possibility that the P, with or without other RABV proteins, is responsible for interacting with one or more mitochondrial components, including Complex I, and in light of our previous findings (Alandijany et al. 2013), resulting in mitochondrial dysfunction. After cells were transfected with plasmids expressing N, M, P, and G, we found that only the P resulted in a significant increase in Complex I activity and increased generation of ROS.
We investigated for evidence of a physical interaction between the RABV P and Complex I, which could result in increased Complex I activity. After immunoprecipitation of Complex I in mitochondrial extracts, we detected abundant P in mitochondrial subfractions providing confirmatory evidence that P directly interacts with Complex I and that this interaction results in an increase in its activity. The signal transducer and activator of transcription 3 (STAT3) protein has been recognized as an important binding partner for RABV P (Lieu et al. 2013), and STAT3 is capable of modulating mitochondrial function depending on its phosphorylation status (Szczepanek et al. 2012; Wegrzyn et al. 2009). Furthermore, deficiency of the STAT3 protein has been shown to decrease Complex I activity (Szczepanek et al. 2012). Although we have demonstrated a direct physical interaction between the RABV P and Complex I, it is unknown if one or more intermediate proteins (e.g., STAT3) may also play a role in Complex I activation.
We have evaluated which part of the P sequence may be responsible for the Complex I interaction resulting in its increased activity. Evaluation of a variety of deletion mutants lacking the termini of P indicated that the region of the amino acid sequence from 139 to 172 was necessary for inducing increased activity of Complex I and also for ROS generation. We do not know whether amino acid residues in this region are important for mitochondrial localization. Both direct and indirect interactions may result in increased activity of Complex I and generation of ROS. This 34-amino-acid region can now be studied in greater detail in order to better define the molecular interactions with Complex I. This would be best approached by evaluating the effects of amino acid mutations in future studies. Major domains of P are shown in Fig. 9, including dimerization (Gerard et al. 2009), the dynein light chain (LC8) (Raux et al. 2000) and STAT (Lieu et al. 2013; Vidy et al. 2005) association sites, and domains that bind to the polymerase (L) (Chenik et al. 1998) and nucleoprotein (N) (Chenik et al. 1994). The 139–172-amino-acid region closely corresponds to the LC8-binding region (138–172). However, we do not know the precise region responsible for the interaction with Complex I, and the involved region may be completely incidental to the known RABV P–dynein interaction.

Sites of important domains of the RABV P. The location of the RABV P domains responsible for dimerization (Dimer) (Gerard et al. 2009), association with the cytoplasmic dynein light chain (LC8) (Raux et al. 2000), and with STAT1, STAT2, and STAT3 (Lieu et al. 2013; Vidy et al. 2005) are shown. The P domains that bind to the polymerase (L) (Chenik et al. 1998) and nucleoprotein (N) (Chenik et al. 1994) are also shown. Numbers refer to amino acid positions within the full-length RABV P
Viral infections are well recognized to interfere with mitochondrial function (Anand and Tikoo 2013; Boya et al. 2004, 2003; Claus and Liebert 2014; Li et al. 2004, 2007; Lichty et al. 2006). This is probably best characterized in hepatitis C virus infection (Li et al. 2007; Quarato et al. 2013; Wang and Weinman 2013). There are numerous examples of reduced activity of mitochondrial complex activities in viral infections (Claus and Liebert 2014), whereas increased activities of complexes have been reported much less frequently, including in both vaccinia virus (Chang et al. 2009) and rubella virus (Claus and Liebert 2014) infections. Another lyssavirus, Mokola virus, is less pathogenic than genotype 1 lyssaviruses that include wild-type (street) and laboratory (e.g., CVS) strains of RABV. Reduced activity of the mitochondrial electron transport system has been reported in Mokola virus infection (Gholami et al. 2008). The Mokola virus matrix protein interacts with subunit I of the cytochrome c oxidase (Complex IV) of the mitochondrial respiratory chain. The mechanism of transport of the matrix protein into mitochondria is uncertain, but it has been speculated that an α-helix in the matrix protein at position 69–82 may act as a mitochondrial import signal and also that heat shock protein-70, which RABV incorporates (Sagara and Kawai 1992), may act as a cytoplasmic chaperone to help maintain the mitochondrial protein in an import-competent state (Gholami et al. 2008; Stojanovski et al. 2003; Young et al. 2003). Furthermore, a 20-amino-acid fragment (from positions 67–86) of the matrix protein inhibited cytochrome c oxidase activity and directed mutagenesis demonstrated that position 77 affected cytochrome c oxidase activity (Larrous et al. 2010). Hence, at least two lyssaviruses in two different genotypes induce mitochondrial dysfunction via different viral proteins and mechanisms.
The therapeutic approach to mitochondrial disorders and, more specifically, Complex I disorders still remains in its infancy. However, because these disorders have been recognized to be fairly common (Papa and De 2013), future advances in therapy of other mitochondrial disorders may also prove to be applicable to rabies virus infection. For example, Parkinson’s disease is thought to be due to reduced activity of Complex I resulting in mitochondrial dysfunction (Schapira and Tolosa 2010). Therapy in an animal model of Parkinson’s disease involving delivery of a truncated form of a 2.7-kb (β2.7) RNA transcript expressed during human cytomegalovirus infection (Reeves et al. 2007), designated p137, functions to prevent mitochondrial-induced cell death and has been shown to have therapeutic efficacy (Kuan et al. 2012). Specific targeted therapeutic approaches may become available after we have a more complete understanding of exactly how rabies virus infection induces mitochondrial dysfunction and oxidative stress. It is not yet known if natural rabies caused by street (wild-type) rabies virus strains in rabies vectors and disease in dead-end hosts (e.g., humans) involve the same pathogenetic mechanisms as in the in vivo mouse model with CVS infection and in CVS infection of cultured neurons. Future studies will be needed to determine whether rabies virus infections caused by street virus strains result in the same mechanisms for neuronal dysfunction and injury and whether the rabies virus proteins induce the same or different patterns of dysfunction. The reports of mitochondrial dysfunction in Mokola virus infection suggests the central importance of mitochondrial involvement in lyssavirus infections, but there may be considerable variations in the exact mechanisms by which lyssavirus proteins interact with mitochondrial complexes or other mitochondrial components and cause injury to infected neurons. More research is needed in order to improve our understanding of the basic mechanisms by which rabies virus infection induces disease in its hosts.
References
Alandijany T, Kammouni W, Roy Chowdhury SK, Fernyhough P, Jackson AC (2013) Mitochondrial dysfunction in rabies virus infection of neurons. J Neurovirol 19:537–549
Anand SK, Tikoo SK (2013) Viruses as modulators of mitochondrial functions. Adv Virol 2013:738794
Blondel D, Regad T, Poisson N, Pavie B, Harper F, Pandolfi PP, De The H, Chelbi-Alix MK (2002) Rabies virus P and small P products interact directly with PML and reorganize PML nuclear bodies. Oncogene 21:7957–7970
Boya P, Roumier T, Andreau K, Gonzalez-Polo RA, Zamzami N, Castedo M, Kroemer G (2003) Mitochondrion-targeted apoptosis regulators of viral origin. Biochem Biophys Res Commun 304:575–581
Boya P, Pauleau AL, Poncet D, Gonzalez-Polo RA, Zamzami N, Kroemer G (2004) Viral proteins targeting mitochondria: controlling cell death. Biochim Biophys Acta 1659:178–189
Chang CW, Li HC, Hsu CF, Chang CY, Lo SY (2009) Increased ATP generation in the host cell is required for efficient vaccinia virus production. J Biomed Sci 16:80
Chenik M, Chebli K, Gaudin Y, Blondel D (1994) In vivo interaction of rabies virus phosphoprotein (P) and nucleoprotein (N): existence of two N-binding sites on P protein. J Gen Virol 75:2889–2896
Chenik M, Schnell M, Conzelmann KK, Blondel D (1998) Mapping the interacting domains between the rabies virus polymerase and phosphoprotein. J Virol 72:1925–1930
Claus C, Liebert UG (2014) A renewed focus on the interplay between viruses and mitochondrial metabolism. Arch Virol 159:1267–1277
Fernyhough P, Gallagher A, Averill SA, Priestley JV, Hounsom L, Patel J, Tomlinson DR (1999) Aberrant neurofilament phosphorylation in sensory neurons of rats with diabetic neuropathy. Diabetes 48:881–889
Gerard FC, Ribeiro EA Jr, Leyrat C, Ivanov I, Blondel D, Longhi S, Ruigrok RW, Jamin M (2009) Modular organization of rabies virus phosphoprotein. J Mol Biol 388:978–996
Gholami A, Kassis R, Real E, Delmas O, Guadagnini S, Larrous F, Obach D, Prevost MC, Jacob Y, Bourhy H (2008) Mitochondrial dysfunction in lyssavirus-induced apoptosis. J Virol 82:4774–4784
Jackson AC (2013a) Current and future approaches to the therapy of human rabies. Antiviral Res 99:61–67
Jackson AC (ed) (2013b) Rabies: scientific basis of the disease and its management, 3rd edn. Elsevier Academic Press, Oxford
Jackson AC (2013c) Therapy of human rabies. In: Jackson AC (ed) Rabies: scientific basis of the disease and its management, 3rd edn. Elsevier Academic Press, Oxford, pp 573–587
Jackson AC, Fu ZF (2013) Pathogenesis. In: Jackson AC (ed) Rabies: scientific basis of the disease and its management, 3rd edn. Elsevier Academic Press, Oxford, pp 299–349
Jackson AC, Kammouni W, Zherebitskaya E, Fernyhough P (2010) Role of oxidative stress in rabies virus infection of adult mouse dorsal root ganglion neurons. J Virol 84:4697–4705
Kuan WL, Poole E, Fletcher M, Karniely S, Tyers P, Wills M, Barker RA, Sinclair JH (2012) A novel neuroprotective therapy for Parkinson’s disease using a viral noncoding RNA that protects mitochondrial complex I activity. J Exp Med 209:1–10
Larrous F, Gholami A, Mouhamad S, Estaquier J, Bourhy H (2010) Two overlapping domains of lyssavirus matrix protein acting on different cell death pathways. J Virol 84:9897–9906
Li D, Wang XZ, Yu JP, Chen ZX, Huang YH, Tao QM (2004) Cytochrome C oxidase III interacts with hepatitis B virus X protein in vivo by yeast two-hybrid system. World J Gastroenterol 10:2805–2808
Li Y, Boehning DF, Qian T, Popov VL, Weinman SA (2007) Hepatitis C virus core protein increases mitochondrial ROS production by stimulation of Ca2+ uniporter activity. FASEB J 21:2474–2485
Lichty BD, McBride H, Hanson S, Bell JC (2006) Matrix protein of vesicular stomatitis virus harbours a cryptic mitochondrial-targeting motif. J Gen Virol 87:3379–3384
Lieu KG, Brice A, Wiltzer L, Hirst B, Jans DA, Blondel D, Moseley GW (2013) The rabies virus interferon antagonist P protein interacts with activated STAT3 and inhibits Gp130 receptor signaling. J Virol 87:8261–8265
Lin MT, Beal MF (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443:787–795
Nadin-Davis SA, Fehlner-Gardiner C, Sheen M, Wandeler AI (2010) Characterization of a panel of anti-phosphoprotein monoclonal antibodies generated against the raccoon strain of rabies virus. Virus Res 152:126–136
Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M (2000) Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 404:787–790
Obrosova IG, Van HC, Fathallah L, Cao XC, Greene DA, Stevens MJ (2002) An aldose reductase inhibitor reverses early diabetes-induced changes in peripheral nerve function, metabolism, and antioxidative defense. FASEB J 16:123–125
Obrosova IG, Pacher P, Szabo C, Zsengeller Z, Hirooka H, Stevens MJ, Yorek MA (2005) Aldose reductase inhibition counteracts oxidative–nitrosative stress and poly(ADP-ribose) polymerase activation in tissue sites for diabetes complications. Diabetes 54:234–242
Papa S, De RD (2013) Complex I deficiencies in neurological disorders. Trends Mol Med 19:61–69
Quarato G, Scrima R, Agriesti F, Moradpour D, Capitanio N, Piccoli C (2013) Targeting mitochondria in the infection strategy of the hepatitis C virus. Int J Biochem Cell Biol 45:156–166
Raux H, Flamand A, Blondel D (2000) Interaction of the rabies virus P protein with the LC8 dynein light chain. J Virol 74:10212–10216
Reeves MB, Davies AA, McSharry BP, Wilkinson GW, Sinclair JH (2007) Complex I binding by a virally encoded RNA regulates mitochondria-induced cell death. Science 316:1345–1348
Reynolds A, Laurie C, Mosley RL, Gendelman HE (2007) Oxidative stress and the pathogenesis of neurodegenerative disorders. Int Rev Neurobiol 82:297–325
Roy Chowdhury SK, Zherebitskaya E, Smith DR, Akude E, Chattopadhyay S, Jolivalt CG, Calcutt NA, Fernyhough P (2010) Mitochondrial respiratory chain dysfunction in dorsal root ganglia of streptozotocin-induced diabetic rats and its correction by insulin treatment. Diabetes 59:1082–1091
Russell JW, Golovoy D, Vincent AM, Mahendru P, Olzmann JA, Mentzer A, Feldman EL (2002) High glucose-induced oxidative stress and mitochondrial dysfunction in neurons. FASEB J 16:1738–1748
Sagara J, Kawai A (1992) Identification of heat shock protein 70 in the rabies virion. Virology 190:845–848
Schapira AH, Tolosa E (2010) Molecular and clinical prodrome of Parkinson disease: implications for treatment. Nat Rev Neurol 6:309–317
Scott CA, Rossiter JP, Andrew RD, Jackson AC (2008) Structural abnormalities in neurons are sufficient to explain the clinical disease and fatal outcome in experimental rabies in yellow fluorescent protein-expressing transgenic mice. J Virol 82:513–521
Stojanovski D, Johnston AJ, Streimann I, Hoogenraad NJ, Ryan MT (2003) Import of nuclear-encoded proteins into mitochondria. Exp Physiol 88:57–64
Szczepanek K, Chen Q, Larner AC, Lesnefsky EJ (2012) Cytoprotection by the modulation of mitochondrial electron transport chain: the emerging role of mitochondrial STAT3. Mitochondrion 12:180–189
Vidy A, Chelbi-Alix M, Blondel D (2005) Rabies virus P protein interacts with STAT1 and inhibits interferon signal transduction pathways. J Virol 79:14411–14420
Wang T, Weinman SA (2013) Interactions between hepatitis C virus and mitochondria: impact on pathogenesis and innate immunity. Curr Pathobiol Rep 1:179–187
Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, Derecka M, Szczepanek K, Szelag M, Gornicka A, Moh A, Moghaddas S, Chen Q, Bobbili S, Cichy J, Dulak J, Baker DP, Wolfman A, Stuehr D, Hassan MO, Fu XY, Avadhani N, Drake JI, Fawcett P, Lesnefsky EJ, Larner AC (2009) Function of mitochondrial Stat3 in cellular respiration. Science 323:793–797
Young JC, Hoogenraad NJ, Hartl FU (2003) Molecular chaperones Hsp90 and Hsp70 deliver preproteins to the mitochondrial import receptor Tom70. Cell 112:41–50
Zhang G, Wang H, Mahmood F, Fu ZF (2013) Rabies virus glycoprotein is an important determinant for the induction of innate immune responses and the pathogenic mechanisms. Vet Microbiol 162:601–613
Acknowledgments
We thank Peyman Ezzati and Dr. John Wilkins (Manitoba Centre for Proteomics and Systems Biology, Winnipeg, Manitoba, Canada) for their assistance with the proteomics studies. We thank Dr. Zhen Fu (University of Georgia, Athens, GA, USA) for the plasmids expressing the rabies virus proteins and Drs. Ludger Klewes and Sabine Mai (Genomic Centre for Cancer Research and Diagnosis, Manitoba Institute of Cell Biology, Winnipeg, MB, Canada) for their assistance with colocalization studies using confocal microscopy.
Funding statement
This work was supported by Canadian Institutes of Health Research/Manitoba Regional Partnership Program with the Manitoba Health Research Council (to A.C. Jackson and P. Fernyhough).
Conflict of interest
The authors declare they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kammouni, W., Wood, H., Saleh, A. et al. Rabies virus phosphoprotein interacts with mitochondrial Complex I and induces mitochondrial dysfunction and oxidative stress. J. Neurovirol. 21, 370–382 (2015). https://doi.org/10.1007/s13365-015-0320-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13365-015-0320-8