
Abstract
Soybean (Glycine max L.) is an annual, self-pollinated species, whose genetic base in Brazil is the result of several cycles of selection and effective recombination among a relatively small number of genotypes selected from the USA cultivars. This frequent selection, admixed population, and the crossing of a small number of cultivars can lead to increase the genetic relationship and affect the patterns of population structure. These factors affect the patterns of linkage disequilibrium (LD) blocks, which can be an effective approach for the screening of target loci for agricultural traits in cultivars of tropical soybean. The objectives of this research were to analyze LD blocks, estimate population structure and relatedness through of genotyping of 169 cultivars of tropical soybean by using a BARCSoySNP6K of Illumina iScan platform. The genotyping revealed a high genetic relatedness and population structure among the cultivars of soybean in Brazil, suggesting the existence of a shared genetic base. Our results provide a help to understand the distribution of genetic variation contained within the Brazilian soybean cultivar collection. We showed that the extensive use of a small number of elite genotypes in Brazilian breeding program further reduced the genetic variability, generate extensive LD and probably increase the haplotype block size. These results are in agreement with results of USDA soybean collection, mainly with accessions of north American, when compared with wild and landraces accessions. We constructed a small haplotype block maps (941 blocks), which may be useful for association studies aimed at the identification of genes controlling traits of economic importance in soybean.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Soybean (Glycine max L.) is an annual, self-pollinated species with a genome size of 1115 Mpb (Schmutz et al. 2010). The specie is believed to have originated from wild soybean Glycine soja, considering that both have 20 chromosomes (2n = 40), hybridize easily, exhibit normal meiotic chromosome pairing, and generate viable fertile hybrids (Kim et al. 2010). The exact region of origin of soybean is still unknown, but southern China, the Yellow River valley of central China, northeastern China, and several other regions are all candidate sources because G. soja grows naturally in far eastern Russia, China, Korea and Japan (Carter et al. 2004).
Glycine max is generally considered to have been domesticated from its wild relative (G. soja) 6000–9000 years ago in China (Carter et al. 2004) and may have been introduced to Korea, and then to Japan approximately 2000 years ago, to North America in 1765, and to Central and South America during the first half of the last century. In this process of domestication and selection, a severe genetic bottleneck during soybean domestication was also found in several independent analyses (Xu et al. 2002; Hyten et al. 2006). There is supporting evidence for both single and multiple domestication events (Hymowitz and Kaizuma 1981; Gai et al. 2000; Xu and Gai 2003), which has been accompanied by a reduction in genetic diversity, as well as loss of useful traits reserved in wild relatives. This reduction of genetic diversity is common in crops have been subjected to strong selective pressure directed at genes controlling traits of agronomic importance during their domestication and subsequent episodes of selective breeding (i.e.: Maize-Vigouroux et al. 2002).
The largest resource of soybean germplasm is the Asian landraces of G. max that are the most immediate result of domestication (Hyten et al. 2007). Selection, hybridization and breeding from these landraces have resulted in the release of improved cultivars in north American-USA (Gizlice et al. 1994). These first cultivars developed in USA were introduced and planted in Brazil during the 1960s and 1970s. With the growing importance of soybean, breeders began crossing these cultivars among themselves and with other sources, obtaining the first Brazilian cultivars, such as Industrial, Santa Rosa and Campos Gerais (Hiromoto and Vello 1986). Thus, the current Brazilian soybean germplasm pool, as defined by Hiromoto and Vello (1986), is the result of several cycles of selection and effective recombination among a relatively small number of selections from the USA cultivars.
The frequent selection, admixed population, and the crossing of a small number of cultivars in the Brazilian soybean breeding programs can lead to a reduction in genetic diversity and affect the patterns of linkage disequilibrium (LD). At the moment, few genetic studies have determined the patterns of LD in tropical soybean genotypes. Priolli et al. (2014), using 142 SSR markers and 94 accessions (cultivated and breeding material) obtained of EMBRAPA soybean and USP/ESALQ germplasm that represent soybean breeding lines of public and private institutions, suggest a structure of LD across the soybean genome (LD decay) of approximately 12 cM. In self-pollinated species, as well as soybean, where recombination is less effective than in outcrossing species, LD declines more slowly (Flint-Garcia et al. 2003). Nonetheless, the germplasm that makes up the collection plays a key role in LD variation because the extent of LD is influenced by the level of genetic variation captured by the target population (Soto-Cerda et al. 2013). In soybean, a highly variable pattern of LD has been reported in multiple populations, with variability at different genomic regions (Hyten et al. 2007). In fact, due to the highly variable levels of LD decay in the Landraces and the Elite Cultivars reported for soybean (Hyten et al. 2007; Zhou et al. 2015) and the demands of dense marker sets, it is necessary to determine the LD in tropical soybean cultivars of Brazil that represent the range of photoperiod/temperature latitudinal adaptation as defined by a maturity group (MG) Roman numeral designation.
Most of the process observed in population genetics, as well as domestication, selection, founding events and population subdivision can affect LD decay, however, population structure (admixture) and the mating system of the species (selfing versus outcrossing) can strongly influence patterns of LD (Flint-Garcia et al. 2003). It is known that pairwise LD increases with selfing and can extend very far in highly selfed organisms (Nordborg 2000). For this reason, assume that individuals in a sample are either fully outcrossing may result in spurious inference of population structure in partially selfing populations, as suggested by Falush et al. (2003). To correct spurious evidence for admixture in the presence of partial self-fertilization, Gao et al. (2007) implement a model to accommodate partial selfing and correct the inference of population structure in self-pollinating species as soybean. On the other hand, predict LD decay based on the present-day mating system must be cautious, because the mating system may have changed significantly (Flint-Garcia et al. 2003). For example, G. max and its ancestor, G. soja, differ significantly in their outcrossing rates. The self-pollinating G. max has an outcrossing rate of approximately 1%, whereas G. soja outcrosses at an average rate of 13% (Fujita et al. 1997). The greater amount of outcrossing in G. soja increases the effective recombination rate, leading to the prediction of an 11-fold lower extent of LD in G. soja as compared to G. max (Flint-Garcia et al. 2003).
In this study we genotyped 169 tropical soybean genotypes using high throughput genotyping with SNPs markers. The overall goal was to analyze linkage disequilibrium blocks in a collection of tropical soybean genotypes of Brazil. Our specific goals were: (1) to estimate population structure and assess population relatedness; (2) and to detect the patterns of LD blocks.
Materials and methods
Plant material
A total of 169 cultivars of soybean with commercial use in Brazil were used for genotyping (Table S1). These cultivars represent the core cultivars used for Brazilian farmers from 1990s to 2010s, and some of these were important progenitors in soybean breeding program of Brazil. Additionally, these cultivars were chosen to represent a range of materials developed for the Brazilian production area and representing the range of photoperiod/temperature latitudinal adaptation as defined by a maturity group (MG) Roman numeral designation (Table S1).
DNA extraction and SNPs genotyping
Genomic DNA was extracted from leaf tissues collected from a mix of ten plants of each accession. DNA-easy Plant Kit (Qiagene) was used to DNA extraction. A total of 6000 single nucleotide polymorphism (SNP) was genotyped in the 169 cultivars with an Infinium iSelect HD Custom Genotyping BARCSoySNP6K (Illumina Inc., San Diego, CA, USA) on the Illumina iScan platform. Genotyping was conducted by Deoxi Biotechnology Ltda®, in Araçatuba, Sao Paulo, Brazil. After eliminating: redundant, non-polymorphic SNPs and SNPs with heterozygous alleles considered as missing data, a total of 4949 SNPs remained. In addition, markers with MAF < 0.1 were removed from the genotype data set, leaving 3780 SNPs for the population structure, coancestry and LD analysis.
Linkage disequilibrium
Linkage disequilibrium parameter (r2) for estimating the degree of LD between pair-wise SNPs was calculated using the software TASSEL4.0 for each chromosomal and LD decay graph was plotted with physical distance (Mbp) versus r2 for all intra-chromosomal comparison using nonlinear regression as described by Remington et al. (2001). The expected value of r2 was estimated according to the following equation:
where r2 is the squared correlation coefficient, n is the sample size, and C is a model coefficient for the distance variable (Hill and Weir 1988). The LD decay curve was fitted to predicted r2 values between adjacent markers using the model of Hill and Weir (1988). This model was implemented to determine LD decay as a function of the distance using the ‘nlm’ function in R. To determine the baseline r2 values, a critical value of LD decay was calculated to 50% of its initial value according to Mamidi et al. (2011) and Wen et al. (2015).
Linkage disequilibrium blocks analysis
The pairwise estimates D’ and r2 were calculated by chromosome. LD blocks were estimated by Solid Spine of LD using the software Haploview 4.2 (Barrett et al. 2005). This internally developed method of Haploview searches for a “spine” of strong LD running from one marker to another along the legs of the triangle in the LD chart. A cutoff of 1% was used, meaning that if addition of a SNP to a block resulted in a recombinant allele at a frequency exceeding 1%, then that SNP was not included in the block.
Population structure
Population structure and inbreeding coefficients at population level were estimated under the Markov Chain Monte Carlo (MCMC) algorithm for the generalized Bayesian clustering method implemented in InStruct software (Gao et al. 2007). This method does not assume Hardy–Weinberg equilibrium within loci, and the expected genotype frequencies are estimated based on rates of inbreeding or selfing.
For infer population structure and population selfing rates in soybean, we performed the function (mode) two of InStruct software (Gao et al. 2007). In fact, we implemented one independent run of MCMC sampling for numbers of groups (K parameter) varying from 2 to 10, without prior population information, and burn-in of 5000 with run length periods of 50000 iterations. The best estimate of number of K groups was determined according to the lowest value of Deviance Information Criterion (DIC) among the nine K simulated (Gao et al. 2007). The hierarchical F statistics were used to estimate proportion of genetic variance explained by MG class and company of origin of soybean using ancestry estimates for K = 9 and calculated using the hierfstat R package (Goudet 2005).
Molecular coancestry
Strong relatedness among families, subpopulations and populations can potentially cause spurious association when it is not considered in association mapping model. Relatedness between subpopulations was estimated using Reynolds genetic distance (ϴ), which is given by ϴij = −ln (1 − Fst) for subpopulations i and j (Reynolds et al. 1983), where Fst corresponds to genetic differentiation among subpopulations. Pairwise molecular coancestry between the nine subpopulations of tropical soybean obtained previously with InStruct software was performed in the software Arlequin 3.5 (Excoffier and Lischer 2010) using a total of 3780 SNPs markers.
Results
Tropical soybean genotyping
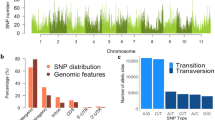
A high coverage of the tropical soybean genome was obtained with the BARCSoySNP6K. In mean 247.5 SNPs markers were found by chromosome, with variation from 198 (chromosome 1) to 323 (chromosome 8). For each chromosome was estimated the ratio between the number of SNPs and the length of each chromosome measured in cM. On average, was found one SNP marker every 0.48 cM, ranging from 0.33 cM (chromosome 4) to 0.60 cM (chromosome 17) by SNP (Table 1). The most marker coverage was found for chromosome 8 that had 323 markers with an average marker density of 0.49 cM. In contrast, the chromosome 1 had the least number of SNPs markers which is equal to 198, with an average marker density of 0.55 cM. This demonstrates that Illumina Infinium platform of genotyping identified SNPs that were well distributed throughout the tropical soybean genome.
Some loci were found in heterozygosity. The percentage of heterozygosity was ranging from 3% (BRSMT Crixás, CD 205, P98Y70 and Celeste) to 41% (BMX Titan RR), with a mean of 9% among the 169 cultivars. Seventy-six percent of the cultivars (129) had fewer than 10% of heterozygosity; 10% (33) was between 10 and 30% and 4% had more than 35% of heterozygosity (Fig. 1).

Frequency of observed heterozygosity in 169 cultivars of soybean, using 4949 SNPs markers
Population structure and molecular coancestry
The genetic structure of the 169 tropical soybean cultivars was estimated using a bayesian clustering approach to infer the number of strongly differentiated genetic subpopulations. According to the lowest DIC value obtained of the posterior bayesian clustering analysis implemented in InStruct, the most probable number of subpopulations was 9 (Fig. 2; Fig. S1). Fst values indicate that 43% of all genotypes present more than 50% of membership to their respective groups. Each subpopulation (K = 9) contained admixed cultivars that come from different soybean genetic breeding programs of Brazil (Table 2). Among the nine subpopulations, none had individuals exclusively from one company or maturity group (Tables 3, 4; Table S1). In fact, this result confirms the shared genetic base among the public and private breeding programs of soybean in Brazil.

Bar plot of the estimated population structure of 169 cultivars of soybean (k = 9). The y-axis is the subgroup membership, and the x-axis is the genotype. The groups go from G1 to G9 from left to right
Based on the alleles of 4949 SNPs markers, and considering the nine subpopulations obtained with InStruct, the average molecular coancestry among the pairwise subpopulation comparisons was 0.234 in the tropical soybean collection as a whole. Approximately 60% of the pairwise coancestry estimates were lower than 0.23 (Mean = 0.196), 30% ranged from 0.24 to 0.3 (Mean = 0.264), and 10% was higher than 0.31 (Mean = 0.332) (Fig. 3). According to the pairwise coancestry estimates most cultivars had moderate relatedness among subpopulations of tropical soybean collection.

Global pairwise molecular coancestry estimates of the 169 tropical soybean cultivars that represent nine subpopulations of Brazil
LD blocks analysis and LD-decay by chromosome
The SNPs with MAF > 0.1 distributed over the soybean genome (3780) has permitted to identify 941 linkage disequilibrium blocks in the tropical soybean material, with 3086 SNPs constituting the haplotype LD block (62% from total SNPs) (Table 1). In mean, the number of blocks by chromosome was 47.05, ranging from 32 (chromosome 1) to 74(chromosome 18) (Table 1). The quantity of SNP in linkage disequilibrium in each block ranged between 2 and 9, with an average of 2.69 SNPs per block. Among the blocks in LD, 64% presented two or three markers, and less than 3% presented seven or more SNPs (Fig. S2). The length of the blocks was very similar by chromosome, and most of these were represented among 51–500 kb. Length blocks larger than 500 kb was not found or was in a very low proportion. There was no relationship between the number of SNPs markers and the increase in LD blocks, indicating that these blocks are randomly localized in the genome. The average length of blocks was 252.4 kb, ranging among 1 (chromosome 4) to 499 kb (chromosome 11). More than 70% of LD blocks showed a length lower than 200 kb (Fig. S3). The sums of the lengths for LD blocks were 237,535 kb, and represents 20% of soybean genome, which have a length of 1.1 gb.
To understand the specific LD block patterns in cultivated tropical soybean, we used Haploview to carry out an LD analysis. It was performed using 3780 SNPs and a nonlinear regression model was used to estimate the LD decay. LD decays reaching at r2 value of 0.2 after 8.5 Mb (Fig. 4).

LD decay among 169 cultivars of tropical soybean
Discussion
SNP genotyping BARCSoySNP6K is a promising method for characterizing soybean genetic diversity and linkage disequilibrium, and for constructing high resolution linkage maps to improve the soybean whole genome sequence assembly (Song et al. 2013). In soybean, Illumina Infinium assay provides a significantly higher level of SNP genotyping capacity respect to Illumina GoldenGate platform, and recent Infinium assays with successful allele calls for nearly 50 k SNPs has been reported by Song et al. (2013). Also, several SNP based marker assays has been developed and validated in soybean, which includes Axiom SoyaSNP array for ~180,000 SNPs (Lee et al. 2015a, b) and the NJAU 355K SoySNP array (Wang et al. 2016). Our SNP genotyping is the first application of Infinium BARCSoySNP6K for tropical soybean. This resulting dataset demonstrate the low to moderate coverage of tropical soybean genome by using this 6 k SNP assay and will assist in the application of genome-wide association studies and high-resolution genetic linkage maps of important traits. At the moment, this assay is being applied to carry out genome-wide association study in important agronomic traits of soybean (Akond et al. 2013; Lee et al. 2015a, b; Contreras-Soto et al. 2017).
Our SNP genotyping represent the first study of soybean genome in tropical cultivars. Our results showed a well distributed number of SNPs into the chromosomes and variable levels of heterozygosity among the 169 cultivars of soybean. In mean, we found 9% of heterozygosity among the cultivars, which may be considered moderately high respect to other studies in soybean (Hyten et al. 2010). In addition it represents an important source of genetic diversity and adaptive evolution.
Some breeding methods for soybean, as single seed descent or back-cross strategies, impose selection on plants that maintain variable levels of heterozygosity during the early generations of the breeding cycle. Haun et al. (2011) following the single seed descent generations in soybean, demonstrate that heterozygous loci may segregate, resulting in genetic heterozygosity within released accessions. Genetic theory predicts, on average, a halving of heterozygous loci with every self-pollination following a given cross. However, heterozygosity may be retained at higher rates if loci confer desirable and selectable phenotypes (Gore et al. 2009), as the case of continuous selections of soybean cultivars in Brazil for different traits.
The varieties used in this study went through dozens of generations of self-fertilization, and each individual plant is very close to, or reaches to 100% homozygous. Heterozygosity, in this case, means a mix of different homozygous genotypes for the locus that was heterozygous when single plants was selected to produce the breeding line, and after that, to advance as a new commercial variety. The level of heterozygosity also depends on the method used to produce the genetic seeds of the new varieties. The pure line method will result in lower level of heterozygosity than bulk method. Our result reveals high levels of heterozygosity in some cultivars of tropical soybean (BMX Titan RR = 41%; NIDERA A 6411 RR = 37%), and it may be useful to promote genetic variability among the genetic base of soybean in Brazil. In fact, a recent study using phenotypic and molecular data (SSR markers) verified the existence of genetic variability among RR soybean® cultivars in public and private soybean breeding companies of Brazil (Villela et al. 2014).
According to DIC value, our population structure analysis supported the existence of nine subpopulations that come from different genetic breeding programs of Brazil. Nearly half of these were considered admixed because the degree of membership within a subpopulation was <0.5. Although 169 cultivars were used in this study, we were only able to obtain the pedigrees for 89 cultivars (Table S1). Due to the Variety Protection Act from 1997 in Brazil, many breeders have not made public the pedigrees of released cultivars, especially recently released varieties. However, our result reveals the existence of shared genetic base among the public and private breeding programs of soybean in Brazil, and showed the high genetic relationship that exist among the commercial cultivars. The same Variety Protection Act from 1997 in Brazil has a clause called “breeders right”. The breeders in Brazil have the right to use, for crossing, any commercial varieties, regardless of whether it is protected or not, and where it originates. This allows the sharing of germplasm between breeding programs.
A previous study conducted by Hiramoto and Vello (1986) indicate that Brazilian soybean ancestors have a narrow genetic base, with only four ancestors (CNS, S-100, Roanoke and Tokyo), that represent approximately 48% of the overall genetic base. Wysmierski and Vello (2013), evaluating 444 cultivars available in the database for the National Cultivar Registry from the Ministry of Agriculture, Livestock and Food Supply of Brazil, showed an increasing in the number of ancestors over time (1971–2009); however the same four main ancestors contribute more than half (55.3%) to the genetic base in soybean and were the same over 1971–2009, showing an increasing on the cumulative relative genetic contribution of ancestors from 46.6 to 57.6%, indicating that the genetic base of Brazilian soybean is still narrow, despite the incorporation of new ancestors.
Company origin and MG may be the principal determinants of population structure within the soybean germplasm collection (Tables 3, 4), however as the genetic base and origin of improved tropical lines are common it’s difficult to explain it. Soybeans are classified into 13 unique MG from very early to very late (000, 00, 0, I, II, III, IV, V, VI, VII, VIII, IX and X), based on temperature and photoperiod response to latitude. Our collection is represented by MG IV to IX and showed admixture population structure among the nine groups. Bandillo et al. (2015) evaluating a diverse soybean MG from the USDA Germplasm Resources Information Network (GRIN) database, reported that near two-thirds of the accessions in the USDA soybean germplasm collection are admixed. Specifically, more than 90% of accessions from America and Europe are admixed. Probably it helps to confirm the admixed genetic structure nature of tropical soybean which has been developed from individuals that have a narrow genetic base of United States. In fact, previous studies demonstrate that the top five ancestors of Brazilian germplasm are the exact same top five ancestors for the soybean genetic base of the southern United States (Wysmierski and Vello 2013).
Simultaneously, as the proportion of individuals for each company and MG within each of the nine subpopulations was not equals, it indicate different degrees of allelic diversity across populations, similar with the results reported by Bandillo et al. (2015) for the USDA soybean germplasm. As expected, individuals of each company of tropical soybean mostly were admixed in all subpopulations as a whole (Table 4). Bandillo et al. (2015), indicates that the analysis of this result is complicated by the fact that ancestors of American soybean, the origin of most of the tropical soybean germplasm (Hiromoto and Vello 1986), contributed at different pedigree levels, coupled with the fact that the American soybean germplasm resulted from a severe population bottleneck when soybeans were introduced to North America (Gizlice et al. 1994) and consequently to Brazil (Hiromoto and Vello 1986). In consequence, company of origin and MG should be explaining a small genetic variation of tropical soybean.
Hierarchical F statistics, calculated using ancestry estimates for K = 9, showed that genetic differentiation explained by MG (~5%) was higher than that explained by Companies (~3%). Similar values of genetic differentiation for MG (MG 000 to X) using ancestry estimates for K = 5 has been reported by Bandillo et al. (2015) for the USDA-GRIN soybean collection. Although the amount of total variation explained is small, these results suggest that population structure in the germplasm collection of Brazil is driven more by MG than companies of origin of soybean cultivars.
At the moment, no information exists about the LD decay in improved tropical soybean lines adapted to Brazil. In addition, most of the studies conducted in soybean, has been used accessions from the U.S. Department of Agriculture (USDA) Soybean Germplasm Resources Information Network (GRIN) database (www.arsgrin.gov). In comparison with the GRIN soybean germplasm resource, with similar MG (Wen et al. 2015; Vuong et al. 2015) our improved tropical soybean showed a higher LD decay (Fig. 4). The difference of LD patterns may be attributed to low genome coverage of markers and fewer genotypes used at the present study. Consequently, as suggested by Song et al. (2015) for soybean, most of the studies conducted for LD evaluation have been limited in terms of sample size and/or the number of loci analyzed, in fact, probably is necessary to evaluate the germplasm of tropical soybean with a greater number of markers.
We found that LD declined below 0.2 at ~8.5 Mb (Fig. 4). In improved cultivars that represent public and private breeding programs for the north central of the United States (MG 0 and early I), LD declined below 0.1 at 7.0 Mb, 5.9 Mb and 8 Mb in studies conducted in the years 2005, 2006 and 2013, respectively (Mamidi et al. 2011, 2014). In Elite cultivars of a single breeding program of Canada LD dropped below 0.1 at ~2.8 Mb (Bastien et al. 2014). Hyten et al. (2007), reported a declined LD decay to 0.1 at 574 kb in north American Elite Cultivars. In fact, highly variable pattern of LD have been reported in multiple soybean populations, and photoperiod sensitivity (maturity) has been proposed how a factor that may have contributed to increase LD in soybean, because their effect resulted in population subdivision in elite soybean cultivars (Hyten et al. 2007). Bastien et al. (2014), suggest that their results of less extensive LD is likely a reflection of the broader scope of the genotypes as it comprised genetically-modified, conventional, and food-type soybeans belonging to Maturity Groups 000 to II. In contrast, our tropical soybean collection showed high relationship among them, and this maybe explains our more extensive LD decay respect to others studies conducted with germplasm of soybean. It is not surprising to find high levels of LD in cultivars with high genetic relationship. In fact, the stringent cleistogamy and relatively long generation time of soybeans suggested that there would be high LD in the soybean genome (Lam et al. 2010).
It is known that LD increases with selfing and can extend very far in highly selfed organisms (Nordborg 2000). Nordborg and Donnelly (1997) showed that the degree of selfing that a species exhibits is related to effective recombination rate. This is because recombination is less effective in selfing species where individuals are more likely to be homozygous at a given locus than in outcrossing species. In the current study, tropical soybean cultivars showed selfing rates equal to s = 0.966 (data not shown). This relationship between recombination rate and selfing can extend to LD, because effective recombination is reduced severely in highly selfing species, as soybean, and consequently LD will be more extensive.
Cultivars contain specific sequence blocks in their chromosomes, which may be associated with artificially selected phenotypic variations from many generations of breeding (Kim et al. 2014; Song et al. 2015). The current study identified an extensive LD, with a set of 941 LD blocks, with most of the SNPs (3086 or 62% from total SNPs number) constituting the haplotype LD blocks. Song et al. (2015) recently provided the first high-resolution haplotype maps based on the largest sample size and the largest number of loci reported in soybean thus far, and they identified that the extent of LD and the average haplotype block sizes were the greater in the north American cultivar population, respect to wild and landraces populations. Our results were similar with the result reported for north American cultivars, and probably this corroborate that the extensive use of a small number of elite genotypes in Brazilian breeding program further reduces genetic variability. In fact, domestication and artificial selection have led to extensive LD and haplotype structure.
This study provides the first comprehensive sequencing data of tropical soybean genome and explored approximately 20% of soybean genome, considering that the sum of lengths for LD blocks were 237,535 kb. According to Schmutz et al. (2010), the soybean genome has about 1.1–1.15 gb, which means that this study was used one marker by 222 kb for evaluate the LD decay and LD haplotype blocks in tropical soybean. Our results showed small differences in length and number of LD blocks and demonstrate that the frequency of occurrence of LD blocks of lengths <500 kb is predominant in cultivated soybean of Brazil. Lam et al. (2010) reported that the frequency of occurrence of LD blocks of lengths <20 kb was higher in wild soybeans than in cultivated soybeans, and indicate that LD blocks of wild soybeans was about half that of cultivated soybeans. In fact, the genetic material used in this study maybe supported the relatively long LD blocks reported here.
Our results of high genetic relatedness and population structure in cultivars of tropical soybean, demonstrate that the nature of soybean fertilization, which results in high inbreeding and thus a reduction in recombination, may have promoted low genome diversity in the tropical soybean and high LD. According to Lam et al. (2010) the presence of high LD in the soybean genome indicates that soybeans would serve as a good model for studying the genomes of crops with extreme LD. Additionally, the information provided by the present study about population structure, genetic relatedness and LD haplotype block location and distribution for cultivated soybean genome, can facilitate the identification of genes of interest. For breeding applications, our identification of the high LD nature in tropical soybean genome indicates that marker-assisted breeding and association mapping studies are better choices for soybean improvement, whereas mapbased cloning using genetic populations will be challenging.
Abbreviations
- DIC:
-
Deviance information criterion
- LD:
-
Linkage disequilibrium
- MAF:
-
Minor allele frequency
- MCMC:
-
Markov Chain Monte Carlo
- QTL:
-
Quantitative trait loci
- SNP:
-
Single nucleotide polymorphism
References
Akond M, Liu S, Schoener L, Anderson JA, Kantartzi SK, Meksem K, Song Q, Wang D, Wen Z, Lightfoot DA, Kassem MA (2013) A SNP-based genetic linkage map of soybean using the SoySNP6K Illumina Infinium BeadChip genotyping array. J Plant Genome Sci 1(3):80–89. doi:10.5147/jpgs.2013.0090
Bandillo N, Jarquin D, Song Q, Nelson R, Cregan P, Specht J, Lorenz A (2015) A population structure and genome-wide association analysis on the USDA soybean germplasm collection. Plant Genome 8(3):1–13. doi:10.3835/plantgenome2015.04.0024
Barrett JC, Fry B, Maller J, Daly MJ (2005) Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21(2):263–265. doi:10.1093/bioinformatics/bth457
Bastien M, Sonah H, Belzile F (2014) Genome wide association mapping of sclerotinia sclerotiorum resistance in soybean with a genotyping-by-sequencing approach. Plant Genome 7(1):1–13. doi:10.3835/plantgenome2013.10.0030
Carter TE, Nelson R, Sneller CH, Cui Z (2004) Genetic diversity in soybean. In: Boerma HR, Specht JE (eds) Soybeans: improvement, production and uses. Am Soc Agron, Madison
Contreras-Soto RI, Mora F, Oliveira MAR, Higashi W, Scapim CA, Schuster I (2017) A genome-wide association study for agronomic traits in soybean using SNP markers and SNP-based haplotype analysis. PLoS ONE e0171105
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567. doi:10.1111/j.1755-0998.2010.02847.x
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164(4):1567–1587
Flint-Garcia S, Thornsberry JM, Bukler ES (2003) Structure of linkage disequilibrium in plants. Annu Rev Plant Biol 54:357–374. doi:10.1146/annurev.arplant.54.031902.134907
Fujita R, Ohara M, Okazakia K, Shimamoto Y (1997) The extent of natural crosspollination in wild soybean (Glycine soja). J Hered 88(2):124–128. doi:10.1093/oxfordjournals.jhered.a023070
Gai JY, Xu DH, Gao Z et al (2000) Studies on the evolutionary relationship among eco-types of G. max and G. soja in China. Acta Agron Sinica 26:513–520
Gao H, Williamson S, Bustamante CD (2007) A Markov Chain Monte Carlo approach for joint inference of population structure and inbreeding rates from multilocus genotype data. Genetics 176:1635–1651. doi:10.1534/genetics.107.072371
Gizlice Z, Carter TE, Burton JW (1994) Genetic base for North American public soybean cultivars released between 1947 and 1988. Crop Sci 34(95):1143–1151. doi:10.2135/cropsci1994.0011183X003400050001x
Gore MA, Chia JM, Elshire RJ, Sun Q, Ersoz ES, Hurwitz BL, Peiffer JA, McMullen MD, Grills GS, Ross-Ibarra J et al (2009) A first-generation haplotype map of maize. Science 326:1115–1117. doi:10.1126/science.1177837
Goudet J (2005) HIERFSTAT, a package for R to compute and test hierarchical F-statistics. Mol Ecol Notes 5:184–186
Haun WJ, Hyten DL, Xu WW, Gerhardt DJ, Albert TJ, Richmond T, Jeddeloh JA, Jia G, Springer NM, Vance CV, Stupar RM (2011) The composition and origins of genomic variation among individuals of the soybean reference cultivar Williams 82. Plant Physiol 155:645–655. doi:10.1104/pp.110.166736
Hill WG, Weir BS (1988) Variances and covariances of squared linkage disequilibria in finite populations. Theor Popul Biol 33:54–78
Hiromoto DM, Vello NA (1986) The genetic base of Brazilian soybean (Glycine max (L.) Merrill) cultivars. Brazil J Genet 9:295–306
Hymowitz T, Kaizuma N (1981) Soybean seed protein electrophoresis profiles from 15 Asian countries or regions: hypotheses on paths of dissemination of soybeans from China. Econ Bot 35(1):10–23. doi:10.1007/BF02859210
Hyten DL, Song Q, Zhu Y, Choi IY, Nelson RL et al (2006) Impacts of genetic bottlenecks on soybean genome diversity. Proc Natl Acad Sci USA 103:16666–16671. doi:10.1073/pnas.0604379103
Hyten DL, Choi IY, Song Q et al (2007) Highly variable patterns of linkage disequilibrium in multiple soybean populations. Genetics 175:1937–1944. doi:10.1534/genetics.106.069740
Hyten DL, Cannon SB, Song Q, Weeks N, Fickus EW, Shoemaker RC, Specht JE, Farmer AD, Maya GD, Cregan PB (2010) High-throughput SNP discovery through deep resequencing of a reduced representation library to anchor and orient scaffolds in the soybean whole genome sequence. BMC Genomics 15:38. doi:10.1186/1471-2164-11-38
Kim MY, Lee S, Van K, Kim TH, Jeong SC, Choi IY et al (2010) Whole-genome sequencing and intensive analysis of the undomesticated soybean (Glycine soja Sieb. and Zucc.) genome. PNAS 107(51):22032–22037. doi:10.1073/pnas.1009526107
Kim YH, Park HM, Hwang TY, Lee SK, Choi MS, Jho S et al (2014) Variation block-based genomics method for crop plants. BMC Genomics 15(1):477. doi:10.1186/1471-2164-15-477
Lam HM, Xu X, Liu X, Chen W, Yang G, Wong FL, Li MW, He W, Qin N, Wang B, Li J, Jian M, Wang J, Shao G, Wang J, Suna SS, Zhang G (2010) Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat Genet 42:1053–1059. doi:10.1038/ng.715
Lee S, Freewalt KR, McHale LK, Song Q, Jun TH, Michel AP, Dorrance AE, Mian MAR (2015a) A high-resolution genetic linkage map of soybean based on 357 recombinant inbred lines genotyped with BARCSoySNP6K. Mol Breed 35:58. doi:10.1007/s11032-015-0209-5
Lee YG, Jeong N, Kim JH, Lee K, Kim KH, Pirani A, Ha BK, Kang ST, Park BS, Moon JK, Kim N, Jeong SC (2015b) Development, validation and genetic analysis of a large soybean SNP genotyping array. Plant J 81(4):625–636
Mamidi S, Chikara S, Goos RJ, Hyten DL, Annam D et al (2011) Genome-wide association analysis identifies candidate genes associated with iron deficiency chlorosis in soybean. Plant Genome 4:154–164. doi:10.3835/plantgenome2011.04.0011
Mamidi S, Lee RK, Goos JR, McClean PE (2014) Genome-Wide Association studies identifies seven major regions responsible for iron deficiency chlorosis in soybean (Glycine max). PLoS ONE 9:e107469. doi:10.1371/journal.pone.0107469
Nordborg M (2000) Linkage disequilibrium, gene trees and selfing: an ancestral recombination graph with partial self-fertilization. Genetics 154:923–929
Nordborg M, Donnelly P (1997) The coalescent process with selfing. Genetics 146(3):1185–1195
Priolli RHG, Campos JB, Stabellini NS, Pinheiro JB, Vello NA (2014) Association mapping of oil content and fatty acid components in soybean. Euphytica 203:83–96. doi:10.1007/s10681-014-1264-4
Remington DL, Thornsberry JM, Matsuoka Y, Wilson LM, Whitt SR, Doebley J, Kresovich S, Goodman MM, Buckler ES (2001) Structure of linkage disequilibrium and phenotypic associations in the maize genome. PNAS 98:11479–11484. doi:10.1073/pnas.201394398
Reynolds J, Weir BS, Cockerham CC (1983) Estimation of the coancestry coefficient: basis for a short-term genetic distance. Genetics 105:767–779
Schmutz J, Cannon SB, Schlueter J, Ma J et al (2010) Genome sequence of the palaeopolyploid soybean. Nature 463:178–183. doi:10.1038/nature08670
Song Q, Hyten DL, Jia G, Quigley CV, Fickus EW et al (2013) Development and evaluation of SoySNP50K, a high-density genotyping array for soybean. PLoS ONE 8:e54985. doi:10.1371/journal.pone.0054985
Song Q, Hyten DL, Jia G, Quigley CV, Fickus EW, Nelson RL, Cregan PB (2015) Fingerprinting soybean germplasm and its utility in genomic research. G3, Genes/Genomes/Genetics 5(9):1–17. doi:10.1534/g3.115.019000
Soto-Cerda B, Diederichsen A, Ragupathya R, Cloutier S (2013) Genetic characterization of a core collection of flax (Linum usitatissimum L.) suitable for association mapping studies and evidence of divergent selection between fiber and linseed types. BMC Plant Biol 13:78. doi:10.1186/1471-2229-13-78
Vigouroux Y, McMullen M, Hittinger CT, Houchins K, Schulz L, Kresovich S, Matsuoka Y, Doebley J (2002) Identifying genes of agronomic importance in maize by screening microsatellites for evidence of selection during domestication. Proc Natl Acad Sci USA 99:9650–9655. doi:10.1073/pnas.112324299
Villela OT, Unêda-Trevisoli SH, Da Silva FM, Bárbaro LS, Di Mauro AO (2014) Genetic divergence of roundup ready (RR) soybean cultivars estimated by phenotypic characteristics and molecular markers. Afr J Biotech 13:2613–2625. doi:10.5897/AJB2014.13661
Vuong TD, Sonah H, Meinhardt CG, Deshmukh R, Kadam S, Nelson RL, Shannon JG, Nguyen HT (2015) Genetic architecture of cyst nematode resistance revealed by genome-wide association study in soybean. BMC Genomics 16:593. doi:10.1186/s12864-015-1811-y
Wang J, Chu S, Zhang H, Zhu Y, Cheng H, Yu D (2016) Development and application of a novel genome-wide SNP array reveals domestication history in soybean. Sci Rep 9(6):20728
Wen Z, Boyse JF, Song Q, Cregan PB, Wang D (2015) Genomic consequences of selection and genome-wide association mapping in soybean. BMC Genomics 16:671. doi:10.1186/s12864-015-1872-y
Wysmierski PT, Vello NA (2013) The genetic base of Brazilian soybean cultivars: evolution over time and breeding implications. Genet Mol Biol 36:547–555. doi:10.1590/S1415-47572013005000041
Xu DH, Gai JY (2003) Genetic diversity of wild and cultivated soybeans growing in China revealed by RAPD analysis. Plant Breed 122:503–506. doi:10.1046/j.0179-9541.2003.00911.x
Xu DH, Abe J, Gai JY, Shimamoto Y (2002) Diversity of chloroplast DNA SSRs in wild and cultivated soybeans: evidence for multiple origins of cultivated soybean. Theor Appl Genet 105:645–653. doi:10.1007/s00122-002-0972-7
Zhou Z, Jiang Y, Wang Z, Gou Z et al (2015) Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nat Biotechnol 33(4):408–414. doi:10.1038/nbt.3096
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Contreras-Soto, R.I., de Oliveira, M.B., Costenaro-da-Silva, D. et al. Population structure, genetic relatedness and linkage disequilibrium blocks in cultivars of tropical soybean (Glycine max). Euphytica 213, 173 (2017). https://doi.org/10.1007/s10681-017-1966-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10681-017-1966-5