
Abstract
The work describes systematic development of nanomicellar cationic supersaturable self-nanoemulsifying drug delivery systems (CS-SNEDDS) for augmenting oral biopharmaceutical performance of raloxifene hydrochloride. Plain SNEDDS formulation containing Capryol 90, Cremophor RH 40, and Transcutol HP was optimized using D-optimal mixture design. SNEDDS were characterized for emulsification time, globule size, in vitro drug release, and ex vivo permeation. The CS-SNEDDS formulation was prepared from the optimized SNEDDS by adding oleylamine as the cationic charge inducer and HPMC as the polymeric precipitation inhibitor. Evaluation of CS-SNEDDS was carried out through in vitro cell line studies on Caco-2 and MCF-7 cells, in situ perfusion, and in vivo pharmacokinetic studies, which indicated significant improvement in biopharmaceutical attributes of the drug from CS-SNEDDS over plain drug.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Drug therapy through oral route is considered as one of the most preferred routes of drug delivery due to ease in manufacturing, robust formulation nature, low-cost therapy, and higher patient compliance [1, 2]. In this regard, oral delivery of anticancer drugs has been gaining considerable attention over the conventional chemotherapeutic strategies through a parenteral route [3, 4]. However, oral delivery of anticancer drugs is associated with multiple challenges like poor water solubility, first-pass metabolism, limited intestinal permeability, poor gastric stability, and efflux by P-gp transporters, leading eventually to low oral bioavailability and sub-maximal therapeutic response [5].
Raloxifene hydrochloride is a selective estrogen receptor modulator administered through an oral route for the prevention and treatment of breast cancer and osteoporosis [6]. Despite its effectiveness for breast cancer treatment, the oral delivery of raloxifene is considered to be highly challenging owing to its poor oral bioavailability (i.e., 2%), which may be attributed to the poor aqueous solubility, extensive hepatic first-pass effect, and high P-gp efflux [7,8,9]. Many drug delivery systems including solid dispersions [10, 11], inclusion complexes [12], microspheres [13], co-grinding [14], and nanoencapsulation [15,16,17] have been investigated for enhancing the oral bioavailability of raloxifene. None of these formulations are found to be highly satisfactory ostensibly owing to the limited improvement in dissolution rate and drug absorption characteristics.
Of late, self-nanoemulsifying drug delivery systems (SNEDDS) are the lipid-based nanoformulations extensively investigated for augmenting oral absorption of highly lipophilic drugs, as these not only facilitate improvement in the dissolution rate and permeability of the drugs, but also help in circumventing hepatic first-pass effect and P-gp efflux-like problems [18, 19]. SNEDDS upon oral administration produces oil-in-water emulsion with globule size less than 100 nm in the presence of aqueous phase or gastric fluid, which increases the dissolution rate and potentiates absorption of the drugs through intestinal lymphatic pathways [18]. Despite several merits of SNEDDS, these systems possess hiccups like low drug loading potential, drug precipitation during storage, gastric irritation, and toxicity by higher concentration of surfactants, etc. [20]. In this regard, several modifications have been made in SNEDDS for avoiding aforementioned challenges [21]. Among these, supersaturable SNEDDS have been considered as one of the novel formulation approaches over conventional SNEDDS for augmenting the biopharmaceutical performance of the drugs [22]. These primarily contain water-soluble cellulosic polymeric precipitation inhibitors (PPIs) for improving the drug stability by maintaining the supersaturated state under in vivo conditions [23]. In supersaturable formulations, the drug concentration exceeds equilibrium solubility, where PPIs tend to reduce the precipitation of the drug by maintaining the thermodynamic equilibrium state [24].
For the selected drug, raloxifene hydrochloride, only one literature report is available on bioavailability enhancement using SNEDDS, where authors have reported improved drug loading potential of the SNEDDS containing triethanolamine as an alkalinizing agent [25]. However, no mechanistic details on the improvement in biopharmaceutical performance and adequate data with respect to safety evaluation of the formulation have been furnished. This calls for the development of novel cationic supersaturable SNEDDS (CS-SNEDDS) of raloxifene hydrochloride for augmenting its biopharmaceutical performance. Unlike conventional SNEDDS, the CS-SNEDDS formulation contains a cationic charge inducer (i.e., oleylamine) for augmenting drug loading and permeability of the drug, while the polymeric precipitation inhibitor (i.e., HPMC) helps in maintaining thermodynamically the stable supersaturation state of the drug for prolonged periods of time.
In this regard, CS-SNEDDS were optimized using an experimental design and evaluated for emulsification efficiency, globule size, zeta potential, in vitro drug release and supersaturation study, cellular cytotoxicity and uptake studies, ex vivo permeation study, in situ intestinal perfusion study, and in vivo pharmacokinetic and biocompatibility studies for corroborating the underlying mechanistics responsible for improvement in the biopharmaceutical performance of the drug.
Materials and methods
Raloxifene hydrochloride was provided by M/s Zydus Cadila, Ahmedabad, India. Capryol 90 and Transcutol HP were gifted by M/s Gattefosse, Saint Priest, France. Cremophor RH 40 and PVP (K25 and K30) were obtained from M/s BASF, Mumbai, India. HPMC premium grades (i.e., E5LV and E15LV) were received as gift samples from M/s Colorcon Asia Pvt. Ltd, Verna, India. Empty gelatin HPMC capsules (size 00) were obtained as generous gift samples from M/s ACG Capsules, Mumbai, India. All other materials and chemicals used during the present work were of analytical reagent grade and used as obtained.
Excipient screening and phase diagram construction
The excipients were screened on the basis of determining solubility of the drug, where excess amount of the drug was added in lipids, surfactants, and cosolvents. The mixtures kept in vials were shaken up to 72 h at 37 ± 0.5 °C by placing in a water bath shaker [26]. The equilibrated samples were centrifuged at 5000 rpm (1398×g) for 10 min to remove the undissolved drug. The supernatant fraction was separated, and the drug was extracted in methanol. The drug concentration was estimated spectrophotometrically at λmax of 291 nm. The excipients with maximal drug solubility were selected, and phase titration studies were performed. Further, surfactant/cosolvent mixtures (Smix), i.e., 1:0, 1:1, 1:2, 2:1, 1:3, and 3:1, were prepared and mixed with lipidic phase in the range of 1:9 and 9:1 ratio [27]. The mixtures were titrated with water to delineate the boundaries of the nanoemulsion region in the pseudoternary phase diagrams. The ratio with maximal nanoemulsion region was selected for further studies.
Preparation of SNEDDS as per the experimental design
The SNEDDS formulations were prepared by mixing drug with lipidic and emulsifying excipients at ambient temperature to obtain a homogeneous mixture. SNEDDS formulations were subjected to systematic optimization studies employing a D-optimal mixture design. Design-Expert® 9.0.1 (Stat-Ease, Minneapolis, USA) software was used, and amounts of excipients, i.e., Capryol 90 (i.e., lipid), Cremophor RH 40 (i.e., surfactant), and Transcutol HP (i.e., cosolvent) were selected as the factors. Supplementary data Table S1 summarizes the design matrix containing the composition of SNEDDS as per the experimental design. The prepared formulations were evaluated for various response variables, viz., emulsification time (Temul), globule size (Dnm), percentage of drug release in 15 min (Rel15min), and percent of drug permeated in 45 min (Perm45min).
Characterization of the SNEDDS
Emulsification time
Aliquot 1.0 g of each of the SNEDDS prepared was added to 500 mL of simulated gastric fluid (SGF; pH 1.2) under continuous stirring at 50 rpm using USP Apparatus 2 (DS 8000, M/s Labindia, Mumbai, India) maintained at 37 ± 0.5 °C. The time required for emulsification of the SNEDDS preconcentrate to form nanoemulsion was recorded as emulsification time in seconds.
Globule size
Aliquots 1.0 g of each of the SNEDDS prepared as per the experimental design were diluted with 250 mL of simulated gastric fluid and observed for globule size distribution using dynamic light scattering (ZS 90, Malvern, Worcestershire, UK).
In vitro drug release
The in vitro drug release study was carried out as per the dialysis bag method. SNEEDS formulations containing a drug equivalent to 10 mg of raloxifene hydrochloride were kept in a dialysis bag with a molecular cutoff weight of 12,000 Da. The bag was placed in USP Apparatus 2 (DS 8000, M/s Labindia, Mumbai, India) containing 500 mL of SGF and 0.5% w/v SLS, and tied with a paddle. The paddle rotation was fixed at 50 rpm, and the release medium was maintained at 37 ± 0.5 °C. Aliquots (5 mL) were periodically withdrawn at periodic time intervals (i.e., 15, 30, 45, 60, and 120 min), followed by replenishment with an equal volume of fresh dissolution medium. The samples after suitable dilutions were analyzed spectrophotometrically at λmax of 291 nm, and data analysis was carried out by applying the correction factor for volume and drug losses during sampling [28].
Ex vivo permeation
Healthy unisex Sprague-Dawley rats (weighing between 200 and 250 g) were procured from the Central Animal House, Panjab University, Chandigarh, after prior approval of the study protocol (no. PU/IAEC/S/14/106). The rats were sacrificed by cervical dislocation, and the abdomen was opened by mid-line incision to collect the small intestine. The entire length of the small intestine was carefully washed and kept in ice-cold Kreb’s Ringer Buffer (KRB) solution with continuous aeration. For investigational purposes, the medial jejunum segment was isolated and closed at one end with a thread holding a 1-g weight to avoid peristalsis movement of the sac during experiment. Accurately 1 g of the SNEDDS formulation-loaded drug was poured in the sac and placed in the KRB solution (50 mL) as the outer medium maintained at 37 ± 0.5 °C using a water jacket. Aliquot 1 mL of the sample was withdrawn from the outer medium at periodic time intervals followed by replenishment with an equal volume of KRB solution. The samples were analyzed spectrophotometrically at λmax of 289 nm to determine the percent drug permeated in 45 min (Perm45min).
Selection of the optimized formulation and validation studies
The optimized SNEDDS formulation was identified after data analysis, where obtained experimental data for each response variable was analyzed. Multiple linear regression analysis was used applied for fitting the data with a second-order quadratic polynomial model. Interaction terms corresponding to each of the factors were also taken into consideration, and statistical validity of the model was confirmed. Response surface mapping was carried out for understanding the relationship among the studied factors on the response variables. Optimum formulation was identified conducted by numerical desirability function and demarcation of it within the design space. Further, an experimental model was validated by analyzing the closeness between the predicted and observed values of the responses.
Formulation of cationic SNEDDS
The cationic SNEDDS (C-SNEDDS) were prepared from optimized liquid SNEDDS. Aliquot 1.0 g of the formulation was taken in a glass vial, and cationic charge inducer (i.e., oleylamine) was changed. Different concentrations of the charge-inducing agent were investigated for obtaining maximal drug solubilization in the cationic SNEDDS. The prepared C-SNEDDS were evaluated for drug content, particle size, and zeta potential.
Selection of cationic charge inducer concentration
Oleylamine was added to the optimized SNEDDS formulation at different concentrations (i.e., 1 to 3% w/w), followed by addition of excess amount of drug for attaining the supersaturation state of drug in the system. The prepared formulations were evaluated for drug loading efficiency by analyzing the drug content spectrophotometrically at λmax of 291 nm. C-SNEDDS with the highest drug loading capacity was selected for further studies.
Fourier transform infrared spectroscopy
The FTIR studies were performed for the pure drug and physical mixture of raloxifene with oleylamine using a FTIR spectrometer (M/s Perkin-Elmer, Wisconsin, USA). The samples were mixed with KBr in a mortar-pestle and triturated well. The mixture was pressed to form a pellet, which was subjected for FTIR analysis, and overlaid diffractograms were prepared.
Formulation of cationic supersaturable SNEDDS
Aliquot 1.0 g of the formulation was taken in a glass vial, and cationic charge inducer (i.e., oleylamine) was added. Further, HPMC and PVP were added as the PPIs in incremental order for attaining the supersaturation state of the drug.
Selection of PPI and its concentration
The selected PPIs including HPMC (E5, E15) and PVP (K25, K30) were employed at two different concentrations (i.e., 5% and 10% w/w) for preparing the CS-SNEDDS. The PPIs were added in the specified concentrations to the previously optimized C-SNEDDS formulation followed by mixing under magnetic stirring to achieve a homogeneous mixture. All the prepared formulations were stored under room temperature until characterization studies.
Characterization of the CS-SNEDDS
In vitro supersaturation test
The in vitro supersaturation test was performed as per the experimental procedure described in literature reports [29]. An in-house fabricated assembly containing a round-bottom flask heater (M/s Omega, Mumbai, India), a three-neck round bottom flask, and a digital mechanical stirrer was used for conducting the test. The test formulations (i.e., CS-SNEDDS containing 60 mg drug) were super seeded in 100 mL SGF containing 0.5% w/v SLS in a 250-mL round-bottom flask and subjected to continuous stirring at 100 rpm and 37 ± 0.5 °C. Aliquot 1 mL of the sample was withdrawn at periodic time intervals (i.e., 0.25, 0.5, 0.75, 1, 1.5, 2, and 3 h) without replacement and analyzed spectrophotometrically at λmax of 291 nm for drug concentration determination. Further, an in vitro supersaturation test was carried out for 24 h to determine equilibrium solubility (Seq) and subsequently the degree of supersaturation (So) for various PPIs. Based on the in vitro supersaturation test results, the suitable type and concentration of PPI was selected. The precipitates collected at the end of test were subjected to various characterization studies including optimal microscopy, field emission scanning electron microscopy (FESEM), differential scanning calorimetry (DSC), and power X-ray diffraction (PXRD).
Optical microscopy
A small quantity of the precipitated sample was placed on a slide, and a cover slip was mounted. The morphological structure of precipitate was visualized by optical microscopy using a Nikon Eclipse 80i microscope (M/s Nikon Corporation, Chiyoda-ku, Japan).
Field emission scanning electron microscopy
The morphology structure of precipitate was also visualized using FESEM Type II (Model-S-4800, M/s Hitachi, Tokyo, Japan). The surface morphology was analyzed at a working distance of 8.7 mm and accelerating voltage of 1.0 kV was applied to generate the secondary electrons from the sample surface. The images were captured at different magnifications using the detector fixed with the instrument.
Differential scanning calorimetry
Different scanning calorimetry (DSC) analysis was performed for pure drug and precipitate obtained after an in vitro supersaturation test using DSC Q20 (M/s TA Instruments, Detroit, USA). Around 2 mg sample was crimped in the aluminum pans and heated from 30 to 300 °C at a rate of 10 °C/min under an N2 environment (100 mL/min). An empty aluminum pan was used as a reference, and thermograms were recorded using in-built Platinum™ software.
Powder X-ray diffraction
PXRD was performed on pure drug and precipitate obtained after the in vitro supersaturation test for analyzing the crystal morphology using an X’Pert PRO diffractometer (M/s PANalytical, Almelo, The Netherlands). A Cu Kα radiation source was used with an applied voltage of 45 kV and current of 40 mA. Approximately 10 mg sample was placed on the sample holder, and the stage was rotated over the range of 2θ from 5° to 50°. The overlaid diffractograms were prepared by in-built X’Pert High Score version 2.2.4 software.
Dissolution in biorelevant media
In vitro drug release from CS-SNEDDS was carried out in fasted-state simulated gastric fluid (FaSSGF), fed-state simulated gastric fluid (FeSSGF), fasted-state small intestinal fluid (FaSSIF), and fed-state small intestinal fluid (FeSSIF) for simulating drug release profile with various regions of the GI tract [30]. Analogous experimental procedure and study conditions were employed as per the method described in the “In vitro drug release” section. The drug release was measured in the aforesaid media at periodic time intervals, and aliquot (5 mL) samples were withdrawn by replacement with an equivalent amount of fresh medium. The samples were suitably diluted and analyzed spectrophotometrically at a λmax of 291 nm for estimating the amount of drug release and cumulative percent drug release at the studied time points.
Caco-2 cell culture studies
A standard protocol was designed for culture, growth, and maintenance of Caco-2 cells. The cells were grown in culture flasks (75 cm2) and kept under incubation condition with 95% O2 and 5% CO2 at 37 ± 0.5 °C. The culture medium containing Dulbecco’s Modified Eagle’s culture medium, 20% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin was regularly changed for 20 days. After the specified time period, cells were harvested after trypsinization with 0.25% trypsin-EDTA solution.
Cytotoxicity study
A cytotoxicity study was carried out on Caco-2 cells by MTT assay as per the procedure described in literature reports [31, 32]. The cells were seeded in 96-well plate (1 × 104 cells/well) and incubated for 24 h. The positive control (Triton-X 100) and treatment formulations (plain SNEDDS and CS-SNEDDS) were added to the culture plates containing cells in the concentration of 1, 2, 3, 5, and 10 μg/mL. After a 24-h incubation period, cells were washed with phosphate buffer solution (pH 7.4) and 150 μL of MTT solution (500 mg/mL in media) was added to each well. After a 3-h incubation period, 200 μL of dimethylsulfoxide was added to solubilize the formazan crystals and optical density of the resulting solution was measured at 540 nm using an ELISA microplate reader (M/s BioTek, Vermont, USA). Based on the results of optical density, percent cell viability was calculated using Eq. (1).
Uptake study
The qualitative uptake was carried out after seeding the Caco-2 cells at a density of 5 × 105 cells/well in a 96-well plate and incubated overnight for cell attachment. The cells were then incubated with free coumarin-6 dye, and dye-loaded plain SNEDDS and CS-SNEDDS (equivalent to 1 μg/mL) for 2 h, respectively. The culture medium was aspirated, and cells were washed with phosphate buffer solution (pH 7.4). One drop of the cell suspension was placed on a glass slide, fixed with 2.5% v/v glutaraldehyde, and a cover slip was placed to visualize cells under the CLSM.
MCF-7 cell culture studies
Like Caco-2 cells, an analogous standard procedure was adopted for culture of MCF-7, except the growth medium containing Eagle’s medium supplemented with Earle’s salts, L-glutamine, nonessential amino acids, sodium bicarbonate, sodium pyruvate, 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin. The cells were grown for 20 days and trypsinized with 0.25% trypsin-EDTA solution.
Cellular cytotoxicity
MTT assay was performed to determine the cellular cytotoxicity of drug on the MCF-7 cells. Following the attachment of MCF-7 cells, fresh media containing pure drug, plain SNEDDS, and CS-SNEDDS were seeded on 1 × 104 cells/well in 96-well plates in concentrations of 0.1, 1, 10, and 20 μg/mL, equivalent of raloxifene hydrochloride and incubated for 24 h, and the cell viability was calculated as per the protocol mentioned above in the “Cytotoxocity study” section.
Cellular uptake
For qualitative uptake, the optimized plain SNEDDS and CS-SNEDDS formulations loaded with coumarin-6 dye were evaluated through CLSM imaging (Olympus FV1000i, M/s Olympus, Tokyo, Japan). The cells were incubated with dye solution (equivalent to 1 μg/mL free coumarin-6) for 3 h and analyzed as per the protocol mentioned in the “Uptake study” section.
Animal studies
All the animal studies performed in the present research work were conducted after prior approval of the study protocol by the Institutional Animal Ethics Committee, Panjab University, Chandigarh, India (Protocol no. PU/IAEC/S/14/106). The care and maintenance of animals were taken as per the guidelines of the Committee for Prevention, Control and Supervision of Experimental Animals (CPCSEA), Govt. of India. During the study period, the animals were housed in polypropylene cages under standard laboratory conditions at 25 ± 2 °C and 55 ± 5% RH with free access to standard diet and water ad libitum.
In situ single-pass intestinal perfusion studies
In situ single-pass intestinal perfusion (SPIP) studies were performed in Sprague-Dawley rats (weighing 250–300 g), previously abstained for 12 h from the solid food and free access to water ad libitum. The treatment formulations include pure drug suspension, marketed (MKT) formulation, optimized plain SNEDDS, and CS-SNEDDS (raloxifene hydrochloride in the dose of 0.16 mg/kg equivalent to body weight of rats) [26, 33]. The animals were anesthetized using intraperitoneal injection of thiopental sodium (50 mg/kg). A mid-line incision in the abdomen was made, and the proximal part of the jejunum was cannulated with plastic tubing to make inlet and outlet. Further, the intestine was perfused with KRB solution maintained at 37 ± 0.5 °C until the perfusate was clear. After attaining steady-state equilibrium in perfusion at a rate of 0.2–0.3 mL/min, the treatment formulations were injected and aliquot (3 mL) samples were periodically collected at regular intervals of 15, 30, 45, and 60 min. The samples were suitably diluted and analyzed by UV spectrophotometric method at λmax of 291 nm to determine the drug concentration from the previously developed linear calibration plot of raloxifene hydrochloride in KRB solution. Various permeability parameters such as effective permeability (Peff) and wall permeability (Pw) and absorption parameters such as fraction absorbed (Fa) and absorption number (An) were calculated for each of the treatment as per the equations given in literature reports [26, 33].
In vivo pharmacokinetic studies
The single dose and parallel design study was carried out in Sprague-Dawley rats (weighing 200–250 g) under fasting condition. The animals were randomly divided into three groups (i.e., A, B, C), with six rats in each group. The animals in group A were orally administered with pure drug suspension, while animals in groups B and C received optimized plain SNEDDS and CS-SNEDDS, respectively. Each formula contained 0.16 mg/kg dose of raloxifene hydrochloride equivalent to the body weight of rats. All the formulations were orally administered to the rats with the help of a feeding cannula. The pure drug suspension was administered by suspending in 1 mL of 0.25% solution of sodium carboxymethylcellulose in water, while the treatment formulations were administered directly without any dilutions. Blood samples (~ 0.2 mL) were withdrawn from the retro-orbital plexus at periodic time intervals in plastic tubes coated with heparin. Plasma was harvested from blood by centrifugation at 10,000 rpm (5590×g) for 15 min, and subsequently, the drug was extracted by treatment with acetonitrile solvent followed by vortexed mixing for 5 min. The samples were again subjected to centrifugation at 10,000 rpm for 15 min, and the supernatant fraction was collected and filtered through a 0.22-μm membrane filter. The analysis of the drug content was carried out using the previously developed and validated bioanalytical HPLC method of raloxifene hydrochloride in rat plasma [34].
Computer-based pharmacokinetic modeling and data analysis was carried out using WinNonlin Version 5.0 (M/s Pharsight, California, USA) employing Model 3 for the Wagner-Nelson method for a one-compartment body model (1-CBM) with zero lag time for peroral administration. Noncompartmental pharmacokinetic analysis was also carried out by selecting Model 200 for extravascular input. Various pharmacokinetic parameters, viz., Cmax, Tmax, AUClast, Cmax/AUC, Ka, and MRT, were calculated for different treatment formulations.
In vitro/in vivo correlations
The attempts were made for establishing point-to-point correlation (level A in vitro/in vivo correlations (IVIVC)) between percent drug release in vitro with the corresponding percent in vivo drug absorption for the plain SNEDDS, CS-SNEDDS, and pure drug suspension.
Statistical data analysis
All the experimental data from the in vitro and in vivo studies were subjected to statistical data analysis by GraphPad Prism software ver 6.01 (M/s GraphPad Inc., California, USA) using two-way ANOVA followed by Tukey–Kramer multiple-comparison test at the 5% level of significance.
Results and discussion
Excipient screening and construction of phase diagrams
A maximum solubilized fraction of raloxifene was observed in Capryol 90 (0.09 mg/mL), Cremophor RH 40 (2.74 mg/mL), and Transcutol HP (9.12 mg/mL). The detailed data on equilibrium solubility of raloxifene hydrochloride in lipids, surfactants, and cosolvents is illustrated in supplementary data Figs. S1–S3. Based on the excipients with higher solubility, the phase titration studies were carried out to construct the pseudoternary phase diagrams at the Smix ratio, i.e., 1:1, 1:2, 2:1, and 3:1, respectively (Fig. 1). The results revealed an increase in the nanoemulsion region with an increase in the amount of surfactant in the Smix ratio from 1:1 to 2:1, followed by no change in the area for Smix ratio 3:1. Among all the Smix combinations, the maximal nanoemulsion region was observed for the 2:1 ratio of Smix. Hence, this ratio was finally selected for further formulation development studies.

Pseudoternary phase diagrams depicting the nanoemulsion region formed after titration of Capryol 90 with Smix containing Cremophor RH 40: Transcutol HP (THP) at varying ratios i.e., a 1:0, b 1:1, c 2:1, and d 3:1 using water as titrant
Preparation and characterization of SNEDDS
All the SNEDDS formulations prepared as per the D-optimal design were evacuated for various response variables like Temul, Dnm, Rel15min, and Perm45min. Emulsification times (Temul) of the all the formulations were found to be ranging between 54 and 174 s, thus confirming faster emulsification efficiency of the SNEDDS to produce ultrafine dispersion with the globule size (Dnm) in the nanosize range between 58 and 99 nm. The drug release studies showed that all the prepared formulations exhibited complete drug release within 30 min of the time period, followed by complete drug release in 60 min. Analogous results were also observed for Perm45min indicating more than 70% drug permeated within 45 min of time course for all the prepared formulations. The observed results revealed that a variation in the values of aforesaid parameters from the prepared formulations was attributed plausibly owing to the difference in the amount of lipid, surfactant, and cosolvent, respectively. The obtained experimental data for each of the response variable was subjected for fitting with the quadratic polynomial model. The coefficient analysis was carried out for the model equation generated for the responses, which revealed an excellent goodness of fit of the experimental data to the selected model. Moreover, statistical parameters were high values of R ranging between 0.902 and 0.999 (p < 0.001 in all the cases), insignificant lack of fit, and low values of PRESS.
Optimization data analysis and response surface mapping
The optimization data analysis using the second-order quadratic polynomial model shown in Eq. (2) revealed excellent goodness of fit of the experimental data to the selected model. Moreover, a high degree of statistical significance was observed for all the selected model terms with p < 0.05, high values of R ranging between 0.943 and 0.979, and insignificant lack of fit.
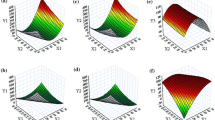
Response surface analysis was carried out for understanding the relationship among the studied factors on the responses. The 3D- and 2D-response surface plot for Temul is depicted in Fig. 2a, b, which revealed a significant increase in the values with increasing levels of Capryol 90 and Transcutol HP, whereas a declining trend was observed with an increase in the levels of Cremophor RH 40. A lower Temul value, however, was observed at higher levels of Capryol 90 and Transcutol HP, and at the intermediate levels of Cremophor RH 40. Figure 2c, d illustrates the 3D-and 2D-response surface plots indicating prominent influence of Cremophor RH 40 on Dnm over Capryol 90 and Transcutol HP. An increase in the values of Dnm was observed with increase in the levels of Capryol 90, whereas Transcutol HP showed a negligible impact on Dnm. Lower values of Dnm were observed at intermediate levels of Capryol 90, high level of Cremophor RH 40, and low levels of Transcutol HP, as is clearly evident from the “region of minimum.” Like Temul, Fig. 2e, f portrays the 3D-and 2D-response surface plots for Rel15min. A faster drug release profile was observed at lower levels of Capryol 90, and intermediate to higher levels of Cremophor RH 40 and Transcutol HP, respectively. This can be attributed to the combined effect of Cremophor RH 40 and Transcutol HP, leading eventually to a marginal reduction in surface interfacial tension and faster drug release characteristics. Further, as shown in Fig. 2g, h, the response surface plots for Perm45min exhibited a typical descending fashion with increasing Capryol 90 and Cremophor RH 40 from intermediate to higher levels. On the contrary, Transcutol HP revealed an increase in the values of Perm45min. A higher Perm45min value was observed at lower levels of Capryol 90, and intermediate levels of Cremophor RH 40 and Transcutol HP, respectively.

3D- and 2D-response surface plots depicting the influence of a Capryol 90, b Cremophor RH 40, and c Transcutol HP on the response variables a, b Temul, c, d Dnm, e, f Rel15min, and g, h Perm45min
Selection of the optimized formulation and validation studies
The optimized SNEDDS formulation selected numerical optimization by assigning desired objectives, i.e., minimization of Temul and Dnm, and maximization of Rel15min and Perm45min. Based on these objectives, the selection criterion was fixed to search the optimized formulation with Temul < 180 s, Dnm < 100 nm, Rel15min, and Perm45min > 80%. Figure 3 depicts the overlay plot design space with the optimized SNEDDS formulation containing Capryol 90 (389 mg), Cremophor RH 40 (415 mg), and Transcutol HP (195 mg) with Temul of 147.7 s, Dnm of 79.6 nm, Rel15min of 77.5%, and Perm45min of 74.3%, respectively. Further, validation studies revealed a close proximity among the predicted values of the responses with observed ones for the prepared checkpoint formulations, which indicated a high degree of prognostic ability of the experimental design used for optimization of SNEDDS.

Overlay plot depicting the design space (yellow color region) and optimized SNEDDS formulation
Formulation and characterization of the cationic SNEDDS
The optimization of cationic SNEDDS was carried out by varying the concentration of the charge inducer (i.e., oleylamine). Table 1 enlists various formulation attributes of cationic SNEDDS prepared by varying concentrations of oleylamine. The physical appearance of the formulations was described by visual observation, which indicated a transparent appearance of cationic SNEDDS containing oleylamine up to 2% w/w, followed by hazy appearance at 2.5 and 3% w/w concentration. Evaluation of drug loading efficiency of cationic SNEDDS showed drastic from 1 to 6% w/w, globule size from 85.1 to 108 nm, and zeta potential from 17.5 to 33.6 mV with increase in the concentration of oleylamine from 1 to 3% w/w. The increase in drug loading efficiency can be ascribed to the positive influence of oleylamine for increasing the drug solubilization in SNEDDS plausibly by electrostatic interactions [32, 35].
Fourier transform infrared spectroscopy
Fourier transform infrared (FTIR) spectroscopy was used for evaluating the underlying mechanism responsible for solubility enhancement of the drug in the lipidic vehicle by altering the physiochemical properties of the drug. Figure 4 illustrates the FTIR spectra of pure drug with functional groups at 3141, 1030–1250, 1641, and 1465 cm−1 corresponding to hydroxyl, oxo, keto, and aromatic rings, respectively. However, the FTIR spectra of the physical mixture of raloxifene with oleylamine shows disappearance of the carbonyl peak (C=O) at 1641 cm−1.

FTIR spectra of a pure drug-raloxifene and b physical mixture of raloxifene with oleylamine
Formulation optimization of the CS-SNEDDS
Based on these results, oleylamine at 2% w/w was selected as the optimum concentration for further studies. Various PPIs were used at their respective grades, i.e., HPMC (HPMC E5 and E15) and PVP (PVP K25 and K30), in the concentrations of 5 and 10% w/w for preparing the CS-SNEDDS, which were evaluated extensively for identifying the optimum type of PPI and its concentration for the purpose.
Characterization of the optimized CS-SNEDDS
Globule size and polydispersity index
The globule size distribution of optimized CS-SNEDDS shows a mean globule size of 185.7 nm and a polydispersity index of 0.23. These results clearly vouch the presence of nanoemulsion globules with a monodisperse nature of their distribution, thus fulfilling the requisite of SNEDDS as per the published literature reports [36]. Moreover, globule size of the optimized CS-SNEDDS showed a slight increase in size as compared to the plain SNEDDS (i.e., 79.6 nm) and cationic SNEDDS (i.e., 140.5 nm).
Zeta potential
The zeta potential distribution of the diluted optimized CS-SNEDDS shows the zeta potential value of 29.8 mV for C-SNEDDS. On the contrary, the plain SNEDDS shows a zeta potential of − 22.4 mV, which indicated a sharp drift in the value plausibly owing to the presence of oleylamine. The observed results for the SNEDDS fulfill the zeta potential requirement for the thermodynamically stable nature of the prepared formulations [36, 37].
Transmission electron microscopy
The transmission electron microscopy (TEM) study of optimized plain SNEDDS and CS-SNEDDS shows the presence of spherical dark nanoemulsion globules. These visual observations further supported the results of globule size distribution analysis.
In vitro supersaturation test
The selection of PPI at its suitable concentration was performed using an in vitro supersaturation test. The CS-SNEDDS were prepared employing the optimized cationic SNEDDS exhibiting maximal drug content, followed by addition of the various grades of HPMC (i.e., HPMCE5 and E15) and PVP (i.e., PVP K25 and K30) as PPIs in the concentrations of 5 and 10% w/w for maintaining the supersaturation state of the drug inhibiting drug precipitation. Supplementary data Fig. S4 illustrates the supersaturation effect of different concentrations of PPIs for inhibiting the precipitation of drug from the CS-SNEDDS.
Interestingly, the results showed that the HPMC grade, i.e., E5, was found to be better over E15 LV in reducing the drug precipitation, while the PVP grade, i.e., K30, was found to be markedly superior over K25. The studies also revealed a insignificant difference in the effect of increasing the concentration of both the PPI grades from 5 to 10% w/w (p > 0.05). This indicated that the suitability of 5% w/w concentration of PPIs was suitable for inhibiting the precipitation of the drug after attaining supersaturation kinetics. Further, comparative effectiveness of the PPIs was evaluated on the basis of parameters like apparent drug concentration (Rc), equilibrium solubility (Seq), and degree of supersaturation (So).
Apparent drug concentration (Rc)
The performance of PPIs was evaluated by comparing the obtained apparent concentration of the drug (Rc) with respect to time, as illustrated in Fig. 5a. Various profiles were observed for Rc with different types of PPIs. It indicates that the initial value for Rc was found to be 0.3 mg mL−1 (based on the dilution factor of 100, i.e., C-SNEDDS, containing 30 mg of raloxifene hydrochloride dissolved in 100 mL of the medium) before starting the supersaturation experiment. After initiating the experiment, Rc was found to be 0.086 and 0.052 mg mL−1 in the case of HPMC grades, i.e., for HPMC E5 and E15 LV at the 3-h time period, respectively. However, in the case of PVP grades (i.e., K30 and K25), Rc was found to be significantly reduced to 0.029 and 0.08 mg mL−1, respectively. Moreover, the PVP grades also showed Rc values between 0.216 and 0.195 mg mL−1 at t = 5 min, which undergoes rapid declining values in the values from 0.029 to 0.007 mg mL−1 at 3 h.

a Apparent drug concentration versus time profile. b Degree of supersaturation for various PPIs used at 5% w/w concentration in CS-SNEDDS. Data represented as mean ± SD (n = 3)
Equilibrium solubility (Seq)
After evaluating Rc for different PPIs, Seq of the drug was calculated for different PPIs, which are enlisted in Supplementary data Table S2. Seq values for the 24-h time course indicated superior values for PVP grades over HPMC grades. The Seq values for HPMC grades were found to be 0.006 and 0.008 mg mL−1 for E5 and E15, respectively. However, Seq values for PVP K25 and K30 were found to be 0.013 and 0.018 mg mL−1, respectively. These results vouch that the lower Seq values for HPMC grades were highly effective for maintaining drugs in the supersaturated state by reducing precipitation during the in vitro test over PVP grades [38].
Degree of supersaturation (S°)
Supplementary data Table S2 summarizes the data on S° of raloxifene hydrochloride in various PPIs used for formulating the CS-SNEDDS. It was observed that HPMC E5 has the highest value for S° (46.9) as compared to that of HPMC E15 (31.3). On the other hand, the value of S° was found to be quite less for PVP grades, i.e., 11.5 and 14.19, in the case of PVP K25 and PVP K30, respectively. Moreover, the So pattern of the drug from various PPIs used for preparing CS-SNEDDS over the 24-h time period is illustrated in Fig. 5b. Analogous observations were discerned from the graph indicating a sharp descending trend for So from PVP grades as compared to that of the HPMC grades, thus construing superior precipitation inhibiting characteristics of the HPMC by sustaining precipitation of the drug by maintaining the supersaturated state. Between the two different HPMC grades, HPMC E5 was found to be more effective than HPMC E15, which can be assigned to the medium viscosity of E5 helpful in maintaining optimal supersaturation kinetics of the drug over E15 having a higher viscosity [22, 35, 39].
Additionally, the supersaturation index (σ) was also calculated in order to determine the supersaturation state. Supplementary data Table S2 illustrates the values of σ obtained for different PPIs used for preparing the CS-SNEDDS. It can be observed from the data that all the polymers have effectively maintained the supersaturated state of the drug, which can be well supported with the values of S° > 1 and σ > 0. Among the HPMC grades, E5 showed a higher value of σ over E15, and thus is more effective in sustaining the supersaturation state of the drug over PVP grades (K25 and K30). Based on the lower values of Seq, and higher values of S° and σ, HPMC E5 was finally selected as the suitable PPI for the CS-SNEDDS.
Characterization of precipitate of the CS-SNEDDS
Optical microscopy
Figure 6a, b depicts the optical microscopy images of pure drug and precipitate of the drug obtained after the supersaturation test. The pure drug shows presence of spear-shaped crystals, while precipitates obtained from optimized CS-SNEDDS show absence of the crystalline structure collected after subjecting to in vitro supersaturation test for 24 h. This indicated change in the crystal morphology of the drug from crystalline to amorphous structure in the precipitates collected from CS-SNEDDS, which might be responsible for enhancing the equilibrium solubility of the drug [40, 41].

a, b Optical photomicrographs. c, d FESEM images of pure drug and precipitate obtained from CS-SNEDDS after in vitro supersaturation test
Field emission scanning electron microscopy
Figure 6c portrays the surface photomicrographs of the pure drug indicating its highly crystalline nature with appearance of flat sheet-shaped crystals. On the contrary, Fig. 6d reveals the amorphous appearance of drug in the precipitates collected from CS-SNEDDS, which indicated significant change in the crystal habit of the drug.
Differential scanning calorimetry
The DSC thermograms of pure drug and precipitates obtained from the CS-SNEDDS using HPMC E5 as PPI are shown in Fig. 7a. The DSC thermogram for the pure drug exhibits a sharp melting endothermic peak initiating from 266 to 267.67 °C [42, 43]. The precipitates obtained from the CS-SNEDDS, however, show no sharp peak of the drug, confirming the conversion of crystalline to amorphous state. Besides, the enthalpy value of the drug was found to be 67.54 J g−1, while the precipitate of the drug obtained from the CS-SNEDDS showed an enthalpy value of 27.66 J g−1. The reduction in the enthalpy of the drug clearly indicated transformation of the drug from crystalline to amorphous state during precipitation from the CS-SNEDDS formulation [44].

a DSC thermograms. b PXRD spectra of pure drug and precipitates obtained from the CS-SNEDDS after in vitro supersaturation test
Powder X-ray diffraction
Figure 7b depicts the PXRD patterns of the pure drug and precipitates of the drug obtained from the CS-SNEDDS. The PXRD pattern of the pure drug shows sharp peaks indicating the crystalline nature of the drug. On the contrary, the precipitates collected from the CS-SNEDDS formulations reveals amorphous appearance with absence of a characteristic PXRD pattern. The data therefore suggest that CS-SNEDDS are effective in controlling the precipitation and maintaining adequate drug stability under a supersaturated state. Thus, HPMC plays a pivotal role in inhibiting the nucleation of drugs, thereby maintaining the supersaturation state [45, 46]. The results were found to be in line with the reports stating the applicability of cellulosic polymers for inhibiting the precipitation of the drugs from supersaturable formulation [29, 41, 47, 48].
Rheological studies
Supplementary data Fig. S5 illustrates the rheological profiles of plain SNEDDS and optimized CS-SNEDDS. A linear correlation was observed between shear rate (0 to 100 s−1) and shear stress (0 to 10 Pa) over a wide range, thus unequivocally indicating Newtonian-type flow characteristics of both SNEDDS and CS-SNEDDS formulations. Moreover, the viscosity of both the formulations was found to be ranging between 0.01 and 0.09 (Pas), which revealed good flow characteristics. As the values are within the desirable limits of viscosity, i.e., 0.1–1.0 Pas, the developed SNEDDS formulation fulfilled the requisite rheological criteria for filling in hard gelatin capsules [49].
In vitro drug release in biorelevant media
In vitro drug release studies of the optimized CS-SNEDDS performed in various biorelevant dissolution media like FaSSGF, FeSSIF, FaSSIF, and FeSSGF are shown in Fig. 8. The results showed more than 95% drug release within 30 min from the CS-SNEDDS in FaSSGF and FaSSIF, whereas in the case of FeSSIF and FaSSIF, only 60% drug release was observed in 30 min with a maximum of 82% drug release in the 2 h time course. These studies indicated faster drug release characteristics from FaSSGF and FeSSGF over FaSSIF and FeSSIF.

Dissolution profile of raloxifene hydrochloride from CS-SNEDDS in the presence of different biorelevant dissolution media. Data represented as mean ± SD (n = 3)
Caco-2 cell culture studies
Cellular uptake
Figure 9a depicts the CLSM images of Caco-2 cells incubated with coumarin-6 dye (as control) and CS-SNEDDS loaded with coumarin-6 dye. The images of cells treated with free coumarin-6 dye revealed miniscule fluorescence, while higher fluorescent intensity was observed for the cells incubated with C-6-loaded CS-SNEDDS. These observations indicated higher cellular uptake of the nanoemulsion globules formed from CS-SNEDDS. Moreover, the fluorescent intensity was compared from both the control cells and cells treated with formulation through line analysis (data not shown), which further substantiated the supremacy of the CS-SNEDDS for attaining a higher cellular uptake of CS-SNEDDS. This can be assignable to the presence of lipidic and emulsifying excipients in CS-SNEDDS which helps for faster permeation, while the nanosized structure of the emulsion globules facilitates in the uptake across the membrane of Caco-2 cells [26, 31].

a CLSM images of Caco-2 cells indicating uptake of free Coumarin-6 dye and CS-SNEDDS under different channels. b Percent cell viability of Caco-2 cells treated with SNEDDS, CS-SNEDDS, and Triton-X 100. Data represented as mean ± SD (n = 3)
Cellular cytotoxicity
Figure 9b depicts the concentration versus percent cell viability data for the Caco-2 cells incubated with optimized plain SNEDDS, CS-SNEDDS, and Triton-X 100 (as negative control). The percent cell viability > 95% was found to be observed for the cells treated with both the type of SNEDDS, at all the studied concentrations without any statistically significant difference (p > 0.05). Triton-X 100, however, showed a concentration-dependent reduction in the percent cell viability. The results demonstrated that the optimized SNEDDS formulations were found to be biocompatible and safe for oral administration. Also, the cytotoxicity studies justified the selection of the appropriate type and concentration of excipients for formulating the SNEDDS.
MCF-7 cell culture studies
Cellular uptake
Figure 10a shows the uptake of plain coumarin-6 dye (as control), coumarin-6-loaded plain SNEDDS, and CS-SNEDDS across the MCF-7 cells. The uptake studies indicated higher fluorescence intensity from the CS-SNEDDS, while the plain dye showed presence of miniscule fluorescence. This can be clearly evident from fluorescent images along with intensity measurement through horizontal line analysis.

a CLSM images of MCF-7 cells indicating uptake of free Coumarin-6 dye and CS-SNEDDS under different channels. b Percent cell viability of MCF-7 cells treated with SNEDDS, CS-SNEDDS, and Triton-X 100. Data represented as mean ± SD (n = 3)
Cellular cytotoxicity
Figure 10b illustrates the concentration-dependent percent decrease in cell viability of MCF-7 cells treated with pure drug and CS-SNEDDS. The CS-SNEDDS showed significant reduction in the cell viability at all the studied concentrations of CS-SNEDDS vis-à-vis the pure drug (p < 0.01) within 6 h of the time course. The obtained results, therefore, construed supremacy of the prepared CS-SNEDDS in accomplishing more than 75% cell growth inhibition within the studied time period.
In situ single-pass intestinal perfusion studies
The SPIP studies indicated remarkable improvement in the magnitudes of permeability and absorption parameters from the optimized CS-SNEDDS, plain SNEDDS, and MKT formulation vis-à-vis the pure drug suspension. Figure 11a illustrates the effective permeability (Peff) and wall permeability (Pwall) parameters, where a nearly 3.84- and 2.32-fold enhancement in the values of Peff was observed for the optimized CS-SNEDDS and plain SNEDDS as compared to the pure drug suspension, while MKT formulation showed only 1.13-fold augment in permeability. Likewise, Pwall also showed considerable increase in the values by 6.51- and 3.47-fold from the optimized CS-SNEDDS and plain SNEDDS, while MKT formulation shows only 2.6-fold enhancement vis-à-vis the pure drug suspension.

a Plot depicting percent increase in permeability parameters. b Absorption parameters (i.e., Fa and An) of optimized CS-SNEDDS and SNEDDS vis-à-vis the MKT formulation obtained from intestinal perfusion studies. Data represented as mean ± SD (n = 3)
Likewise, Fig. 11b also illustrates the values of An and Fa, where nearly a 9.11- and 7.84-fold increase in An was observed from the optimized CS-SNEDDS and plain SNEDDS with reference to the pure drug suspension, while the MKT formulation exhibited 3.61-fold increase in the values of absorption parameters. Similarly, a remarkable increase (i.e., 8.65- and 6.32-fold) in the magnitude of Fa was observed from the optimized CS-SNEDDS and plain SNEDDS vis-à-vis the pure drug suspension, whereas the MKT formulation exhibited only 3.82-fold improvement in drug absorption parameters. The obtained results indicated appreciable improvement in the extent of drug absorption from the intestine [50, 51].
In vivo pharmacokinetic studies
Figure 12 depicts the mean plasma concentration versus time profiles of raloxifene hydrochloride observed after peroral administration of a single dose of optimized C-SNEDDS, plain SNEDDS, and pure drug suspension. A highly statistically significant difference (p < 0.0001) in plasma concentration was observed for the C-SNEDDS and plain SNEDDS vis-à-vis the pure drug suspension at all the time-points. The plasma level time profile from both the SNEDDS formulations showed sharp increasing trends and superimposability during the absorptive phase at the initial time points (i.e., < 1 h) construing faster drug absorption by both the formulations over pure drug suspension. Further, pharmacokinetic data analysis explored using the compartmental model indicated best model fitting as per the one-compartment body model, which was statistically ratified using AIC, SBC, SSR, and R. As per the selected model, significant changes were observed for rate and extent of drug absorption parameters including Cmax, AUC, Cmax/AUC ratio, Tmax, and Ka from the prepared SNEDDS formulations. Table 2 enlists the pharmacokinetic parameters of the drug from different treatment groups, while Fig. 12 (inset) illustrates the percent alteration in these pharmacokinetic parameters from the C-SNEDDS and plain SNEDDS as compared to that of the pure drug suspension in the 24-h time course.

Plasma concentration versus time profile graph of raloxifene hydrochloride at various time-points from the optimized CS-SNEDDS and plain SNEDDS and pure drug suspension. Data expressed in mean ± S.D. (n = 6), while inset depicts a bar chart with comparative change in pharmacokinetic parameters in case of the optimized CS-SNEDDS and plain SNEDDS vis-à-vis the pure drug suspension
The values of Cmax and AUC indicating extent of drug absorption showed a considerable increase from the prepared SNEDDS formulations vis-à-vis the pure drug suspension. Among the prepared SNEDDS formulations, a higher magnitude of Cmax of raloxifene hydrochloride was observed for C-SNEDDS (i.e., 6.10-fold) and plain SNEDDS (i.e., 2.12-fold) over the pure drug suspension. Likewise, multi-fold enhancement in the AUC (i.e., 4.8- and 2.3-fold) was observed for CS-SNEDDS and plain SNEDDS vis-à-vis the pure drug suspension, respectively. The values of Cmax/AUClast also showed mild alteration by 1.2- and 0.64-fold for the CS-SNEDDS and plain SNEDDS as compared to that of pure drug suspension, respectively. Also, the magnitude of Ka showed significant improvement with nearly 4.6- and 4.0-fold augmentation from the CS-SNEDDS and plain SNEDDS vis-à-vis the pure drug suspension, respectively. On the contrary, the values of Tmax indicating the rate of drug absorption showed drastic reduction by 67 and 35% for CS-SNEDDS and plain SNEDDS with respect to the pure drug suspension, respectively.
Overall, the pharmacokinetic studies construed exclusively higher values of Cmax, AUClast, Cmax/AUC, Ka, and lower values of Tmax in the case of the optimized CS-SNEDDS vis-à-vis the pure drug suspension indicating a remarkable increase in the rate and extent of absorption of the investigated drug from the latter one. These results also demonstrated superiority of CS-SNEDDS over the plain SNEDDS in improving the drug absorption characteristics.
In vitro/in vivo correlations
Figure 13 illustrates level A IVIVC for CS-SNEDDS, plain SNEDDS, and pure drug suspension. High magnitudes of R for plain SNEDDS (i.e., 0.967), C-SNEDDS (i.e., 0.973), and pure drug suspension(i.e., 0.983) as per the linear model fitting between the in vitro drug release and in vivo drug absorbed confirmed the prevalence of level A IVIVC [52, 53]. The prepared formulations showed good correlation for in vitro drug release and in vivo drug absorption up to 45 min, while the pure drug suspension exhibited a linear relationship from 30 min and 2 h. Moreover, the close proximity in the R values of the prepared formulations with the pure drug suspension did not reveal any significant difference in the dissolution and absorption behavior.

Level A IVIVC for the optimized a CS-SNEDDS, b plain SNEDDS, and c pure drug suspension
Discussion
The research work involved development and characterization of nanomicellar self-nanoemulsifying formulations of raloxifene hydrochloride. The preformulation studies including equilibrium solubility profile analysis was carried out on various oils, surfactants, and cosurfactants. Among the studied excipients, Capryol 90, Cremophor RH 40, and Transcutol HP exhibited maximal solubility of the drug in it. Being highly lipophilic in nature, Capryol 90 with a HLB value of 5 contains propylene glycol monocaprylate, which are long-chain triglycerides of caprylic acid. Many literature reports have demonstrated the usefulness of Capryol 90 in developing nanoemulsion systems and good solubilization potential [54]. Cremophor RH 40 is a nonionic solubilizer and emulsifying agent commonly used in diverse ranges of pharmaceutical products. It is primarily constituted of glyceryl polyethylene glycol oxystearate, thus providing excellent self-emulsification property when coming in contact with gastrointestinal fluids. However, Cremophor is reported to possess toxicity to the biological system above the safety limit for oral/parenteral use, ostensibly owing to its ability to produce hypersensitivity reactions [55]. However, we have judiciously selected the concentration of Cremophor in our formulation. On the contrary, Transcutol HP is used in formulations as solubilizer and permeation enhancer. It is primarily constituted of diethylene glycol monoethyl ether, and reported in many literature studies for development of self-nanoemulsifying formulations with adequate safety profile [27, 56].
Based on the equilibrium solubility profile, a pseudoternary phase titration study was performed by the water/oil titration method using various Smix combinations. Among the Smix combinations explored during phase titration, the ratio 2:1 was selected for further study. This indicated that the particular ratio is suitable for producing the desired emulsification capacity and nanoemulsion region for further exploration study. The boundaries of the nanoemulsion region obtained from the corresponding pseudoternary phase diagrams were taken as the ranges of the factors, and systematic optimization was performed using experimental design. D-optimal mixture designs are best suited for optimizing the composition of formulations where the sum total of excipients needs to be 100%. Moreover, such designs are also helpful in the actual prediction of the design space, leading eventually to the identification of optimum formulation with quantities of all excipients within the permissible daily intake limit. Several literature reports have demonstrated the use of optimal mixture designs for the development of self-nanoemulsifying formulations [32, 57]. Response surface analysis indicated the presence of a complex interaction among the factors taken for experimental design optimization. Capryol 90, Cremophor RH 40, and Transcutol HP exhibited significant influence on the formulation CQAs, Temul, Dnm, Rel15min, and Perm45min.
Using mathematical modeling of data, the optimum SNEDDS formulation was selected from the design space by meeting the desired objectives. The optimum SNEDDS was converted into the cationic SNEDDS by adding oleylamine at varying concentrations (1–3% w/w) as the cationic charge inducer. The charge inducer concentration was optimized on the basis of the loading capacity of the drug in the SNEDDS. Among the different concentrations of charge inducer, maximal solubility was observed at 3% w/w of oleylamine. The increase in drug loading efficiency of cationic SNEDDS can be ascribed to the positive influence of oleylamine for increasing the drug solubilization in SNEDDS plausibly owing to the presence of electrostatic forces [32, 35]. Other vital mechanisms plausibly responsible for solubility improvement could be attributed to the highly lipophilic characteristic of oleylamine and associated electrostatic interaction by surface positive charge. Moreover, the surfactant-like properties of oleylamine also help in improving wettability of drug in the lipophilic vehicle, thus helping in improving the drug loading potential of the developed nanoformulation. Several literature reports have demonstrated the use of oleylamine as a surfactant, solvent, solubilizer, and reducing agent for development of nanoformulations [58,59,60,61]. Further, we investigated the impact of charge inducer on the globule size, which showed a mild increase in the values owing to the long-chain structure of oleylamine. Similar observations were also observed for zeta potential, where a drastic change in potential was observed as compared to the plain SNEDDS [62]. The FTIR analysis performed on the cationic SNEDDS formulation further confirmed the potential interaction of drug with cationic lipid, which was reflected by characteristic change in the peak of drug due to formation of hydrogen bonding between drug and oleylamine. This helped in accentuating the solubilization capacity of the drug for the purpose.
Upon fixing the charge inducer concentration, PPIs (i.e., HPMC E5/E15, PVP K25/K30) were added to prepare the cationic supersaturable SNEDDS (CS-SNEDDS) of the drug. The developed formulation was subjected to extensive characterization through in vitro, ex vivo, in situ, and in vivo studies. Globule size analysis indicated the presence of nanoemulsion globules with monodisperse nature, thus fulfilling the requisite of SNEDDS as per the several published literature reports [36]. The comparison of the globule size of plain SNEDDS and CS-SNEDDS revealed a mild increase in particle size of the latter one. This can be attributed to the presence of oleylamine and PPI, which increases the globule size by their adherence on the surface of nanoemulsion globules. The observed results were in consonance with several literature reports published on supersaturated SNEDDS [38, 56, 63]. Like globule size, the parameter zeta potential also indicated a significant difference between plain and CS-SNEDDS. The sharp drift in the zeta potential of the modified formulation over the plain formulations can be attributed to the presence of oleylamine [32]. The observed results for the CS-SNEDDS fulfill the zeta potential requirement for the thermodynamically stable nature of the prepared formulations. The particle morphology of the nanoemulsion assessed through TEM imaging confirmed the presence of spherical globules, which indicated the formation of nanoemulsion globules after the self-emulsification process.
An in vitro supersaturation test was performed to check the performance of different PPIs used for inhibiting the precipitation and maintaining the supersaturation stability of the drug within the formulation system. Among the studied carriers, the results showed that HPMC grades were found to be superior in inhibiting the precipitation of the drug as compared to the PVP grades, which can be clearly observed from the sharp declining trends in the precipitation rate of the drug during in vitro supersaturation test for PVP grades over HPMC grades. This can be attributed to the cellulosic nature of the HPMC which upon contact with the aqueous medium undergo swelling to form gel phase and reduces the precipitation rate of the drugs [24, 38, 47, 56, 64]. Moreover, literature reports have described the “spring-parachute” mechanism as a potential approach of the cellulosic nature of HPMC, which helps in retarding drug precipitation plausibly owing to the thermokinetic approach [23, 40, 65]. In this regard, PPIs act as a parachute to slow down the precipitation rate of the drug from its supersaturation state in optimized CS-SNEDDS. Several literature reports have described the above mechanism for improving the thermodynamic stability of the drugs [46, 65].
After the in vitro supersaturation test, the precipitates obtained from pure drug and CS-SNEDDS were examined through optical microscopy. This revealed phase transition from crystalline to amorphous state of the drug, which helped in providing desired drug solubility. Moreover, the FESEM imaging, DSC, and PXRD analysis revealed analogous observation regarding the physical transitions of the drug. The dissolution performance evaluated in biorelevant media indicated a quite analogous release pattern of the drug in the studied media. The presence of bile salt and lecithin in these media is considered to be responsible for facilitating micellar solubilization of the drug and quite faster drug release characteristics. The faster drug release nature is required for attaining faster absorption of drug into the systemic circulation for early onset of action [66]. Moreover, the rational selection of biorelevant dissolution media simulating the in vivo conditions helped in better understanding the actual drug release behavior.
The cell line studies indicated absence of any toxic effect of the prepared CS-SNEDDS and plain SNEDDS formulations on Caco-2 cells, while on MCF-7 cells, a significant effect on reduced percent cell viability was observed. Moreover, both the cell lines exhibited good uptake potential when examined through CLSM imaging study. The increase in fluorescence intensity can be attributed as a function of enhanced uptake of nanoemulsion globules containing lipids, emulgents, and cationic charge inducers, which particularly increases the permeability and uptake across the cell membrane [32, 57].
Evaluation of the in vivo drug absorption behavior from the CS-SNEDDS through intestinal perfusion study indicated significant improvement in the intestinal drug permeation and absorption profile over the plain SNEDDS. This could be primarily attributed owing to the increase in intestinal permeability by lipids, emulgents, and charge inducer [67, 68]. Also, the pharmacokinetic study revealed superior drug absorption parameters from the CS-SNEDDS formulation over the plain SNEDDS. Various mechanisms could be responsible for augmenting the oral bioavailability of drugs from CS-SNEDDS. These include improvement in dissolution rate, gastric permeability and absorption, inhibition of P-gp efflux, avoidance of hepatic first-pass effect, and metabolism by gut cytochromes owing to the presence of functional lipids, emulsifying agents, and cationic charge inducers [69,70,71,72]. Moreover, we observed a high degree of correlation between the in vitro drug release and in vivo drug absorption parameters. This testifies the predictive ability of dissolution apparatus, drug release medium, dissolution conditions, etc., selected for establishing the relationship. Also, establishment of level A IVIVC ratifies significant dependability of the in vivo drug absorption parameters on in vitro drug release parameters in the present work. Overall, the establishment of level A IVIVC vouches the enhanced biopharmaceutical performance of drug in the developed formulations.
Conclusions
The present studies vouch the systematic development of optimized CS-SNEDDS with enhanced biopharmaceutical attributes for a BCS class II drug, raloxifene hydrochloride, exhibiting low solubility, high hepatic first pass, and poor oral bioavailability. Equilibrium solubility and pseudoternary phase diagram studies revealed selection of apt concentrations of lipidic and emulsifying agents for formulating SNEDDS with faster emulsification efficiency, nanosized globules, stable zeta potential, immediate drug release, and permeation profile. Use of a cationic charge inducer along with a polymeric precipitation inhibitor indicated remarkable improvement in drug loading and stability of the drug in the CS-SNEDDS as compared to that of plain SNEDDS formulation. Cell line studies on Caco-2 and MCF-7 cells also indicated significant improvement in cellular uptake along with absence of any cytotoxic effect of the optimized CS-SNEDDS, which was further ratified through histopathological studies. Further, extensive evaluation through in situ perfusion and in vivo pharmacokinetic studies indicated multi-fold enhancement in permeability and absorption parameters of the drug from the CS-SNEDDS vis-à-vis the pure drug suspension. The outstanding findings of the current studies, therefore, ratified formulation of CS-SNEDDS as one of the promising alternatives over plain SNEDDS, which can be applicable to other BCS class II and IV drugs exhibiting identical challenges for limited drug absorption from conventional SNEDDS.
References
Singh B, Garg B, Chaturvedi SC, Arora S, Mandsaurwale R, Kapil R, et al. Formulation development of gastroretentive tablets of lamivudine using the floating-bioadhesive potential of optimized polymer blends. J Pharm Pharmacol. 2012;64:654–69.
Gupta H, Bhandari D, Sharma A. Recent trends in oral drug delivery: a review. Recent Pat Drug Deliv Formul. 2009;3:162–73.
Thanki K, Gangwal RP, Sangamwar AT, Jain S. Oral delivery of anticancer drugs: challenges and opportunities. J Control Release. 2013;170:15–40.
Mazzaferro S, Bouchemal K, Ponchel G. Oral delivery of anticancer drugs I: general considerations. Drug Discov Today. 2012;18:25–34.
Stuurman FE, Nuijen B, Beijnen JH, Schellens JHM. Oral anticancer drugs: mechanisms of low bioavailability and strategies for improvement. Clin Pharmacokinet. 2013;52:399–414.
Lee WL, Chao HT, Cheng MH, Wang PH. Rationale for using raloxifene to prevent both osteoporosis and breast cancer in postmenopausal women. Maturitas. 2008;60:92–107.
Snyder KR, Sparano N, Malinowski JM. Raloxifene hydrochloride. Am J Health Syst Pharm. 2000;57:1669–75. quiz 1676-8
Heringa M. Review on raloxifene: profile of a selective estrogen receptor modulator. Int J Clin Pharmacol Ther. 2003;41:331–45.
Chen Y, Jia X, Chen J, Wang J, Hu M. The pharmacokinetics of raloxifene and its interaction with apigenin in rat. Molecules. 2010;15:8478–87.
Tran TH, Poudel BK, Marasini N, Chi SC, Choi HG, Yong CS, et al. Preparation and evaluation of raloxifene-loaded solid dispersion nanoparticle by spray-drying technique without an organic solvent. Int J Pharm. 2013;443:50–7.
Tran TH, Poudel BK, Marasini N, Woo JS, Choi HG, Yong CS, et al. Development of raloxifene-solid dispersion with improved oral bioavailability via spray-drying technique. Arch Pharm Res. 2013;36:86–93.
Wempe MF, Wacher VJ, Ruble KM, Ramsey MG, Edgar KJ, Buchanan NL, et al. Pharmacokinetics of raloxifene in male Wistar-Hannover rats: influence of complexation with hydroxybutenyl-beta-cyclodextrin. Int J Pharm. 2008;346:25–37.
Jha RK, Tiwari S, Mishra B. Bioadhesive microspheres for bioavailability enhancement of raloxifene hydrochloride: formulation and pharmacokinetic evaluation. AAPS Pharm SciTech. 2011;12:650–7.
Jagadish B, Yelchuri R, K B, et al. Enhanced dissolution and bioavailability of raloxifene hydrochloride by co-grinding with different superdisintegrants. Chem Pharm Bull. 2010;58:293–300.
Fontana MC, Beckenkamp A, Buffon A, Beck RC. Controlled release of raloxifene by nanoencapsulation: effect on in vitro antiproliferative activity of human breast cancer cells. Int J Nanomedicine. 2014;9:2979–91.
Kushwaha AK, Vuddanda PR, Karunanidhi P, et al. Development and evaluation of solid lipid nanoparticles of raloxifene hydrochloride for enhanced bioavailability. Biomed Res Int. 2013;2013:584549.
Tran TH, Ramasamy T, Cho HJ, Kim YII, Poudel BK, Choi HG, et al. Formulation and optimization of raloxifene-loaded solid lipid nanoparticles to enhance oral bioavailability. J Nanosci Nanotechnol. 2014;14:4820–31.
Singh B, Bandopadhyay S, Kapil R, Singh R, Katare O. Self-emulsifying drug delivery systems (SEDDS): formulation development, characterization, and applications. Crit Rev Ther Drug Carrier Syst. 2009;26:427–521.
Porter CJ, Trevaskis NL, Charman WN. Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat Rev Drug Discov. 2007;6:231–48.
Kohli K, Chopra S, Dhar D, Arora S, Khar RK. Self-emulsifying drug delivery systems: an approach to enhance oral bioavailability. Drug Discov Today. 2010;15:958–65.
Singh B, Beg S, Khurana RK, Sandhu PS, Kaur R, Katare OP. Recent advances in self-emulsifying drug delivery systems (SEDDS). Crit Rev Ther Drug Carrier Syst. 2014;31:121–85.
Gao P, Morozowich W. Development of supersaturatable self-emulsifying drug delivery system formulations for improving the oral absorption of poorly soluble drugs. Expert Opin Drug Deliv. 2006;3:97–110.
Mathews CDC, Sugano K. Supersaturable formulations. Drug Deliv Systems. 2010;25:371–4.
Brouwers J, Brewster ME, Augustijns P. Supersaturating drug delivery systems: the answer to solubility-limited oral bioavailability? J Pharm Sci. 2009;98:2549–72.
Elsheikh MA, Elnaggar YS, Gohar EY, Abdallah OY. Nanoemulsion liquid preconcentrates for raloxifene hydrochloride: optimization and in vivo appraisal. Int J Nanomedicine. 2012;7:3787–802.
Singh B, Singh R, Bandyopadhyay S, Kapil R, Garg B. Optimized nanoemulsifying systems with enhanced bioavailability of carvedilol. Colloids Surf B Biointerfaces. 2013;101:465–74.
Beg S, Jena SS, Patra Ch N, et al. Development of solid self-nanoemulsifying granules (SSNEGs) of ondansetron hydrochloride with enhanced bioavailability potential. Colloids Surf B Biointerfaces. 2013;101:414–23.
Singh B, Kaur T, Singh S. Correction of raw dissolution data for loss of drug during sampling. Ind J Pharm Sci. 1997;59:196–9.
Gao P, Rush BD, Pfund WP, Huang T, Bauer JM, Morozowich W, et al. Development of a supersaturable SEDDS (S-SEDDS) formulation of paclitaxel with improved oral bioavailability. J Pharm Sci. 2003;92:2386–98.
Klein S. The use of biorelevant dissolution media to forecast the in vivo performance of a drug. AAPS J. 2010;12:397–406.
Jain AK, Thanki K, Jain S. Solidified self-nanoemulsifying formulation for oral delivery of combinatorial therapeutic regimen: part I. Formulation development, statistical optimization, and in vitro characterization. Pharm Res. 2013;31:923–45.
Beg S, Sharma G, Thanki K, Jain S, Katare OP, Singh B. Positively charged self-nanoemulsifying oily formulations of olmesartan medoxomil: systematic development, in vitro, ex vivo and in vivo evaluation. Int J Pharm. 2015;493:466–82.
Singh B, Khurana L, Bandyopadhyay S, Kapil R, Katare OOP. Development of optimized self-nano-emulsifying drug delivery systems (SNEDDS) of carvedilol with enhanced bioavailability potential. Drug Deliv. 2011;18:599–612.
Yang ZY, Zhang ZF, He XB, et al. Validation of a novel HPLC method for the determination of raloxifene and its pharmacokinetics in rat plasma. Chromatographia. 2006;65:197.
Chen ZQ, Liu Y, Zhao JH, Wang L, Feng NP. Improved oral bioavailability of poorly water-soluble indirubin by a supersaturatable self-microemulsifying drug delivery system. Int J Nanomedicine. 2012;7:1115–25.
Beg S, Sandhu PS, Batra RS, et al. QbD-based systematic development of novel optimized solid self-nanoemulsifying drug delivery systems (SNEDDS) of lovastatin with enhanced biopharmaceutical performance. Drug deliv 2014; 1–20.
Tripathi CB, Beg S, Kaur R, Shukla G, Bandopadhyay S, Singh B. Systematic development of optimized SNEDDS of artemether with improved biopharmaceutical and antimalarial potential. Drug Deliv. 2016;23:3209–23.
Gao P, Akrami A, Alvarez F, Hu J, Li L, Ma C, et al. Characterization and optimization of AMG 517 supersaturatable self-emulsifying drug delivery system (S-SEDDS) for improved oral absorption. J Pharm Sci. 2009;98:516–28.
Xu S, Dai WG. Drug precipitation inhibitors in supersaturable formulations. Int J Pharm. 2013;453:36–43.
Singh G, Pai RS. In vitro and in vivo performance of supersaturable self-nanoemulsifying system of trans-resveratrol. Artif Cells Nanomed Biotechnol. 2016;44:510–6.
Gao P, Guyton ME, Huang T, Bauer JM, Stefanski KJ, Lu Q. Enhanced oral bioavailability of a poorly water soluble drug PNU-91325 by supersaturatable formulations. Drug Dev Ind Pharm. 2004;30:221–9.
Khanfar M, Sheikh Salem M, Hawari R. Formulation factors affecting the release of ezetimibe from different liquisolid compacts. Pharm Dev Technol 2012
Gulsun T, Gursoy RN, Oner L. Design and characterization of nanocrystal formulations containing ezetimibe. Chem Pharm Bull (Tokyo). 2011;59:41–5.
Chen Y, Chen C, Zheng J, Chen Z, Shi Q, Liu H. Development of a solid supersaturatable self-emulsifying drug delivery system of docetaxel with improved dissolution and bioavailability. Biol Pharm Bull. 2011;34:278–86.
Thomas N, Holm R, Garmer M, et al. Supersaturated self-nanoemulsifying drug delivery systems (super-SNEDDS) enhance the bioavailability of the poorly water-soluble drug simvastatin in dogs. AAPS J. 2012;15:219–27.
Warren DB, Benameur H, Porter CJ, et al. Using polymeric precipitation inhibitors to improve the absorption of poorly water-soluble drugs: a mechanistic basis for utility. J Drug Target. 2010;18:704–31.
Raghavan SL, Trividic A, Davis AF, Hadgraft J. Effect of cellulose polymers on supersaturation and in vitro membrane transport of hydrocortisone acetate. Int J Pharm. 2000;193:231–7.
Thomas N, Holm R, Mullertz A, et al. In vitro and in vivo performance of novel supersaturated self-nanoemulsifying drug delivery systems (super-SNEDDS). J Control Release. 2012;160:25–32.
Marques RC, Cole E, Kruep D, et al. Liquid-filled gelatin capsules. USP Pharmacopeial. Forum. 2009;35:1029–41.
Madan J, Chawla G, Arora V, Malik R, Bansal AK. Unbiased membrane permeability parameters for gabapentin using boundary layer approach. AAPS J. 2005;7:E224–30.
Oh DM, Curl RL, Amidon GL. Estimating the fraction dose absorbed from suspensions of poorly soluble compounds in humans: a mathematical model. Pharm Res. 1993;10:264–70.
Takekuma Y, Takenaka T, Kiyokawa M, Yamazaki K, Okamoto H, Kitabatake A, et al. Evaluation of effects of polymorphism for metabolic enzymes on pharmacokinetics of carvedilol by population pharmacokinetic analysis. Biol Pharm Bull. 2007;30:537–42.
Polli JE, Rekhi GS, Augsburger LL, Shah VP. Methods to compare dissolution profiles and a rationale for wide dissolution specifications for metoprolol tartrate tablets. J Pharm Sci. 1997;86:690–700.
Rao MRP, Aghav S, Sukre G, et al. Determination of required HLB of Capryol 90. J Dispers Sci Technol. 2012;35:161–7.
Rao Z, Si L, Guan Y, Pan H, Qiu J, Li G. Inhibitive effect of cremophor RH40 or tween 80-based self-microemulsiflying drug delivery system on cytochrome P450 3A enzymes in murine hepatocytes. J Huazhong Univ Sci Technol. 2010;30:562–8.
Bandyopadhyay S, Katare OP, Singh B. Development of optimized supersaturable self-nanoemulsifying systems of ezetimibe: effect of polymers and efflux transporters. Expert Opin Drug Deliv. 2014;11:479–92.
Sharma G, Beg S, Thanki K, Katare OP, Jain S, Kohli K, et al. Systematic development of novel cationic self-nanoemulsifying drug delivery systems of candesartan cilexetil with enhanced biopharmaceutical performance. RSC Adv. 2015;5:71500–13.
Farahmandjou M. Effect of oleic acid and oleylamine surfactants on the size of FePt nanoparticles. J Supercond Novel Magn. 2012;25:2075–9.
Mourdikoudis S, Liz-Marz án LM. Oleylamine in nanoparticle synthesis. Chem Mater. 2013;25:1465–76.
Chokprasombata K, Sirisathitkula C, Hardinga P, et al. Monodisperse magnetic nanoparticles: effects of surfactants on the reaction between iron acetylacetonate and platinum acetylacetonate. Revista Mexicana de Fısica. 2013;59:224–8.
Harris RA, Shumbula PM, van der Walt H. Analysis of the interaction of surfactants oleic acid and oleylamine with iron oxide nanoparticles through molecular mechanics modeling. Langmuir. 2015;31:3934–43.
Mourdikoudis S, Liz-Marzán LM. Oleylamine in nanoparticle synthesis. Chem Mater. 2013;25:1465–76.
Nan Z, Lijun G, Tao W, Dongqin Q. Evaluation of carbamazepine (CBZ) supersaturatable self-microemulsifying (S-SMEDDS) formulation in-vitro and in-vivo. Iran J Pharm Res. 2012;11:257–64.
Song WH, Yeom DW, Lee DH, et al. In situ intestinal permeability and in vivo oral bioavailability of celecoxib in supersaturating self-emulsifying drug delivery system. Arch Pharm Res. 2013;37:626–35.
Brewster ME, Vandecruys R, Verreck G, Peeters J. Supersaturating drug delivery systems: effect of hydrophilic cyclodextrins and other excipients on the formation and stabilization of supersaturated drug solutions. Pharmazie. 2008;63:217–20.
Feeney OM, Crum MF, McEvoy CL, et al. 50 years of oral lipid-based formulations: provenance, progress and future perspectives. Adv Drug Deliv Rev. 2016;101:167–94.
Chen Y, Li G, Wu X, Chen Z, Hang J, Qin B, et al. Self-microemulsifying drug delivery system (SMEDDS) of vinpocetine: formulation development and in vivo assessment. Biol Pharm Bull. 2008;31:118–25.
Sun M, Zhai X, Xue K, Hu L, Yang X, Li G, et al. Intestinal absorption and intestinal lymphatic transport of sirolimus from self-microemulsifying drug delivery systems assessed using the single-pass intestinal perfusion (SPIP) technique and a chylomicron flow blocking approach: linear correlation with oral bioavailabilities in rats. Eur J Pharm Sci. 2011;43:132–40.
Beg S, Swain S, Singh HP, Patra CN, Rao MEB. Development, optimization, and characterization of solid self-nanoemulsifying drug delivery systems of valsartan using porous carriers. AAPS PharmSciTech. 2012;13:1416–27.
Garg B, Katare OP, Beg S, Lohan S, Singh B. Systematic development of solid self-nanoemulsifying oily formulations (S-SNEOFs) for enhancing the oral bioavailability and intestinal lymphatic uptake of lopinavir. Colloids Surf B Biointerfaces. 2016;141:611–22.
Sandhu PS, Beg S, Mehta F, Singh B, Trivedi P. Novel dietary lipid-based self-nanoemulsifying drug delivery systems of paclitaxel with p-gp inhibitor: implications on cytotoxicity and biopharmaceutical performance. Expert Opin Drug Deliv. 2015;12:1809–22.
Bandyopadhyay S, Beg S, Katare OP, Sharma G, Singh B. QbD-oriented development of self-nanoemulsifying drug delivery systems (SNEDDS) of valsartan with improved biopharmaceutical performance. Curr Drug Deliv. 2015;12:544–63.
Acknowledgements
The authors deeply acknowledge the support provided by M/s Zydus Cadila (Ahmedabad, India), M/s Gattefosse (Saint Priest, France), M/s BASF (Mumbai, India), M/s Colorcon Asia Pvt. Ltd. (Verna, India), M/s ACG Capsules (Mumbai, India), and M/s Stat-Ease (Minneapolis, USA) in conducting the present research work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
ESM 1
(DOCX 70 kb)
Rights and permissions
About this article

Cite this article
Jain, A., Kaur, R., Beg, S. et al. Novel cationic supersaturable nanomicellar systems of raloxifene hydrochloride with enhanced biopharmaceutical attributes. Drug Deliv. and Transl. Res. 8, 670–692 (2018). https://doi.org/10.1007/s13346-018-0514-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13346-018-0514-8