
Abstract
Research on host-pathogen interactions (HPIs) has evolved rapidly during the past decades. The more humans discover new pathogens, the more challenging it gets to find a cure and prevent infections caused by those pathogens. Many experimental techniques have been proposed to predict the interactions but most of them are highly costly and time-consuming. Fortunately, computational methods have been proven to be efficient in overcoming such limitations. In this study, we propose utilizing Deep Learning methods to predict HPIs using protein sequences. We use the monoMonoKGap (mMKGap) algorithm with K = 2 to extract features from the sequences. We also used the Negatome Database to generate negative interactions. The proposed method was performed on three separate balanced human-pathogen datasets with 10-fold cross-validation. Our method yielded very high accuracies of 99.65%, 99.52%, and 99.66% (mean accuracy of 99.61%). To further evaluate the performance of the deep Network, we compared it with other classification methods, which were the Random Forest (RF) as multiple Decision Tree, the Support Vector Machine (SVM), and Convolutional Neural Network (CNN). We also tested the Dipeptide Composition algorithm as another feature extraction method to compare the results with the mMKGap method. The experimental results prove that the proposed method is very accurate, robust, and practical and could be used as a reliable framework in HPI research.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Background
Host-pathogen protein-protein interaction (HP-PPI) plays a vital role in proteomics and the pathogenesis of systems biology research [1]. Discovery and characterization of protein-based interactions are of high importance, regarding the fact that infectious diseases are still among the major death causes in humans. By understanding how the pathogens interact with their host for successful invasion, we may have a chance to discover appropriate preventive measures and develop new drugs against many infections and diseases [2]. A pathogen can interact with the host in several ways, including proteins, small molecules, nucleic acids, and metabolites; with direct protein-protein interaction being the most common interaction type [3]. Various studies have applied experimental approaches to detect HP-PPIs. For example, for virus-host protein-protein interactions, they have used methods such as Y2H, AP-MS, GST-pull-down, luminescence, SPR, and protease assay, to name a few [4]. Some high-throughput experimental techniques have successfully led to the development of several HPI databases including PHISTO [5], HPIDB [6], and PHI-base [7]. As promising as these datasets may seem, they only comprise a limited number of pathogens since experimental methods are usually costly and highly time-consuming. In fact, given that at least hundreds of different species can infect the host, thousands of interactions are still unknown.
To enrich host-pathogen interactome and construct comprehensive HPI networks, we should try utilizing computational models [8]. Numerous studies have used computational methods to build large-scale HPI networks, especially for viruses and bacteria [9,10,11]. Amongst the different computational methods that have been proposed throughout the years, machine learning-based methods are extensively popular. For example, Kshirsagar et al. [12] have used multitask learning to predict host-pathogen protein interactions. It is worth mentioning that Support Vector Machines (SVMs) are among the most popular machine learning-based methods.
Although common machine-learning approaches yield promising results, some drawbacks should be considered. One of the main challenges is that the features have to be predefined before they are fed to the machine and the appropriate choice of features affects the outcome of the prediction result. On the other hand, there are limitations in changing or updating the models. Considering these issues, we can use Deep Learning techniques that reduce the feature engineering phase; meaning the model itself extracts the features while fitting the parameters to the training data [13].
Homology-based information, structural information, evolutionary information, and physicochemical information are amongst the variety of protein features that researchers have used to train their machine learning algorithms. These methods have some limitations that need to be considered though. For example, methods that use genomic information by calculating the presence or absence of genes, only work on fully sequenced data [14]. As for homology-based approaches, they work only if the sequence similarity is high. Hence, methods that directly extract information from protein sequences have recently gained attention. For example, Amino Acid Composition (AAC) and Sequence Encoding are some of the most common and simple methods. In this paper, we use a technique called monoMonoKGap (mMKGap) from the PyFeat library [15] which is also a sequence-based feature extraction method.
One of the main obstacles to predicting host-pathogen interactions through computational methods is the lack of data about non-interacting proteins. Almost all available HPI datasets consist of experimentally detected interactions between host and pathogen proteins. For a classification model to work, we also need a negative dataset (no-interaction dataset) that shows which host-pathogen protein pairs will not interact with each other. In most articles, researchers have suggested algorithms to create the negative dataset from the positive data itself. But there are some limitations to these methods that we will cover in the upcoming sections of this paper. We tested three different methods to create negative interactions, one of which uses a database called Negatome [16]. This database consists of experimentally derived non-interacting protein pairs and protein families.
Applying computational methods to identify large-scale HP-PPIs has been used in various recent studies. Fisch et al. proposed HRMAn, which is an image analysis platform based on machine learning algorithms and deep learning [17]. In another study, Lian et al. developed a Random Forest-based predictor of Yersinia pestis PPIs by incorporating host-network properties [18]. Zhang et al. compared various machine learning methods to separate infectious from non-infectious pathogens [19]. Asimet et al. introduces a novel approach for generating detailed statistical representations of viral-host protein sequences. This is achieved by combining local and global residue contextual information. These representations are then used in a machine learning framework called LGCA-VHPPI, based on a deep forest model. This framework effectively extracts important feature correlations for distinguishing interactive viral-host protein pairs from non-interactive pairs, even in the presence of complex data characteristics such as non-linearity, noise, and limited training set size [20]. Kaundal et al. introduced the deepHPI web server, which is the first of its kind to utilize convolutional neural network (CNN) models for predicting host-pathogen interactions (HPI). deepHPI provides quantitative answers and offers enhanced visualization of the resulting host-pathogen network. Additionally, it includes links to protein annotation resources for further exploration. The deepHPI web server offers four distinct host-pathogen model types, covering plant-pathogen, human-bacteria, human-virus, and animal-pathogen interactions. This broad range of models enables the analysis of various scenarios and facilitates a wide range of HPI analyses [21]. Karan et al. present the development of four computational models for predicting protein-protein interactions (PPIs) on a genome-wide scale between rice and M. grisea. The four models include the interolog, domain-based, GO, and phylogenetic prediction approaches. By intersecting the results obtained from these four methods, high-confidence PPIs are identified. The study also introduces a filtering method to analyze and identify potential candidate proteins involved in interactions. Furthermore, the study shows that the SVM model using amino acid composition (AAC) and conjoint triad features (CT) of protein sequences showed better accuracy [22].
In this study, we propose utilizing Deep Learning methods to predict HPIs using protein sequences. The proposed model introduces a Deep Learning-based approach that achieves high accuracy in predicting HP-PPIs (Host-Pathogen Protein-Protein Interactions). It effectively addresses the challenge of creating a dataset of non-interacting host-pathogen pairs, known as the negative set. To overcome this challenge, the model utilizes the Negatome Database, which provides a vast collection of non-interacting protein families. By leveraging this resource, the model improves the selection of non-interacting pairs.
Additionally, the model selects a golden standard positive set from the HPIDB interactions, consisting of known interacting human-bacteria protein pairs. This positive set serves as a reliable benchmark for evaluating the model’s performance. By incorporating these contributions, the proposed model enhances the accuracy and effectiveness of HP-PPI prediction. It successfully overcomes the challenges associated with the negative set and utilizes valuable resources like the Negatome Database and HPIDB interactions, leading to improved prediction outcomes.
Methods
Datasets
We used HPIDB as the main source for the positive interaction’s dataset. It consists of almost 70 thousand interaction samples between different host and pathogen species. Since we wanted to limit research to humans as hosts and bacteria and viruses as pathogens, we had to filter the data to remove non-related interactions. In order to achieve this objective, the initial list was refined by excluding certain taxa, namely FUNGI, AMOEBOZOA, PROTOZOA, and ARCHAEA, originating from the pathogen and the PLANT from the host. After cleaning the dataset and removing homologue data, we ended up with 45,892 interactions which we labeled as the golden standard. Here we should mention that goal was to create a balanced dataset, meaning the number of samples in the positive set was supposed to be equal to the number of samples in the negative set (no-interaction set).
To create a negative dataset, we examined three different methods: In the first method, we tried to create the negative set from new proteins, meaning that we used random host-pathogen proteins that neither the host nor the pathogen was present in the positive set. At first, this method seemed promising but when we tested the trained model with new protein pairs, we realized that the machine has become biased towards the host proteins that were present in the positive set and did not care about the pathogen proteins.
In the second method, we created a negative set directly from positive samples. This method was on the presumption that if no report so far has shown a host protein x will interact with a pathogen protein y, there is a good chance that y will never infect x and the x-y pair can be added to the negative set. After training a simple Decision Tree to test presumption, we concluded that this method does not generate a reliable negative set because the difference between the positive samples and negative samples is not significant enough for the prediction machine to learn how to distinguish between interacting and non-interacting pairs.
In the third method, we referred to the Negatome Database as a reliable source of non-interacting protein families. According to Negatome, two protein families PF00091 and PF02195 do not interact. We chose human proteins (hosts) from PF00091 and bacteria proteins (pathogens) from PF02195. This gave us 136 host proteins and 27,856 pathogen proteins to choose from, yielding 136 * 27,856 or 3,788,416 potential non-interacting protein pairs. We then randomly selected 45,892 of those protein pairs, equal to the number of positive samples we had in golden standard data. We repeated this random selection two more times to obtain three separate negative sets of protein pairs. This was necessary to make sure that the results of the experiment were not dependent on the negative set selection. By combining the positive set with these three negative sets, we created 3 separate datasets to train and test models.
Every classification algorithm must have access to both interacting and non-interacting protein pairs to learn how to distinguish between the two classes. For positive data (interacting proteins), we used HPIDB which includes almost 70 thousand samples of interacting host-pathogen proteins. Many of these samples are obtained from other famous databases like VirHostNet [23], IntAct [24], and MINT [25].
To create the negative data (non-interacting proteins), many previous studies have focused on generating samples from the positive dataset. Urquiza et al. proposed a hierarchical k-means clustering based on the statistical concept of mutual information using the mRMR criterion [26]. In terms of performance, their study shows that this method is better than randomly selecting a negative dataset. In another study, Ben-Hur and Noble found that annotations of cellular localization - which is a very common method for choosing negative examples - lead to biased estimates of prediction accuracy [27]. They demonstrated the effects of this bias in the context of both sequence-based and non-sequence-based features.
To overcome the biases mentioned above, we decided to use the Negatome database, which contains lots of protein families that have been experimentally proven to be non-interacting. This helped us create very reliable negative datasets of protein pairs that were derived from humans and bacteria.
Feature extraction
Many aspects of proteins can be used to derive features for predictors and the features extract directly from protein sequences. They mainly use physicochemical information or amino acid sequence information. Shen et al. [28] grouped the 20 naturally occurring amino acids into seven classes based on their dipole and side-chain volumes. The features were then extracted based on amino acid classes of protein pairs. In another study, Chou [29] proposed a Pseudo Amino Acid Composition which extracts a set of discrete numbers from a protein’s amino acid sequence, while preserving the sequence-order information as well.
After examining different options like the ones explained above, we decided to use monoMonoKGap as the main feature extraction algorithm which is also a sequenced-based technique. This method counts the number of X_X, X__X, X___X, etc. in a protein’s sequence with X being an amino acid and the distance between these Xs will be determined by the K parameter. When implementing the feature extraction algorithm with a gap size (K) set at 2, the output will consist of 32 features for both DNA and RNA sequences and 800 features for protein sequences. Specifically, these features correspond to individual nucleotides represented by letters A, C, G, or T within each respective sequence type. Features will be numbers of A_A, A_C, A_G, A_T, C_A, C_C, C_G, C_T, G_A, G_C, G_G, G_T, T_A, T_C, T_G, T_T, A__A, A__C, A__G, A__T, C__A, C__C, C__G, C__T, G__A, G__C, G__G, G__T, T__A, T__C, T__G, and T__T of the whole sequence of DNA respectively [15]. The resulting feature vectors would then be fed to the deep learning algorithm for further processing. We compare this method with the Dipeptide Composition algorithm which comes from the Amino Acid Composition feature group.
Classification
To determine whether a pair of proteins would interact or not, a machine learning algorithm has to be trained with the available data and their labels, which is called supervised learning. Many methods have been used in the field of HP-PPI throughout the years but the most common ones are as follows. Naïve Bayes is a statistical classifier that calculates conditional probability without taking into account the dependence between the features. Another famous algorithm is Random Forest (RF) [30] which uses an ensemble of classification trees. This method is heavily dependent on the number of trees and the number of randomly selected features. RF can be useful classifiers for host-pathogen protein-protein interaction prediction tasks because they can handle nonlinear relationships and complex hierarchical structures well. However, the effectiveness of multiple decision trees depends on various factors such as the quality of training data, feature selection, pruning strategies, and hyperparameter optimization. We optimized the maximum number of trees to 50, while the optimized learning rate was set to 0.01. SVM is one of the most commonly used classifiers in HP-PPI prediction. The most challenging part of using SVM is choosing a good kernel function. It is also not so suitable for large datasets due to the rapid increase of the support vector. We have used Support Vector Machines with a radial basis function kernel. Due to the computational costs of SVM, we only utilized one-fifteenth of the training samples to optimize parameters. In this study, we set C = 20 and γ = 0.1.
Artificial Neural Networks are a field in machine learning that is evolved from the idea of simulating the human brain. They can detect complex relations between dependent and independent variables. A typical neural network consists of an input layer, an output layer, and a few hidden layers between them. As the number of hidden layers increases, the network can solve more complex nonlinear problems. This leads to the notion of deep neural networks and in general, Deep Learning. For deep learning models to work robustly, they need to be trained with a lot of data. An increase in the number of hidden layers or the number of input features would most definitely require a larger dataset. This is one of the reasons that deep learning methods have not been used widely in HP-PPI prediction research so far.
Tools
We used the Python programming language in different stages. PyFeat is a Python library that we used for feature extraction [15]. PyFeat is a Python library that provides a set of tools for feature engineering, which is the process of creating new input features from raw data to improve the performance of machine learning models. It provides lots of different feature extraction methods like zCurve, gcContent, atgcRation, pseudoKNC, etc. These algorithms can be used to extract features from DNA, RNA, and protein sequences. We utilized its monoMonoKGap method to generate the protein features used in the classification algorithm. We also used the Tensorflow [31] and Propy [32] libraries; the first one was for creating Convolutional Neural Network (CNN) models and the other one was for extracting Dipeptide Composition features from the protein sequences.
Another tool is a stand-alone toolkit called H2O (https://h2o-release.s3.amazonaws.com/h2o/rel-3.46.0/2/index.html) which provides state-of-the-art machine-learning algorithms with a sophisticated UI/UX [33]. In this study, the tool is installed locally, allowing users to fully utilize its capabilities on their own machines. It also has tools to work with big data technologies like Hadoop and Spark. We used its implementation of Deep Neural Networks as classification algorithm. It helped us analyze results by generating a variety of charts and graphs.
monoMonoKGap
As we discussed earlier, we used monoMonoKGap as the main feature extraction algorithm. By changing the parameter K in the algorithm, the number of features in the output will differ. monoMonoKGap yields 400 features by setting K equal to 1. If we increment K by 1 each time, we will get 400 more features, meaning we will have 800 and 1200 features for K = 2 and K = 3 respectively. Having more features in the output is on one hand beneficial because it lets the prediction machine find more complex patterns. But on the other hand, it makes the training process more time-consuming and might even lead to overfitting the machine. For determining the best value for K, we referred to the results of [34]. The paper analyzes different feature extraction methods, one of which is monoMonoKGap. It compares the results of different values for the K parameter and concludes that K = 2 is the very best option. By setting K equal to 2 we get 800 features that will be fed to Deep Learning algorithm for further processing.
Deep learning
Neural Networks are nonlinear statistical classifiers that extract complex relationships between variables. The use of Neural Networks in proteomics is a relatively new concept and still has room to grow. One of the first researches that utilized it was [35] in which the writers predicted protein secondary structure. Xue et al. [36] introduced DeepT3 as a Deep Convolutional Neural Network to identify gram-negative bacterial type 3 secreted effectors. Ahmed et al. [37] predicted human-bacillus anthracis protein-protein interactions by using Multilayer Neural Networks. Furthermore, it is worth mentioning that there are recent studies [38,39,40,41] that specifically focus on this approach. These studies contribute to the growing body of research in this area and provide valuable insights into the topic.
A typical Deep Learning method receives the input data, extracts feature from it and then classifies the result. Another type of Deep Neural Network is called Auto-encoder which usually gets the input and represents it with a smaller set of features. This is normally applied because classification algorithms like SVM are sensitive to the size of feature vectors. We utilized a typical Deep Learning algorithm. In order to optimize the performance of our convolutional neural network (CNN) model, we chose the hyperparameters based on extensive experimentation and validation.
Neural Networks (ANNs) are characterized by their relatively shallow architecture, typically comprising a single or a few hidden layers. They learn to approximate complex functions through the connections between neurons, relying on manual feature engineering and limited depth. While ANNs provide a basic understanding of neural network principles, they may struggle with complex patterns in large datasets due to their shallow architecture and limited feature representation capabilities.
In contrast, Deep Neural Networks (DNNs) are distinguished by their depth, containing many hidden layers that enable them to learn hierarchical representations of data. DNNs excel at representation learning, automatically extracting features directly from raw data without the need for manual feature engineering. Despite being more computationally intensive to train, DNNs have demonstrated superior performance in tasks involving large datasets and complex relationships, making them a significant advancement in neural network technology. The optimized hyperparameters are given in Table 1.
These hyperparameters were carefully selected to strike a balance between model complexity and generalization ability, leading to improved accuracy and outperformance relative to previous approaches.
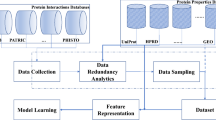
Finally, Fig. 1 shows a graphical abstract of methods.

A graphical abstract of the framework. To create the input dataset, the negative data created from the Negatome database is combined with the positive data obtained from HPIDB. Then the mMKGap feature extraction algorithm is applied to create the feature vectors. Finally, a Feed Forward Neural Network (Deep Learning algorithm) is used for classification
Evaluation criteria
The following criteria, such as Accuracy, Precision, Recall, and F1 score were utilized to assess the work [42]. Accuracy is a ratio of correctly predicted observations to the total observations. It describes the overall system error. Since dataset is symmetric, accuracy can be used as a reliable metric. Precision is the ratio of correctly predicted positive observations to all the observations that we have labeled as positive. The Recall metric is the ratio of correctly predicted positive observations to all the actual positive cases. Finally, the F1 score is a weighted average of Precision and Recall, which works better than Accuracy in situations where the dataset is not symmetric. These indicators are defined below:
Where TP is the number of correctly predicted interacting pairs. FP is the number of falsely predicted interacting pairs. TN is the number of correctly predicted noninteracting pairs. Finally, FN is the number of falsely predicted noninteracting pairs.
Results
Results of the proposed method
We allocated 80% of the data for training, 10% for validation, and the last 10% for testing the model. Furthermore, a 10-fold cross-validation method was used in the training phase, so that predictions would be independent of the training data. To implement the Deep Learning phase, we utilized a stand-alone toolkit named H2O which will be explained in the upcoming sections. We tried to keep the network as simple as possible so we just used 3 hidden layers of sizes 512, 256, and 128 respectively, with the Rectifier algorithm as the activation function. We also set the number of iterations (epochs) to 10.
We took three different approaches to create the negative dataset. In the first one, we used completely new proteins for both the host and the pathogen and created random host-pathogen pairs. This technique yielded a 90.81% accuracy on the test data. The main problem with this approach was that the system became biased towards human proteins and did not care about the pathogen that much. This led to wrong predictions in cases where the host was chosen from database but the pathogen was chosen from a different dataset.
The second approach was to randomly create host-pathogen pairs from the positive set. We were already skeptical of this approach because such random data creations usually need further preprocessing or else the machine won’t be able to distinguish between the positive class and the negative class. We tested the algorithm and it resulted in a nearly 50% accuracy which was not acceptable at all.
Finally, we assessed the third approach in which we utilized the Negatome database to create negative interactions. Table 2 depicts the results summary of 10-fold cross-validation on the validation data of the first dataset. Furthermore, Table 3 shows the prediction performance of the proposed method on the three separate datasets that we created by combining the positive set with the three different negative sets obtained from the Negatome database. Analyzing these data allows concluding that the approach yields a very satisfactory result, where the mean accuracy is above 99.6%. As well as, by changing the threshold, we obtained the ROC curve was calculated. The procedure was repeated 50 times to reduce the variation introduced by the selection of the negative testing data. For this purpose, the ROC curve and the AUC for comparing the results of three different approaches is shown in Fig. 2. Also, in Table 4, we have shown a prediction performance comparison of the three approaches that we used to create the negative dataset.

ROC curve for comparing the three different approaches. (A) Random pairs from new proteins (First approach), (B) Random pairs from the positive set (Second approach), and (C) Random pairs from the Negatome database (Third approach)
Comparison with other approaches
To highlight the feasibility of Deep Learning method, we compared it with other classification algorithms, which were RF, SVM, and CNN. We also compared the main feature extraction algorithm (mMKGap) with the Dipeptide Composition method which comes from the commonly used AAC feature group.
In the first step of the comparison phase, we wanted to make sure whether Deep Learning methods yield better results compared to the RF and SVM, which are more commonly used classification methods in HP-PPI studies. For this experiment to be fair, we applied the latter 2 classifiers to the same 3 datasets that we used for Deep Learning method. The same feature extraction method (mMKGap with K = 2) was applied to the experiments as well. We used Python’s Scikit-Learn [43] library to implement both the RF and SVM algorithms.
The classification results of the RF, SVM, and Deep learning classifiers on three datasets are listed in Table 5. As we can see, the average result of the RF method achieved 99.54% accuracy, 99.88% precision, 99.20% recall, and 99.54% f1 score. The average results of the SVM method yielded 99.45% accuracy, 99.94% precision, 98.95% recall, and 99.45% f1 score. Finally, the average results of the Deep Learning method achieved 99.61% accuracy, 99.96% precision, 100% recall, and 99.61% f1 score. Taking into account the interpretability and ease of implementation, the RF method may be a more suitable choice compared to Deep Learning, even though the Deep Learning method achieved a slightly higher average accuracy.
The AUC value for the Random pairs from new proteins (First approach) is 0.999533, for Random pairs from the positive set (Second approach) is 0.984489, and for Random pairs from the Negatome database (Third approach) is 0.925098. Also, by comparing the AUC values of the three approaches, it can be concluded that deep learning network results are better in comparison between the three approaches.
These data show that the Deep Learning model yields better results compared to the other two models.
While all these algorithms performed impressively, Deep Learning proved to be the most suitable for our problem due to its superior performance metrics, especially in recall and precision. This indicates that Deep Learning can effectively capture complex patterns in the data, which is crucial for accurate prediction in HP-PPI studies. Additionally, the scalability and ability to handle large datasets make Deep Learning a more robust and versatile choice for our specific application.
In the second step of the comparison phase, we wanted to examine whether more sophisticated deep networks such as CNNs yield better results compared to simple “Feed Forward” deep network. We also needed to test whether the mMKGap feature extraction algorithm is the right choice for the proposed approach. Hence, we decided to compare it to the Dipeptide Composition (DPC) method. We used Python’s Tensorflow library (27) to implement the CNN algorithm and the Propy library (28) to implement the DPC method. We compared four different approaches:
-
1
Feed Forward neural network (FFNN) + mMKGap
-
2
FFNN + DPC
-
3
CNN + mMKGap
-
4
CNN + DPC
Figures 3 and 4 shows CNN models for the mMKGap and DPC approaches respectively. mMKGap with K = 2 yields 800 features per protein, while the DPC method yields 400 features per each. Since we combine the feature vectors of the host and pathogen before feeding them to the classifier, we end up with feature vectors of sizes 1600 and 800 respectively. CNN filters are usually N*N, so the features which are fed to it should be N*N as well. We can reshape the vectors of size 1600 to a 40 * 40 array, but this does not apply to the vectors of size 800. To overcome this limitation, we use the “padding” approach to expand the feature vector and reshape it to a 29 * 29 array.

CNN model for the mMKGap approach. The input features are of size 40 * 40, equal to the number of features extracted from mMKGap for host and pathogen, combined (1600)

CNN model for the DPC approach. We applied padding to the input features and increased their size from 800 (2 * 400 from DPC) to 841, to create 29 * 29 squares
Table 6 shows the prediction performance comparison of the four different approaches mentioned earlier. The first obvious observation is that CNN does not yield significant results compared to FFNN. This could be because CNN works best when there are spatial relationships in the data. So here we can conclude that there aren’t many significant spatial relationships among the amino acids in the protein sequences.
The next observation is that for FFNN, it does not matter that much whether we use DPC or mMKGap; the performance parameters are almost the same in both approaches. But for CNN, the difference is significant. We can relate this outcome to the “padding” that we had to apply in the DPC approach. It adds some noise to the data and CNN’s sensitivity to noise results in a decrease in the prediction performance. All in all, we can conclude that the approach performs well compared to other methods.
Discussion
One of the most common ways of interaction between hosts and pathogens is through their proteins. Our understanding of protein structures and interaction mechanisms has improved rapidly during the past decades, but there is still much more to be discovered.
Modern researchers have analyzed HP-PPIs using a variety of experimental methods but most of them are highly costly and time-consuming. Even if the results of such experiments are accurate enough, still many more infections are being discovered every day that cannot be analyzed fast enough to prevent pandemics. One of the best solutions to such problems is to use novel computational methods and machine learning algorithms.
Our contributions to this study can be summarized as follows: First, utilizing the Negatome database to create three separate balanced datasets of positive and negative protein-protein interactions. Second, using the monoMonoKGap sequence-based feature extraction algorithm to obtain protein features, which are later used in the classification phase. Third, apply Deep Learning methods to further extract features from data and predict interacting and non-interacting host-pathogen protein pairs. The rest of this section briefly reviews related research in each phase.
Our goal in this study was to find a practical, efficient, and robust approach that could predict HP-PPIs with very high accuracy. With that in mind, we decided to utilize Deep Learning methods which have been proven to be highly accurate and reliable. To use Deep Learning methods, we needed to have a high volume of data to train a model. We addressed this issue by using HPIDB and the Negatome database - two highly valuable datasets available to create a positive set and a negative set (respectively) for predictor. We also used the mMKGap algorithm as a feature extraction method.
The high accuracy, precision, and recall that we obtained from this study proved that with enough data and a good set of features, we can build a machine that can predict HPIs with high accuracy in a short time. Our study introduces a highly accurate and reliable Deep Learning-based framework in HP-PPI research, which has not been proposed in previous studies. This can help researchers validate their findings and predict potential protein infectors in their future works. We also proved that using Deep Learning methods can lead to better results in terms of performance; compared to other famous machine learning methods such as SVMs and RF. We should also note that another reason to use Deep Learning methods over other classification algorithms is that Deep Learning algorithms have been proven to yield better results on Big Data, which is the case in HP-PPI research.
The classification results show that all three machine learning methods - Random Forest (RF), Support Vector Machine (SVM), and Deep Learning - achieved exceptional performance on the given datasets. The Deep Learning method demonstrated the best overall results, with an average accuracy of 99.61%, precision of 99.96%, recall of 100%, and F1-score of 99.61%. The RF and SVM methods also performed very well, with the RF method achieving average accuracy of 99.54% and the SVM method achieving average accuracy of 99.45%. The high and consistent metrics across all three models indicate their strong ability to accurately classify the data. By employing 10-fold cross-validation, the results provide a more reliable and unbiased assessment of the RF, SVM, and Deep Learning models’ performance. The consistently high accuracy, precision, recall, and F1-score across the folds suggest that these models are able to capture the underlying patterns in the data and make accurate predictions, even on data points they have not been trained on.
Although this study shows promising results, some limitations should be considered when using the approach. First, we should note that Deep Learning methods need lots of data for their training process. Fortunately, we had access to a big database of HPIs for bacteria and viruses. But pathogens are not limited to these two main categories. Fungi and parasites are two other groups of pathogens that can cause deadly infections as well. But the amount of data available for these groups is not as big as the other two. On the other hand, we should bear in mind that in this study we used a simple Deep Learning network with only 3 hidden layers. Using more complex Deep networks might need more configurations and could cost more in terms of hardware equipment.
Conclusions
Prediction of HP-PPIs plays a vital role in preventing bacterial and viral infections. A plethora of methods have been proposed throughout the years but they still have lots of room to get improved and yield higher accuracies. The application of artificial intelligence and machine learning methods has gained much attention in recent years. In this paper, we present a Deep Learning-based approach to predict HP-PPIs with high accuracy. One of the most important problems in HP-PPI machine learning approaches is to create a dataset of non-interacting host-pathogen pairs, i.e., the negative set. We tackle this problem by utilizing the Negatome database, which is a large dataset of non-interacting protein families. We select a golden standard (positive) set from the HPIDB interactions and then randomly create 3 sets of noninteracting human-bacteria protein pairs from two noninteracting protein families of the Negatome database. We used a simple 5-layered Deep Learning model with 10-fold cross-validation and obtained mean accuracy, precision, recall, and F1 scores of 99.61%, 99.96%, 100%, and 99.61% respectively. We also compared the prediction performance of Deep Learning method with the RF, SVM, and CNN algorithms and proved that our approach performs well compared to the other approaches. These results suggest that method is very robust and can be used in future studies for developing vaccines and other preventive medicines to cure various pathogenic infections.
Data availability
The datasets supporting the conclusions of this article are available in the Figshare repository, https://figshare.com/articles/dataset/HP-PPI_dataset_after_applying_the_mMKGap_feature_extraction_algorithm/14588937. The python codes are also available in the GitHub repository, https://github.com/shakibaniataha/HP-PPI-Deep-Learning.
Abbreviations
- SVM:
-
Support Vector Machine
- HPI:
-
Host-Pathogen Interaction
- mMKGap:
-
monoMonoKGap
- HP-PPI:
-
Host-Pathogen Protein-Protein Interaction
- CNN:
-
Convolutional Neural Network
- DPC:
-
Dipeptide Composition
- AAC:
-
Amino Acid Composition
- FFNN:
-
Feed Forward Neural Network
References
Chen H, et al. A framework towards data analytics on host–pathogen protein–protein interactions. J Ambient Intell Humaniz Comput. 2020;11:4667–79.
Sen R, Nayak L, De RK. A review on host–pathogen interactions: classification and prediction. Eur J Clin Microbiol Infect Dis. 2016;35:1581–99.
Durmuş S, et al. A review on computational systems biology of pathogen–host interactions. Front Microbiol. 2015;6:235.
Brito AF, Pinney JW. Protein–protein interactions in virus–host systems. Front Microbiol. 2017;8:1557.
Durmuş Tekir S, et al. PHISTO: pathogen–host interaction search tool. Bioinformatics. 2013;29(10):1357–8.
Ammari MG et al. HPIDB 2.0: a curated database for host–pathogen interactions Database, 2016. 2016.
Urban M, et al. PHI-base: the pathogen–host interactions database. Nucleic Acids Res. 2020;48(D1):D613–20.
Arnold R, et al. Computational analysis of interactomes: current and future perspectives for bioinformatics approaches to model the host–pathogen interaction space. Methods. 2012;57(4):508–18.
Tyagi N, Krishnadev O, Srinivasan N. Prediction of protein–protein interactions between Helicobacter pylori and a human host. Mol Biosyst. 2009;5(12):1630–5.
Doolittle JM, Gomez SM. Structural similarity-based predictions of protein interactions between HIV-1 and Homo sapiens. Virol J. 2010;7:1–15.
De Chassey B, et al. Structure homology and interaction redundancy for discovering virus–host protein interactions. EMBO Rep. 2013;14(10):938–44.
Kshirsagar M, Carbonell J, Klein-Seetharaman J. Multitask learning for host–pathogen protein interactions. Bioinformatics. 2013;29(13):i217–26.
Patel H. What is Feature Engineering—Importance, Tools and Techniques for Machine Learning by Towards Data Science. url: https://towardsdatascience.com/what-is-feature-engineering-importance-tools-and-techniques-formachine-learning-2080b0269f10 (visited on 08/31/2022), 2021.
Jansen R, et al. A bayesian networks approach for predicting protein-protein interactions from genomic data. Science. 2003;302(5644):449–53.
Muhammod R, et al. PyFeat: a Python-based effective feature generation tool for DNA, RNA and protein sequences. Bioinformatics. 2019;35(19):3831–3.
Blohm P, et al. Negatome 2.0: a database of non-interacting proteins derived by literature mining, manual annotation and protein structure analysis. Nucleic Acids Res. 2014;42(D1):D396–400.
Fisch D, et al. Defining host–pathogen interactions employing an artificial intelligence workflow. Elife. 2019;8:e40560.
Lian X, et al. Machine-learning-based predictor of human–bacteria protein–protein interactions by incorporating comprehensive host-network properties. J Proteome Res. 2019;18(5):2195–205.
Zhang M, et al. Prediction of virus-host infectious association by supervised learning methods. BMC Bioinformatics. 2017;18:143–54.
Asim MN, et al. LGCA-VHPPI: a local-global residue context aware viral-host protein-protein interaction predictor. PLoS ONE. 2022;17(7):e0270275.
Kaundal R, et al. deepHPI: a comprehensive deep learning platform for accurate prediction and visualization of host–pathogen protein–protein interactions. Brief Bioinform. 2022;23(3):bbac125.
Karan B, et al. Computational models for prediction of protein–protein interaction in rice and Magnaporthe Grisea. Front Plant Sci. 2023;13:1046209.
Guirimand T, Delmotte S, Navratil V. VirHostNet 2.0: surfing on the web of virus/host molecular interactions data. Nucleic Acids Res. 2015;43(D1):D583–7.
Hermjakob H, et al. IntAct: an open source molecular interaction database. Nucleic Acids Res. 2004;32(suppl1):D452–5.
Zanzoni A, et al. MINT: a molecular INTeraction database. FEBS Lett. 2002;513(1):135–40.
Urquiza JM et al. Selecting negative samples for PPI prediction using hierarchical clustering methodology Journal of Applied Mathematics, 2012. 2012.
Ben-Hur A, Noble WS. Choosing negative examples for the prediction of protein-protein interactions. BioMed Central.
Shen J, et al. Predicting protein–protein interactions based only on sequences information. Proc Natl Acad Sci. 2007;104(11):4337–41.
Chou K-C. Pseudo amino acid composition and its applications in bioinformatics, proteomics and system biology. Curr Proteomics. 2009;6(4):262–74.
Denisko D, Hoffman MM. Classification and interaction in random forests. Proc Natl Acad Sci. 2018;115(8):1690–2.
Abadi M et al. Tensorflow: a system for large-scale machine learning. Savannah, GA, USA.
Cao D-S, Xu Q-S, Liang Y-Z. Propy: a tool to generate various modes of Chou’s PseAAC. Bioinformatics. 2013;29(7):960–2.
Aiello S, et al. Machine learning with python and h2o. H2O. ai Inc; 2016.
Dou L, et al. Prediction of m5C modifications in RNA sequences by combining multiple sequence features. Mol Therapy-Nucleic Acids. 2020;21:332–42.
Qian N, Sejnowski TJ. Predicting the secondary structure of globular proteins using neural network models. J Mol Biol. 1988;202(4):865–84.
Xue L, et al. DeepT3: deep convolutional neural networks accurately identify Gram-negative bacterial type III secreted effectors using the N-terminal sequence. Bioinformatics. 2019;35(12):2051–7.
Ahmed I, Witbooi P, Christoffels A. Prediction of human-Bacillus anthracis protein–protein interactions using multi-layer neural network. Bioinformatics. 2018;34(24):4159–64.
Akbar S, et al. cACP-DeepGram: classification of anticancer peptides via deep neural network and skip-gram-based word embedding model. Artif Intell Med. 2022;131:102349.
Akbar S, et al. iAtbP-Hyb-EnC: prediction of antitubercular peptides via heterogeneous feature representation and genetic algorithm based ensemble learning model. Comput Biol Med. 2021;137:104778.
Ahmad A, et al. Deep-AntiFP: prediction of antifungal peptides using distanct multi-informative features incorporating with deep neural networks. Chemometr Intell Lab Syst. 2021;208:104214.
Akbar S, et al. iHBP-DeepPSSM: identifying hormone binding proteins using PsePSSM based evolutionary features and deep learning approach. Chemometr Intell Lab Syst. 2020;204:104103.
Wu H, Meng FJ. Review on evaluation criteria of machine learning based on big data. IOP Publishing.
Pedregosa F, et al. Scikit-learn: machine learning in Python. J Mach Learn Res. 2011;12:2825–30.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
A.N designed and supervised the project. T.SH and M.A participated in designing the project and contributed to data analysis and wrote the manuscript. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by institutional review board and the ethical committee of Baqiyatallah University of Medical science with the code IR.BMSU.REC.1398.018.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Shakibania, T., Arabfard, M. & Najafi, A. A predictive approach for host-pathogen interactions using deep learning and protein sequences. VirusDis. (2024). https://doi.org/10.1007/s13337-024-00882-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13337-024-00882-x