
Abstract
With the rapid development of high-throughput technologies, systems biology is now embracing a great opportunity made possible by the increased accumulation of data available online. Biological data analytics is considered as a critical means to contribute to a better understanding on such data through extraction of the latent features, relationships and the associated mechanisms. Therefore, it is important to evaluate how to involve data analytics from both computational and biological perspectives in practice. This paper has investigated interaction relationships in the proteomics area, which provide insights of the critical molecular processes within infection mechanisms. Specifically, we focused on host–pathogen protein–protein interactions, which represented the primary challenges associated with infectious diseases and drug design. Accordingly, a novel framework based on data analytics and machine learning techniques is detailed for analyzing these areas and we will describe the analytical results from host–pathogen protein–protein interactions (HP-PPI). Based on this framework, which serves as a pipeline solution for extracting and learning from the raw proteomics data, we have firstly evaluated several models from literature using different analytic technologies and performance measurements. An unsupervised deep learning model based on stacked denoising autoencoders, is subsequently proposed to capture higher level feature regarding the sequence information in the framework. The achieved performance indicates a superior capability of the unsupervised deep learning model in dealing with the host–pathogen protein interactions scenario among all of these models. The results will further help to enrich a theoretical and technical foundation for analyzing HP-PPI networks.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Given the high volume and variety of data, many researches are being conducted in data analytics to predict and uncover information and knowledge concerning related domains, including computer vision, economics, online resources and bioinformatics. Based on the availability of data, computational biology methods, including omics fields, biomedical imaging, and biological signal processing (Min et al. 2017), have grown in importance, with pilot studies having been previously conducted in areas such as genomics and proteomics areas (Greene et al. 2014), and biomedical medicine and imaging areas (Savage 2014).
Proteomics is an important branch of system biology in the post-genomics era, with data analytics playing a vital role in understanding and predicting biological knowledge for proteins. Proteomics research focuses on utilising existing experimental data related to the protein interactions in order to elucidate high-fidelity interaction networks for future biological experiments. Predicting protein–protein interactions remains an active research area in bioinformatics (Qi et al. 2010). Among the protein interactions, intra-species protein–protein interactions (PPI) are one type of interactions observed within the same species. Besides these interactions, we are motivated to study inter-species PPIs to reveal interactions between proteins from different species. Specifically, host–pathogen (HP) interactions are considered as key infection processes at the molecular level with the associated infectious diseases representing major worldwide health concerns, which have caused millions of illnesses annually.
There has been an accumulation of experimentally verified PPI data generated through in vitro methods, including small-scale biochemical, biophysical, and genetic experiments, as well as large-scale methods, such as yeast-two-hybrid analysis. However, these methods are time consuming and require substantial biomedical resources. Additionally, many of the methods exhibit high false positive rates, and the occasional large number of potential interactions hinders the deployment of some in vitro methods.
Here, we will describe the development of a new method for HP-PPI prediction. Since host–pathogen protein–protein interactions reveal substantial information concerning HP-specific infection mechanisms, a better understanding on HP-PPIs and the application of computational methods to promote their prediction will assist in vitro experimental design. This study provides the following research contributions:
-
Design of a detailed workflow framework for applying data analytics through curation of the large HP-PPI datasets: multiple databases need substantial reviews, and data processing including different aspects and stages has to be involved.
-
Development of an unsupervised deep learning model is designed to handle the HP-PPI datasets, and the comparison against various supervised machine learning models indicates that our model achieves a best performance: the HP-PPI datasets present both small and large scales, and a highly skewed ratio between different classes exhibits a significant challenge for model learning.
Furthermore, the technical contribution from this study includes the framework implementation, which deals with the processes of data curation, data representation, and data storage, as well as the implemented machine learning models. The framework has detailed a complete life cycle for HP-PPI prediction task, in which our experimental performance emphasizes the potential improvement of recently promising deep learning models and data analytics techniques. In this study, our deep learning based model has also benefited from the utilization of graphic processing unit (GPU) as the primary computing resources, which facilitates a faster training speed of the underlying unsupervised learning model.
The remainder of this paper is organized as follows: Section 2 reviews related work, Section 3 presents the framework associated with the HP-PPI deep learning model, Section 4 discusses the HP-PPI dataset curation process and provides a brief introduction to the supervised machine learning model, Section 5 presents a detailed results analysis and discussion, and Section 6 evaluates the results and pinpoints future HP-PPI research directions.
2 Related work
As PPIs offer insights into molecular interactions and disease genes identification (Masood et al. 2018) for a specific species, such as yeast (Ito et al. 2001), biological experiments are being carried out to reveal or determine the interaction-specific relationships between proteins. In this regard, HP-PPIs could further assist revealing the information concerning infection pathways and providing additional insight from the interactions between host and pathogens (Chen et al. 2016).
However, a database targeting HP-PPI data does not exist yet. A previous review (Chen et al. 2016) detailed the research vision for HP-PPIs and it highlighted the importance of database construction. Several databases, including HPIDB (Kumar and Nanduri 2010), PATRIC (Wattam et al. 2013), PHISTO (Tekir et al. 2013), VirHostNet (Navratil et al. 2009) and VirusMentha (Calderone et al. 2014), represent the most relevant PPI repositories. Owing to these earlier research efforts, these databases provide well sorted and experimentally verified HP-PPI information. Nevertheless, these manually updated databases currently represent only a small quantity of all PPIs.
There have been several recent studies on host–pathogen protein–protein interactions (Kshirsagar et al. 2013a, 2015; Schleker et al. 2015; Kshirsagar et al. 2013b), with each testing a biological hypothesis that ‘similar pathogens target the same critical biological processes in the host’ through the use of learning models. These studies constructed a common structure using the pathway information to compute the similarities between different types of pathogens, with human considered as the primary host. One of these studies constructed a pairwise level multi-task model to combine two different tasks. A potential solution for combining more tasks in the multi-task model has been proposed in Kshirsagar et al. (2013b), where the term ‘Task’ describes a computational model used to predict interactions between a specific pathogen and host.
Since supervised machine learning models have been widely applied for diverse topics of biological data, such as the decision tree for lung carcinoma cancer prediction model (Varadharajan et al. 2018) and an lung cancer diagnosis system based on support vector machine (Prabukumar et al. 2019), the traditional supervised machine learning models have been utilized to facilitate PPI research. A previous study used two pathogen-human datasets as source tasks and a third one as a target task to build a transfer learning model. Two other studies described extreme learning machine (ELM) models, which aimed at obtaining faster training speeds and higher degrees of accuracy (You et al. 2014, 2013). Such a model was deployed via using a balanced intra-species PPI dataset. Additionally, one method using Naïve Bayes classification model was described in Zhang et al. (2012) and the results for a comprehensive study and prediction of PPIs on yeast and humans via three-dimensional structural information were presented. The algorithm (PrePPI) uses Bayesian statistics to derive relationships between structural information and other functional clues. This method yields over 30,000 high-confidence interactions for yeast and over 300,000 for humans (Zhang et al. 2012).
Given the potential in utilizing computational models, especially machine learning models, to facilitate the HP-PPI task, possible solutions have been widely discussed in Sen et al. (2016) and Soyemi et al. (2018). Without positioning verified databases and specific pathogens, a collection of traditional machine learning models has been assessed, including support vector machine, decision tree, Naïve Bayes and so on. Deep learning models, which have shown great power in protein structure prediction task (Gao et al. 2019; Panda and Majhi 2018), have also been included as very important categories of machine learning models for prediction of HP-PPIs. However, a comprehensive framework with detailed artefacts to illustrate data analytics and machine learning models for HP-PPIs is still needed. Meanwhile, how to leverage deep learning model to improve the performance comparing with traditional machine learning models is also lacking.
Regarding the protein information related to host and pathogen species, we mainly focus on protein sequences in this paper, which can be fetched from Uniprot database (UniProt et al. 2008). Since there is a limited amount of protein structure information and domain information, protein sequences information is also the most abundant information available. Nevertheless, the protein sequence information is the raw information, which is important to the subsequent distinct levels of protein structure and model learning. These biological data have allowed the researchers to achieve diverse implementations of encoding scheme (Shen et al. 2007; Dagher et al. 2019). The Uniprot database provides verified details for both hosts and pathogens.
Taking both verified updated databases and the protein sequence information, these data empower the construction of a ‘gold-standard dataset’, which includes positive and negative HP-PPIs, for researchers facilitating data analytics involving similarity reduction and data sampling. Herein, positive HP-PPIs define the physical contacts between proteins from host and pathogen, which activates the protein functions. Conversely, the negative HP-PPIs indicates that the proteins functions are inactivated accordingly. Normally, experimentally verified HP-PPIs from databases only provide the positive HP-PPIs; however, negative HP-PPIs are required for the consideration of supervised machine learning models.
Usually, a balanced dataset, with a nearly 1:1 ratio of positive and negative PPIs, is constructed for traditional model based learning techniques. However, for HP-PPIs, a dataset containing a 1:100 ratio is necessary to prevent a classifier biased towards inaccurate prediction based on a given biological scenario. With regard to these issues, a well-designed ratio is critical for constructions of an “HP-PPIs gold-standard dataset”. A previous study described random sampling of pathogenic and host protein in order to curate a negative HP-PPI dataset (Chen et al. 2016). So far, there is still a big gap in linking these researches.
In the following sections, we will describe our framework for HP-PPI dataset curation and propose a novel method, which achieves the state-of-the-art performance for prediction.
3 HP-PPI framework

A brief illustration of HP-PPI framework
Given the large number of databases, data analytics and learning models can contribute to HP-PPI research. Although previous studies provided a technical workflow for PPI research from various perspectives (Zhang et al. 2012; Kshirsagar et al. 2013a; You et al. 2013; Kshirsagar et al. 2013b; Schleker et al. 2015; Kshirsagar et al. 2015; Mei and Zhu 2015; You et al. 2014), a comprehensive and detailed framework for HP-PPI research involving data analytics, feature representation, and model learning does not exist currently.
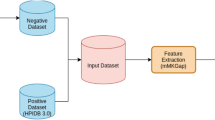
The framework for HP-PPI presented here includes activities related to data collection and manipulation, feature representation, and a machine learning model. We also introduce a new model to be jointly implemented for this framework, which helps boosting the performance. Fig. 1 depicts a brief structure of the framework.
Addressing HP-PPI research as a prediction task, we formulated the framework according to different steps involving data collection, assessment of data redundancy, data sampling, feature representation, and model learning. The framework targets on collecting high-quality data by removing redundancies and homologous data, and sampling negative data to allow construction of a gold-standard dataset. The feature representation and model learning would represent the predictive aspects of the method.
3.1 Data redundancy
Regarding the interdisciplinary nature of HP-PPI research, we have used multiple open-access databases to obtain protein–protein interactions data as well as the corresponding features. These databases play important roles at different stages in the data analytics step.
Several database repositories across both academia and industry, which contain only experimentally verified and positive HP-PPI data, are taken into consideration to prepare the protein interactions data for analyses. Additionally, these HP-PPI database are manually updated. As a few examples herein, HPIDB (Kumar and Nanduri 2010), PATRIC (Wattam et al. 2013), PHISTO (Tekir et al. 2013), VirHostNet (Navratil et al. 2009) and VirusMentha (Calderone et al. 2014) are several main repositories for HP-PPI research. Recently, Human Proteome Organization Proteomics Standards Initiative (HUPO-PSI) has also created the PSI-MI XML format to facilitate storage of PPI data in a single, unified format. In this study, the HP-PPI data was collected using XML format from several database repositories. For exhaustive learning and prediction of HP-PPI data, queries across several different database repositories is necessary to construct a positive HP-PPI datasets. Furthermore, the related protein information queried from Uniprot database is required to construct a negative HP-PPI dataset.
Construction of these datasets considered two levels of data redundancy, which exist from these preliminary database repositories. The first one is regarding the evaluation of redundancy. Since these various databases are maintained by different organizations, it is very likely that they contain duplicated information. These duplication needs to be identified and removed. The second level concerns sequence redundancy, which is more meaningful. Mostly, the homology relationship between different proteins needs to be considered, because the HP-PPI datasets contain the interaction pairs representing different pathogenic proteins interacting with the same host protein.
Sequence redundancy can be determined from various data sources and detected on different protein characteristics. As included in Fig. 1, Uniprot (UniProt et al. 2008), Gene Ontology Consortium (Gene Ontology et al. 2015) and the human protein reference database (HPRD) (Goel et al. 2012) provide protein sequence, gene ontology (GO) and human interactome graph information, respectively. To avoid classifier bias, the introduction of clustering method on these data is necessary to construct a dataset that minimizes the homology redundancy (Li and Godzik 2006). This is achieved by using the sequence information from Uniprot to obtain the protein clusters based on sequence similarity, which is as well termed as ‘CD-HIT’ (Li and Godzik 2006). Sequence redundancy represents the similarity between protein sequences and helps us to avoid the homology redundancy during collection of high-quality data, whereas GO terms allow separation of proteins according to molecular function (F), cellular component (C) and biological process (P). A previous study subsequently used ‘G-Sesame’ (Du et al. 2009) to determine similarities between two individual GO terms, which represented the similarity between two proteins according to these different properties.
3.2 Data sampling and representation
Following the collection of positive protein–protein interactions from various database repositories, the negative protein–protein interactions are also essential to build the supervised machine learning model. In this paper, we used a random sampling method to generate a negative PPI dataset.
As a result of the data analysis, we have obtained a HP-PPI dataset indicating only the identities of interacting proteins between host and pathogen. To input information related to each unique protein interaction into a learning model, feature representation is required, which includes sequence, gene ontology, interactome graph, and gene expression information. Additional to aforementioned several databases, i.e. Uniprot, Gene Ontology Consortium and HPRD, Gene Expression Omnibus (GEO) (Barrett et al. 2013) provide gene expression data, which include micro-array, next-generation sequencing (NGS) and other forms of high-throughput functional genomics data.
Since sequence information includes most information of the corresponding protein and is protein specific, in this study, we primarily use sequence information for feature representation, as described in previously researches (Kshirsagar et al. 2013a, b, 2015; Schleker et al. 2015; Mei and Zhu 2015; You et al. 2014, 2013; Zhang et al. 2012).
For different protein properties, it is required to represent the properties into a numerical form. In the past, numerous studies related to feature representation have been conducted for sequence information (Kshirsagar et al. 2013b; Shen et al. 2007; Guo et al. 2008; Davies et al. 2008; Du et al. 2009). The feature representation remains a hot and ongoing research area for bioinformatics researchers.
As sequence information allow unique information to be imported into the learning model, in this paper, we primarily discuss the feature representation with sequence information. The unique information include the different types of amino acids in different combination and various lengths. As said in ‘The amino acid sequence of a protein determines its three-dimensional structure’ (Berg et al. 2002), it also provides a widely adopted view that knowledge of the sequence information would be adequately feasible to represent a protein.
There are different strategies to categorize the amino acids types, which would thus introduce different feature representation methods. One is based on the differences in their electrostatic and hydrophobic properties. These 20 types of amino acids can be subsequently categorized into seven groups as shown in Fig. 2. Alternative strategy is based on their physicochemical properties. It typically considers the amino acids from seven corresponding physical and chemical characteristics, such as hydrophobicity, volumes of side chains and so on (Shen et al. 2007; Guo et al. 2008).

Groups of amino acids
Since the main goal is to build a supervised learning model for learning the dataset with supervised learning model for prediction, these positive interactions represent a higher quality and less bias dataset based on various well-maintained and manually updated HP-PPI database repositories. A brief introduction about data curation and feature representation methods for selected HP-PPI tasks for several infectious diseases is presented in Sect. 4.
3.3 Model learning
While HP-PPI dataset has been built, in this paper, we consider to deploy both the supervised and unsupervised machine learning models to learn and predict HP-PPIs based on the curated dataset. In addition to improve the performance by introducing new learning models, there have been studies focusing on incorporating more processing and more training on data, including data augmentation and newly developed strategy to obtain extended kernel functions for classification, given a dataset in areas such as cancer, which may benefit HP-PPIs prediction as well (Chaudhari et al. 2019; Wang et al. 2019). Moreover, interpretability is required in some kinds of analytics tasks, such as brain diseases analysis (Tomasiello 2019), to enhance the transparency of the model and retain the performance at the same time.
In this study, we primarily focus on building machine learning model for the binary classification task to infer the interaction relationship of HP-PPIs dataset with high performance. Especially, the benefit of introducing unsupervised deep learning model will be identified and discussed further.
Although supervised machine learning model is considered as the dominant classification model, the unsupervised deep learning model is introduced in this work to build a complementary feature representation, which also helps tuning multi-layer supervised model. As for learning models for comparison, we have simultaneously built several classic supervised machine learning models, including support vector machine (SVM), extreme learning machine (ELM) and Naïve Bayes Model, among others.
4 Learning HP-PPI
To evaluate the feasibility of the framework discussed in section 3, we present a detailed practice in this section. Specifically, two HP-PPI database repositories, PATRIC and PHISTO, were used for construction of the HP-PPI datasets. The benefit from these two databases is that, the hosted positive data are manually extracted and uploaded based on biological literature.
Table 1 shows statistics associated with the bacterium pathogen species used for construction of our datasets and used for model learning. After the data redundancy analysis, we have identified that, these three different bacterium pathogen species were retained containing the enough samples for model training and also in the best interest of infectious diseases for human. These datasets are corresponding to Clostridium difficile, Escherichia coli, and Bacillus anthracis in our study, as shown in Table 1, with the positive protein pairs numbers decreasing after redundancy analyses. Here, Clostridium difficile is the primary cause of the inflammation of the colon, Escherichia coli causes both minor and severe intestines illness and Bacillus anthracis is the etiologic agent of anthrax.
We used relatively small datasets that included 56 and 168 pairs of positive HP-PPIs in this paper, meanwhile, the large size dataset with 6073 pairs of positive HP-PPIs was also exploited. ‘CD-HIT’ was utilised to remove protein pairs with high homology information and as a result, the column under ‘CD-HIT Redundancy Removal’ indicates the final positive protein pairs statistics.
4.1 Feature representation
To avoid a large amount of missing data, we mainly used sequence information to represent protein properties, with Auto Covariance (AC) algorithm (Guo et al. 2008) as the first step of features representation methodology.
As one of the popular feature representation algorithms, AC is capable of transforming numerical vectors to uniform matrices based on sequence information. The representing matrices are having a same dimension after AC transformation regardless of protein sequence length. The steps of AC algorithm for sequence information is listed below.
-
Considering there are 20 different kinds of amino acids and each kind of amino acids exhibits 7 different physicochemical properties, a normalized matrix is acquired to present these information. Sequence information is subsequently translated into numerical values according to this matrix.
-
Given a max distance value D, we represent the numerical sequence information into a uniform matrix by following equation:
$$\begin{aligned} AC(d, j)= & {} \frac{1}{N-d} \sum \limits _{i=1}^{N-d}\left( f_{i,j}-\frac{1}{N}\sum \limits _{i=1}^{N}f_{i,j}\right) \nonumber \\&* \left( f_{i+d,j}-\frac{1}{N}\sum \limits _{i=1}^{N}f_{i,j}\right) \end{aligned}$$(1)
d is the distance between two amino acids and it ranges from 1 to D. \(f_{i,j}\) represents the corresponding jth value of ith amino acid and N is the length of the protein sequence. It calculates the auto cross covariance relationship within the sequence information, and represents the numerical sequence information to a scalar with \(D * 7\) length. In this study, \(D=30\) and the length of each vector was set to 210 for each protein, resulting in a pair-wise feature vector of 420 dimensions for each HP-PPI pair.
Mostly, the AC feature is fed into the following model for learning. However, in host–pathogen–protein interactions scenario, a highly skewed ratio between different classes and different scales of datasets are observed. As the unsupervised deep learning model helps to construct higher level features and initiate a deep neural network in a better state, we are motivated to build an unsupervised deep learning model based on stacked denoising autoencoders to achieve a boost performance comparing with traditional models. The following sections will discuss the details.
4.2 HP-PPI dataset statistics
The ratio of positive and negative pairs was set at 1:100 to align with experiment scenarios, which was normally considered to yield less bias in predictions (Table 1).
We further evaluated the learning models by 10-fold cross validation after dividing the HP-PPI datasets into training and test datasets. Details are listed in Table 2.
4.3 Learning models
We deployed a deep learning model as our primary model for model learning and prediction. Meanwhile, several general supervised learning models were also implemented for comparison, including a linear-kernel support vector machine (SVM), ELM, naïve Bayes and decision tree models.
4.3.1 Support vector machine
SVMs (Cortes and Vapnik 1995) aim to achieve minimal structural risk to achieve optimal performance. It has been successfully applied to many real world problems. In our study, SVM was designed to classify the interaction relationship according to a given dataset of HP-PPIs denoted as \(\{x_i,y_i\}\), \(\hbox {i}=1,2,...,\hbox {N}\), where \(x_i \in R^n\), and \(y_i \in \{+1,-1\}\).
4.3.2 Extreme learning machine
An ELM allows high degrees of accuracy, and also minimizes the running time required to train the classification model. ELM (Huang et al. 2006) is considered to bring these advantages with its operation based on the Moore–Penrose definition of this model.
Given \((x_i,y_i)\), where \(x_i = [x_{i1},x_{i2},...,x_{in}]^T\) and \(y_i = [y_{i1},y_{i2},\) \( ...,y_{im}]\), the learning procedure is presented below with a hidden neuron layer, L:
-
STEP 1
Fix the input weight \(w_i\) and bias \(b_i\), i = 1,...,L
-
STEP 2
Calculate the hidden neurons output H
-
STEP 3
Update \(\beta \) according to \(\beta = H^* Y\), where \(H^*\) is the Moore–Penrose generalized inverse of the hidden neuron output, and Y is the matrix \(y_i\)

The whole model based on SdA
4.3.3 Naïve Bayes model
Naïve Bayes model is a member of a family of simple probabilistic classifiers based on Bayes’ theorem (Zhang 2004; Wikipedia 2017b) and was derived from conditional probability theory.
Given that \(X = (x_1, x_2, x_3, ..., x_n)\), and \(x_i\) represents the \(i_{th}\) feature, Bayes’ model delivers the probability corresponding to the \(k_{th}\) category \(y_k\):
The final prediction is based on the maximum probability assigned to \(y_k\):
In the naïve Bayes model, the features are considered as independent between each other; therefore:
The naïve Bayes model used in this study is Gaussian naïve Bayes. Since we are dealing with continuous data, the data is assumed to distribute according to a Gaussian distribution. Computing \(\mu _k\) and \(\sigma _k^2\) as the mean and variance, respectively, of X associated with \(y_k\), we use the following equation:
4.3.4 Decision tree
A decision tree is considered a non-parametric supervised model (Wikipedia 2017a). It renders a tree-like model capable of predicting an incoming instance based on learned decision rules from known data features. Decision trees are simple to understand and interpret, while it is also capable of handling both numerical and categorical data.
4.3.5 Stacked denoising autoencoder
Deep learning models have achieved good performance on both classification and regression tasks, suggesting their generalized utility for learning relationships from data (LeCun et al. 2015; Min et al. 2017; Yan et al. 2018; Gao et al. 2019; Panda and Majhi 2018). These models have shown that, deep learning models are capable of learning protein structure prediction task in a more efficient way, and can achieve better performance than the other models.
In this study, we are motivated to introduce another group of unsupervised deep learning model, denoising autoencoder (dA), which represents features via a deep neural network. Denoising autoencoder (Vincent et al. 2008) is a training model used for unsupervised learning. It is motivated from general autoencoder and is capable of reconstructing original input from corrupted input. Additionally, the denoising autoencoder could be stacked as stacked denoising autoencoders (SdA) to build a multi-layer network (LeCun et al. 2015).
As a primary unsupervised learning model, a stacked denoising autoencoders can construct higher level features to allow for a better initial state in the deep learning model. Herein, we applied an SdA as the unsupervised model to learn from the curated datasets comprising three different bacterial species, whereas at the top layer, we choose logistic regression (LR) (Hilbe 2009) as our classification model. We subsequently fine-tuned the network to achieve better performance than simply training the network in two separate stages (Chen et al. 2017).
Technically, we corrupted the input by adding small amounts of noise, in which both Gaussian noise and ‘mask’ noise are feasible. The integrated model is depicted in Fig. 3.
We applied this four-layer network to learn and predict from our curated datasets. It has a similar architecture as that of a previously described model (Chen et al. 2017); however, we fine-tuned the network following initial training using LR Layer. The architecture of this network is as follows: input layer (420 input nodes) \(\rightarrow \) dA layer1 (210 neurons) \(\rightarrow \) dA layer2 (210 neurons) \(\rightarrow \) dA layer3 (210 neurons) \(\rightarrow \) LR layer (1 output node).

The denoising autoencoder layer
In Fig. 4, we describe the details of construction of the denoising autoencoder layer. In Fig. 4, the \(\ddot{X}\) is the corrupted input data from X. For our experiments, we ended up with choosing only Gaussian noise as it achieved better performance over \(\ddot{X}\) wit ‘mask’ noise. The encoding process and decoding process is given as:
The dA layer trains each layer as an individual component first, followed by output of the learned data, Y, to subsequent layers. The learned parameters, W, are maintained and will be applied to the entire network during subsequent fine-tuning steps. Each layer is pre-trained using the same process.
The logistic regression layer is our final classification layer. For a binary classification problem, \(y_i = {0,1}\), where i represents the ith example, the LR model returns the result according to the following:
Here, \(\theta \) represents the model parameters. The cost function applied in logistic regression model is:
After pre-training the different layers, we fine-tuned the overall network using a back propagation algorithm. In the next section, we will discuss our experiment evaluation results as well as the compiling environment.
5 Results and discussion
With the curated ‘HP-PPI gold-standard dataset’, we anticipate to evaluate and compare these learning models performance. We applied and implemented the SdA, SVM, ELM, decision tree, naïve Bayes and also logistic regression based on ‘Tensorflow’ (Abadi et al. 2015), ‘libsvm’ (Chang and Lin 2011), ‘hpelm’ (Akusok et al. 2015) and ‘scikit-learn’ (Pedregosa et al. 2011).
Training deep learning model on big datasets highly relies on specific structures, such as GPU/TPU/FPGA, to decrease the running time and finalise the parallel processing tasks. In this regard, our computing resources system is built upon ‘NVIDIA GTX 1080Ti’ GPU and 64GB RAM, which allowed efficient parallelization computing. The working operating system is Ubuntu 16.04. In this study, all framework implementations were written in Python.
5.1 Primary results
To evaluate the performance and robustness of the models, experiments were conducted using 10-fold cross validation. The evaluation results are presented as the mean and variance in terms of precision, recall values, F1 score, and accuracy. It should be noted that the accuracy measurement might not fully reflect the performance of these models, because the datasets are highly skewed. However, we have reported these results for completeness. The precision value represents the fraction of retrieved information relevant to the result, whereas the recall value represents the ratio of successful retrievals by the learning model. These are critical factors necessary to determine system performance, specifically on an imbalanced dataset.
Basic calculations of precision and recall values are as follows:
Here,“TP” represents the true positive number, “FP” is the false positive number and “FN” is the false negative number. The precision and recall values are further used to calculate a harmonic average, which is subsequently termed as F1 score to provide a final measurement for a given model. Normally, the F1 score is ranging between 0 and 1. It reaches the best performance at 1 while worst at 0. The F1 score is calculated as follows:
We initially calculated precision and recall values for all of the models. Table 3 shows the statistics associated with precision results, Table 4 for the recall results, Table 5 for the F1 results and Table 6 for the accuracy results. In these tables, ‘SVM’ refers to linear-kernel SVM, ‘ELM’ represents to extreme learning machine while ‘SdA’ is the stacked denoising autoencoders model, ‘Gaussian NB’ indicates Gaussian Naïve Bayes, ‘DT’ refers to decision tree model and ‘LR’ is logistic regression model.

Learning models ROC Curve on Bacillus anthracis
According to these measurements, the SdA model achieved the best performance on F1 score as well as accuracy for HP-PPI prediction for Clostridium difficile, Escherichia coli and Bacillus anthracis. Specifically, the SdA model outperformed the LR model in terms of F1 score and accuracy, indicating that the unsupervised learning model presented a better feature learning capability and resulted in an improved predictive performance.
Although model performances on different datasets are varied, the SdA model retains the best performance among all the models.
5.2 Area under the receiver operating characteristic (ROC) curve (AUC) Analysis
The results of receiver operating characteristic (ROC) and the area under ROC curve (AUC) value analysis are shown in Fig. 5, Fig. 6, Fig. 7 and Table. 7.

Learning models ROC curve on Clostridium difficile

Learning models ROC curve on Escherichia coli
The ROC results illustrate the classification ability of binary HP-PPI prediction according to various discrimination thresholds. It was plotted based on different settings of TP rates against FP rates. The AUC value ranges between 0 and 1 with higher values indicating a better classification performance.
Moreover, it is worth noting that ELM model achieves better AUC value on smaller datasets based on the comprehensive results from Table7. It achieves AUC values of 0.9997 for C. difficile and 0.9448 for E. coli. However, across all three tasks, the SdA model presents a more stable performance (0.9985 for C. difficile, 0.9431 for E. coli and 0.9250 on B. anthracis). From Table 7, it is observed that the performance of SdA model on B. anthracis specie is much better than the others, including the followings from decision tree model (0.8314) and ELM model (0.8157).
5.3 Learning and convergence curves
Regarding learning and convergence curve, the related comparison results are presented in Fig. 8. The convergence curve represents the relationship between the training epoch and global loss, with a lower global loss suggesting the closeness of the model to the optimal state.

Convergence curve
Fig. 8 shows the convergence curves for logistic regression and SdA model, with pre-training step for the SdA model initially applied in the SdA layers, after which the output of the last SdA layer is used as input for the logistic regression layer. Our results indicated that the training iterations needed for the SdA model for C. difficile and E. coli HP-PPI prediction were much less than those needed for training the LR model. Retaining the parameters from the pre-training step in the SdA layers improved the convergence speed and aided the efficient realization of the optimal state.
6 Conclusion
In this study, we presented a comprehensive framework for HP-PPI prediction and described a SdA-based based deep learning model for HP-PPI datasets. The framework considered information derived from various data sources, and it applied a learning model to build a workflow-like system to predict HP-PPI. Comparison of the SdA model with other models indicated its superiority for this application.
A well-designed framework capable of utilizing open-source resources is critical for HP-PPI specific research and promotes high-fidelity prediction results for biologists. This framework will facilitate the exploration and understanding of HP-PPI networks, and offer critical insights of infectious mechanisms between host and pathogen. Since data continues to accumulate rapidly, a suitable learning model for HP-PPI prediction is demanded. Here, we have evaluated curated datasets using several different supervised learning models. We have found that, the unsupervised SdA model is optimal for the highly skewed and big datasets and is better at feature representation if compared to other models. Additionally, model convergence speed has benefited from the unsupervised learning technique and the usage of GPU. Our results suggested that, the deep learning model was capable of dealing with big HP-PPI datasets.
Our future research will continue to investigate the application of data analytic techniques, including the mentioned data augmentation and new algorithms to deal with data processing, for HP-PPI prediction. This effort will also include the use of larger datasets with a higher degrees of dimensionality in feature representations, including broader use of GO data, interactome data and gene expression data.
References
Abadi M, Agarwal A, Barham P, Brevdo E, Chen Z, Citro C, Corrado GS, Davis A, Dean J, Devin M, et al (2015) Tensorflow: large-scale machine learning on heterogeneous systems, 2015. Software available from tensorflow. org, 1
Akusok A, Björk K-M, Miche Y, Lendasse A (2015) High-performance extreme learning machines: a complete toolbox for big data applications. IEEE Access 3:1011–1025
Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M et al (2013) Ncbi geo: archive for functional genomics data sets–update. Nucleic Acids Res 41(D1):D991–D995
Berg JM, Tymoczko JL, Stryer L (2002) Biochemistry. Freeman, New York. ISBN-10: 0-7167-3051-0
Calderone A, Licata L, Cesareni G (2014) VirusMentha: a new resource for virus-host protein interactions. Nucleic Acids Res 43(D1):D588–D592
Chang C-C, Lin C-J (2011) Libsvm: a library for support vector machines. ACM Trans Intell Syst Technol (TIST) 2(3):27
Chaudhari P, Agarwal H, Bhateja V (2019) Data augmentation for cancer classification in oncogenomics: an improved KNN based approach. Evol Intell. https://doi.org/10.1007/s12065-019-00283-w
Chen H, Shen J, Wang L, Song J (2016) Towards data analytics of pathogen–host protein–protein interaction: a survey. In: 2016 IEEE International Congress on Big Data (BigData Congress), IEEE, pp 377–388
Chen H, Shen J, Wang L, Song J (2017) Leveraging stacked denoising autoencoder in prediction of pathogen–host protein–protein interactions. In: 2017 IEEE International Congress on Big Data (BigData Congress), IEEE, pp 368–375
Cortes C, Vapnik V (1995) Support-vector networks. Mach Learn 20(3):273–297
Dagher GG, Machado AP, Davis EC, Green T, Martin J, Ferguson M (2019) Data storage in cellular DNA: contextualizing diverse encoding schemes. Evol Intell. https://doi.org/10.1007/s12065-019-00202-z
Davies MN, Secker A, Freitas AA, Clark E, Timmis J, Flower DR (2008) Optimizing amino acid groupings for GPCR classification. Bioinformatics 24(18):1980–1986
Du Z, Li L, Chen C-F, Philip SY, Wang JZ (2009) G-SESAME: web tools for GO-term-based gene similarity analysis and knowledge discovery. Nucleic Acids Res 37(Suppl_2):W345–W349
Gao M, Zhou H, Skolnick J (2019) Destini: a deep-learning approach to contact-driven protein structure prediction. Sci Rep 9(1):3514
Gene Ontology C et al (2015) Gene ontology consortium: going forward. Nucleic Acids Res 43(D1):D1049–D1056
Goel R, Harsha H, Pandey A, Prasad TK (2012) Human protein reference database and human proteinpedia as resources for phosphoproteome analysis. Mol BioSyst 8(2):453–463
Greene CS, Tan J, Ung M, Moore JH, Cheng C (2014) Big data bioinformatics. J Cell Physiol 229(12):1896–1900
Guo Y, Yu L, Wen Z, Li M (2008) Using support vector machine combined with auto covariance to predict protein–protein interactions from protein sequences. Nucleic Acids Res 36(9):3025–3030
Hilbe JM (2009) Logistic regression models. CRC Press, USA
Huang G-B, Zhu Q-Y, Siew C-K (2006) Extreme learning machine: theory and applications. Neurocomputing 70(1):489–501
Ito T, Chiba T, Ozawa R, Yoshida M, Hattori M, Sakaki Y (2001) A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc Natl Acad Sci 98(8):4569–4574
Kshirsagar M, Carbonell J, Klein-Seetharaman J (2013a) Multisource transfer learning for host–pathogen protein interaction prediction in unlabeled tasks. NIPS Work Mach Learn Comput Biol 1:3–6
Kshirsagar M, Carbonell J, Klein-Seetharaman J (2013b) Multitask learning for host–pathogen protein interactions. Bioinformatics 29(13):i217–i226
Kshirsagar M, Schleker S, Carbonell J, Klein-Seetharaman J (2015) Techniques for transferring host-pathogen protein interactions knowledge to new tasks. Front Microbiol 6:36
Kumar R, Nanduri B (2010) Hpidb—a unified resource for host–pathogen interactions. BMC Bioinf 11(6):1
LeCun Y, Bengio Y, Hinton G (2015) Deep learning. Nature 521(7553):436–444
Li W, Godzik A (2006) Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22(13):1658–1659
Masood MMD, Manjula D, Sugumaran V (2018) Identification of new disease genes from protein–protein interaction network. J Ambient Intell Human Comput. https://doi.org/10.1007/s12652-018-0788-1
Mei S, Zhu H (2015) A novel one-class svm based negative data sampling method for reconstructing proteome-wide htlv–human protein interaction networks. Sci Rep 5:8034
Min S, Lee B, Yoon S (2017) Deep learning in bioinformatics. Brief Bioinf 18(5):851–869
Navratil V, de Chassey B, Meyniel L, Delmotte S, Gautier C, André P, Lotteau V, Rabourdin-Combe C (2009) Virhostnet: a knowledge base for the management and the analysis of proteome-wide virus–host interaction networks. Nucleic Acids Res 37(suppl 1):D661–D668
Panda B, Majhi B (2018) A novel improved prediction of protein structural class using deep recurrent neural network. Evol Intell. https://doi.org/10.1007/s12065-018-0171-3
Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V et al (2011) Scikit-learn: machine learning in python. J Mach Learn Res 12(Oct):2825–2830
Prabukumar M, Agilandeeswari L, Ganesan K (2019) An intelligent lung cancer diagnosis system using cuckoo search optimization and support vector machine classifier. J Ambient Intell Hum Comput 10(1):267–293
Qi Y, Tastan O, Carbonell JG, Klein-Seetharaman J, Weston J (2010) Semi-supervised multi-task learning for predicting interactions between hiv-1 and human proteins. Bioinformatics 26(18):i645–i652
Savage N (2014) Bioinformatics: big data versus the big c. Nature 509(7502):S66–S67
Schleker S, Kshirsagar M, Klein-Seetharaman J (2015) Comparing human–Salmonella with plant–Salmonella protein–protein interaction predictions. Front Microbiol 6:45
Sen R, Nayak L, De RK (2016) A review on host-pathogen interactions: classification and prediction. Eur J Clin Microbiol Infect Dis 35(10):1581–1599
Shen J, Zhang J, Luo X, Zhu W, Yu K, Chen K, Li Y, Jiang H (2007) Predicting protein–protein interactions based only on sequences information. Proc Natl Acad Sci 104(11):4337–4341
Soyemi J, Isewon I, Oyelade J, Adebiyi E (2018) Inter-species/host–parasite protein interaction predictions reviewed. Curr Bioinf 13(4):396–406
Tekir SD, Çakır T, Ardıç E, Sayılırbaş AS, Konuk G, Konuk M, Sarıyer H, Uğurlu A, Karadeniz İ, Özgür A et al (2013) Phisto: pathogen–host interaction search tool. Bioinformatics 29(10):1357–1358
Tomasiello S (2019) A granular functional network classifier for brain diseases analysis. Comput Methods Biomech Biomed Eng Imaging Vis. https://doi.org/10.1080/21681163.2019.1627910
UniProt C et al (2008) The universal protein resource (uniprot). Nucleic Acids Res 36(suppl 1):D190–D195
Varadharajan R, Priyan MK, Panchatcharam P, Vivekanandan S, Gunasekaran M (2018) A new approach for prediction of lung carcinoma using back propogation neural network with decision tree classifiers. J Ambient Intell Human Comput. https://doi.org/10.1007/s12652-018-1066-y
Vincent P, Larochelle H, Bengio Y, Manzagol P-A (2008) Extracting and composing robust features with denoising autoencoders. In: Proceedings of the 25th international conference on Machine learning, ACM, pp 1096–1103
Wang F, Liu S, Ni W, Xu Z, Qiu Z, Wan Z, Pan Z (2019) Imbalanced data classification algorithm with support vector machine kernel extensions. Evol Intell 12(3):341–347
Wattam AR, Abraham D, Dalay O, Disz TL, Driscoll T, Gabbard JL, Gillespie JJ, Gough R, Hix D, Kenyon R et al (2013) PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res 42(D1):D581–D591
Wikipedia (2017) Decision tree. Accessed 12 Dec 2017
Wikipedia (2017) Naive bayes classifier. Accessed 12 Dec 2017
Yan C, Xie H, Yang D, Yin J, Zhang Y, Dai Q (2018) Supervised hash coding with deep neural network for environment perception of intelligent vehicles. IEEE Trans Intell Transp Syst 19(1):284–295
You Z-H, Lei Y-K, Zhu L, Xia J, Wang B (2013) Prediction of protein-protein interactions from amino acid sequences with ensemble extreme learning machines and principal component analysis. BMC Bioinf 14(8):1
You Z-H, Li S, Gao X, Luo X, Ji Z (2014) Large-scale protein-protein interactions detection by integrating big biosensing data with computational model. BioMed Res Int. https://doi.org/10.1155/2014/598129
Zhang H (2004) The optimality of naive Bayes. AA 1(2):3
Zhang QC, Petrey D, Deng L, Qiang L, Shi Y, Thu CA, Bisikirska B, Lefebvre C, Accili D, Hunter T et al (2012) Structure-based prediction of protein–protein interactions on a genome-wide scale. Nature 490(7421):556–560
Acknowledgements
This work was supported by a scholarship from the China Scholarship Council (CSC) while the first author pursues his PhD degree in University of Wollongong, Australia. The first and second authors were also supported by UGPN RCF 2019 to visit University of Surrey to strengthen the algorithmic part and we wish to extend our deepest gratitude to Prof. Yaochu Jin for his valuable suggestions and supports in this paper.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Chen, H., Shen, J., Wang, L. et al. A framework towards data analytics on host–pathogen protein–protein interactions. J Ambient Intell Human Comput 11, 4667–4679 (2020). https://doi.org/10.1007/s12652-020-01715-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12652-020-01715-7