
Abstract
Inclusion body myositis is the most common acquired myopathy after the age of 50. It is characterized by progressive asymmetric weakness predominantly affecting the quadriceps and/or finger flexors. Loss of ambulation and dysphagia are major complications of the disease. Inclusion body myositis can be associated with cytosolic 5′-nucleotidase 1A antibodies. Muscle biopsy usually shows inflammatory cells surrounding and invading non-necrotic muscle fibers, rimmed vacuoles, congophilic inclusions, and protein aggregates. Disease pathogenesis remains poorly understood and consists of an interplay between inflammatory and degenerative pathways. Antigen-driven, clonally restricted, cytotoxic T cells represent a main feature of the inflammatory component, whereas abnormal protein homeostasis with protein misfolding, aggregation, and dysfunctional protein disposal is the hallmark of the degenerative component. Inclusion body myositis remains refractory to treatment. Better understanding of the disease pathogenesis led to the identification of novel therapeutic targets, addressing both the inflammatory and degenerative pathways.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Inclusion body myositis (IBM) is the most common acquired myopathy after the age of 50, with a varying reported prevalence averaging 24.8 to 45.6/1,000,000 [1]. IBM has a distinctive clinical phenotype and histopathological findings. Despite the inflammatory infiltrate on muscle biopsy, IBM remains refractory to immunotherapy. Although IBM does not usually affect longevity, patients can be markedly disabled at advanced stages, which markedly affects their quality of life and is associated with high economic burden [2]. This resulted in a continuous strive to better understand the disease pathogenesis, and identify novel therapeutic targets.
Clinical Findings
Classically, IBM presents with progressive insidious weakness, often asymmetric, predominantly affecting the quadriceps and/or finger flexors [3] (Fig. 1). Although none of the clinical findings in isolation is specific for IBM, weakness of knee extension more than hip flexion, and finger flexion more than finger extension, strongly raise the suspicion for this disorder. Other commonly involved muscles include the biceps, triceps, anterior leg compartment, and facial and swallowing muscles with dysphagia reported in about half of the patients [4, 5]. Less commonly, IBM can present with respiratory insufficiency, camptocormia, dysphagia, or facial weakness [5,6,7,8].

Clinical characteristics of inclusion body myositis. (A) Patient attempting to make a fist with both hands: asymmetric weakness of finger flexors, severe on the left. (B) Patient in a wheelchair with severe quadriceps weakness and atrophy
Pathological Findings
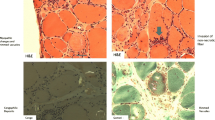
The pathological features of IBM are described in Fig. 2. On muscle biopsy, IBM is characterized by the presence of an inflammatory exudate, predominantly endomysial, where the inflammatory cells surround and focally invade non-necrotic muscle fibers. Besides inflammation, IBM is characterized by the presence of vacuoles rimmed by a membranous cytoplasmic material (rimmed vacuoles), atrophic fibers, as well as congophilic inclusions that may be intra- or extravacuolar. Congophilic inclusions are more easily detected by Congo red staining viewed under rhodamine optics rather than polarized light. Mitochondrial changes, especially an increased number of cytochrome c oxidase negative fibers, are observed in the vast majority of IBM patients [9]. Therefore, the absence of cytochrome c oxidase negative fibers should raise doubts about the diagnosis of IBM. This can be helpful in muscle specimens lacking rimmed vacuoles in which differentiating IBM from other inflammatory myopathies such as dermatomyositis or polymyositis may be challenging [9, 10]. One of the potential issues with this approach is the lack of agreement on upper limit for the percentage of cytochrome c oxidase negative in normally aged muscle. Some experts, however, consider values at least exceeding 2% as the threshold for excessive cytochrome c oxidase negative fibers with aging [11]. Further studies to identify a cutoff value for the percentage of COX−/SDH+ fibers that is sensitive and specific to IBM, as compared to PM, DM, and normally aged muscles, would be extremely helpful and may facilitate future introduction of mitochondrial changes as part of clinical or research diagnostic criteria for IBM. Eosinophilic inclusions can be seen in about half of the specimens [4]. Electron microscopy can help in identifying filamentous inclusions in the proximity of vacuolated fibers and, less commonly, 10 to 14 nm intranuclear inclusions [4].

Histopathological features of inclusion body myositis. (A, B) Hematoxylin & eosin stain: (A) predominantly endomysial inflammatory infiltration; (B) inflammatory cells invading a non-necrotic muscle fiber (arrow) splitting off a small portion of the fiber (arrowhead) and an adjacent necrotic fiber (star). (C) Acid phosphatase stain: mononuclear cells (likely lymphocytes) invading a non-necrotic muscle fiber (arrowhead), backed up by a macrophage (arrow) identified by its acid phosphatase reactivity, as well as myriad endomysial inflammatory cells, some of which are macrophages, surrounding and occasionally focally invading muscle fibers. (D) Trichrome stain: rimmed vacuoles (star). (E) Cytochrome c oxidase stain: multiple cytochrome c oxidase negative fibers (star) in various regions of the specimen. (F) Congo red stain viewed under rhodamine optics: 2 fibers with intravacuolar congophilic inclusions (arrowheads) and 1 fiber with extravacuolar congophilic inclusions (star)
Most proposed IBM diagnostic criteria heavily relied on pathological findings. The Griggs–Barohn 1995 criteria consisted of 2 categories: definite and possible IBM, both requiring the presence of endomysial inflammation with invasion of non-necrotic muscle fibers by mononuclear cells [12]. Further evidence of vacuolated muscle fibers, and either intracellular amyloid deposits or 15 to 18 nm tubulofilaments on electron microscopy, was required for the definite IBM category. In the MRC 2010 criteria, increased MHC-I expression on the surface of intact muscle fibers was added to the pathologic features. While the criteria for definite IBM (pathologically defined IBM) remained unchanged since the Griggs–Barohn 1995 criteria, clinically defined IBM and possible IBM categories required at least 1 of the following pathological features: invasion of non-necrotic fibers by mononuclear cells, rimmed vacuoles, or increased MHC-I expression on the surface of intact muscle fibers [13]. Later on, demonstrating abnormal sarcoplasmic deposition of Tar-DNA binding protein-43 (TDP-43) or p62 via immunohistochemical staining was shown to enhance the sensitivity of a muscle biopsy for the diagnosis of IBM [14,15,16]. Therefore, the ENMC 2011 criteria expanded the pathological criteria to include the “presence of protein accumulation” criterion which can be fulfilled by demonstrating the presence of either intracellular amyloid deposit, or deposit of other proteins demonstrated via immunostaining with antibodies to p62, SMI-31 (phosphorylated tau marker), or TDP-43 [17].
Laboratory Testing
The variability of the clinical and histopathological findings, often resulting in delay in diagnosis, prompted the search for a serological biomarker and the identification of cytosolic 5′-nucleotidase 1A (cN-1A) antibodies [18,19,20]. cN-1A is a protein involved in nucleic acid metabolism. The role of cN-1A antibodies in IBM pathogenesis is unknown. Tawara et al. [21] reported that passively immunized mice with sera from cN-1A-positive IBM patients demonstrate p62-positive sarcoplasmic aggregates associated with macrophages infiltration. It is also unclear whether there is a difference in phenotype or response to immunotherapy in patients with IBM based on their cN-1A serological status [21,22,23]. In a small cohort of 25 patients, cN-1A seropositive patients took longer to get up and stand, whereas there was no significant difference on the 6-min walk with the seronegative group [23]. In this study, the cN-1A seropositive group was reported to have more significant bulbar involvement; however, this finding was not reproduced in a subsequent cohort [21]. A single study evaluated mortality risk based on cN-1A serological status and found a higher adjusted mortality in seropositive IBM patients [24]. Elevated cN-1A antibodies are reported to be 33 to 76% sensitive and 92 to 96% specific for IBM [19, 20]. Despite the initially claimed high specificity, cN-1A antibodies were later reported in non-IBM patients with various autoimmune disorders: Sjögren’s syndrome (23-36%), systemic lupus erythematosus (14-20%), and dermatomyositis (15%) [22, 25]. Therefore, the presence of elevated cN-1A antibodies should be interpreted with caution, taking into consideration the clinical context and histopathological findings.
Creatine phosphokinase levels are very variable ranging from normal to up to 15 times upper limit of normal.
Needle electromyography usually shows increased spontaneous activity and fibrillation potentials, associated with short duration, low-amplitude, motor unit potentials often mixed with long duration, high-amplitude motor unit potentials [4]. Iterative discharges such as complex repetitive discharges and myotonic discharges could also be observed [26]. As muscle involvement can be patchy, we make sure to include needle examination of the deep finger flexors when IBM is suspected.
Diagnosis
To better define inclusion criteria for clinical trials, there have been multiple proposed diagnostic criteria over the years [12, 27, 28]. Despite the lack of effective treatment for IBM, a timely diagnosis is also important in clinical practice for patient’s counseling and to avoid unnecessary immunosuppression, that maybe attempted in patients diagnosed with polymyositis. Lloyd et al. [29] evaluated the sensitivity and specificity of all the published diagnostic criteria: all the categories had very high specificity (98-100%), whereas the sensitivity lagged behind ranging from 11 to 84%. In this study, “probable IBM” category from the ENMC 2011 criteria had the best sensitivity of 84%. The ENMC 2011 criteria consist of 3 diagnostic categories for research purposes: clinicopathologically defined IBM, clinically defined IBM, and probable IBM (Table 1) [17]. Clinically defined IBM category includes patients with weakness in the quadriceps muscles more than hip flexors, as well as in finger flexors more than shoulder abductors. In this case, patients are required to have at least 1 of the following pathological features: endomysial inflammation, rimmed vacuoles, increased MHC-I, 15 to 18 nm filaments, or accumulation of amyloid or other proteins. The sensitivity of “clinicopathologically defined IBM” was reported as 15% and clinically defined IBM as 57% [29]. Clinical guidelines for diagnosis and management of IBM are yet to be published [30].
Pathogenesis
Inflammatory Pathways
IBM is characterized by the presence of inflammatory cells surrounding and focally invading non-necrotic muscle fibers. The muscle fibers are invaded by mostly cytotoxic CD8+ T cells with some macrophages and surrounded by CD4+ T cells and macrophages, indicating a well-orchestrated immune attack [31]. Endomysial T cells display a restricted expression of T-cell receptor gene usage that persists over time [32]. When compared to peripheral blood lymphocytes, endomysial T cells express a significantly higher degree of restriction, suggesting local antigen-driven stimulation recruiting peripheral T-cell lymphocytes to expand in situ [33, 34]. Furthermore, myeloid dendritic cells, serving as antigen-presenting cells, are found in abundance in IBM muscle samples in close proximity to T cells [35]. These findings indicate that the activation of T cells is an antigen-driven response. The role of the humoral response in IBM remained unexplored for a while. Recognition of antigen-directed, clonally expanded plasma cells in IBM muscles [36, 37] was followed, shortly after, by the identification of cN-1A antibodies. Furthermore, expression of MHC-I by non-necrotic muscle fibers led to the discovery of susceptibility regions in the HLA genes as will be detailed in the “Genetics” section.
The association of inclusion body myositis with viral infections such as hepatitis C virus (HCV) or HIV remains controversial. The frequency of HCV antibodies in IBM patients was reported at 28% in Japan but only 3.3% in Brazil (1 out of 30 IBM patients, but it is unclear how many patients were screened for HCV) [38, 39]. Subsequently, Tawara et al. [21] reported that only 4.5% of Japanese IBM patients with positive cN-1A antibodies had concomitant HCV antibodies, compared with 26.5% in the cN-1A seronegative IBM group (p = 0.036). Moreover, the increased incidence of HCV in IBM patients has not been reported yet outside of Japan. Similarly, an association between IBM and HIV infection has been suggested by reported cases of HIV patients who then developed IBM [40, 41], with muscle biopsy showing clonal expansion of viral-specific CD8+ cells in the endomysium [41]. However, these patients displayed the same histopathological features of IBM as in HIV-negative patients, and there was no evidence of expression of viral-specific antigens within the muscle fibers.
In 1 study of 38 patients with IBM, 58% of patients had aberrant populations of large granular lymphocytes in their blood, fulfilling criteria for T-cell large granular lymphocytic leukemia (T-LGL leukemia) [42]. T-LGL leukemia is a rare disorder with a wide spectrum of severity, ranging from benign chronic lymphocytic proliferation to malignancy, and is commonly associated with autoimmune diseases [43]. It is unclear whether the aberrant population of LGL plays a primary role in IBM pathogenesis or is just an innocent bystander resulting from chronic antigenic stimulation [44]. From a hematological perspective, the presence of a clonal expansion of large granular lymphocytes does not necessarily require treatment [45]. Therefore, there is no clear indication yet to routinely screen for T-LGL leukemia in all IBM patients. Nonetheless, a complete blood count with a peripheral blood smear could be considered first looking for cytopenia, anemia, lymphocytosis, or excess of large granular lymphocytes that may warrant further investigation via flow cytometry.
Degenerative Pathways
The pathological evidence of rimmed vacuoles with abnormal protein aggregation and deposition of congophilic inclusions within the muscle fibers, in association with mitochondrial dysfunction, supports the presence of a degenerative component. This is further substantiated by the lack of response to immunomodulatory therapy. Protein inclusions in IBM contain a wide array of proteins, mostly associated with neurodegenerative disorders such as amyloid-β peptides, ubiquitin, phosphorylated tau, TDP-43, and prion protein [14, 46,47,48]. Similar to Alzheimer’s disease, amyloid-β peptides, including amyloid-β42, can aggregate within the muscle fibers, with a potential cytotoxic role suggested by the presence of Aβ42 oligomers in IBM muscles [49,50,51]. However, amyloid-β deposit may be of nonspecific significance, and elevated amyloid-β42 level is also found in the serum of patients with dermatomyositis [52].
Protein aggregation is the result of abnormal protein homeostasis in muscle (proteostasis) which encompasses abnormal protein production, folding, and disposal [53]. Normally, protein disposal, via the proteasomal system and autophagy, is crucial in maintaining proteastasis and avoiding protein accumulation. The 26S proteasome or ubiquitin protease system is responsible of eliminating misfolded/unfolded proteins including amyloid-β and phosphorylated tau, in part via polyubiquitination [54]. In IBM, proteasome 26S and aggregated proteins co-localize on muscle biopsy [55]. Furthermore, there is evidence of decreased 26S proteasomal activity and overexpression of amyloid-β precursor protein in IBM muscle fibers, associated with proteasomal inhibition and further protein aggregation [55].
Autophagy consists of degradation of various molecules in lysosomes. Excessive protein turnover or malfunctioning of the lysosomes can manifest with excess of endosomes, autophagic vacuoles, and autolysosomes, all of which can be commonly found in rimmed vacuoles [56]. Analysis of rimmed vacuoles content via a proteomic approach confirmed that rimmed vacuoles proteins are largely related to protein folding and autophagy [57]. The metabolic regulator mammalian target of rapamycin (mTOR) is a major autophagy mediator. By inhibiting mTOR, rapamycin induces autophagy [58]. In a valosin-containing protein (VCP) inclusion body myopathy mouse model, mTOR signaling was found to be defective, and further inhibition by rapamycin caused exacerbation of the mice muscle weakness and an increase in serum creatine kinase and the number of atrophic and vacuolated fibers [59]. Contrasting findings were reported by another group in which rapamycin-treated VCP mice had improved strength and a decreased number of atrophic and vacuolated fibers [60]. Indeed in IBM, there is evidence for both increased autophagy, as would be expected with the high protein turnover, and dysfunctional autophagy as witnessed by the diminished lysosomal enzymatic activity, indicating lysosomal dysfunction [61,62,63]. p62, also known as sequestosome 1 (SQSTM1), helps in transporting polyubiquinated proteins to both the proteasome and the lysosome [64]. Unlike in dermatomyositis and polymyositis, p62 is overexpressed in IBM [15, 65].
Another important organelle in protein folding is the endoplasmic reticulum (ER). However, ER is very sensitive to disruption of muscle homeostasis [66]. To avoid stress and the accumulation of misfolded protein, the ER heavily relies on chaperone proteins, including heat shock proteins (HSP), which are important for protein–protein interactions and maintaining conformational protein structure [67]. During stress, as a part of a cytoprotective mechanism, there is upregulation of chaperone proteins [68,69,70]. Furthermore, ER stress upregulates the secretion of myostatin precursor protein (MstnPP) and its metabolites [71, 72]. High levels of MstnPP can also induce ER stress, which results in aggregation of high molecular weight MstnPP cleavage products and impaired secretion of mature myostatin [73]. Myostatin, a member of the transforming growth factor β superfamily (TGFβ), is an inhibitor of skeletal muscle mass development [74]. There is also evidence of mitochondrial dysfunction in IBM which is witnessed by the mitochondrial abnormalities observed on the muscle biopsy and the increased amount of mitochondrial DNA rearrangement, deletion, and depletion [75, 76].
Inflammation Versus Degeneration
It remains unclear whether the primary process is immune-mediated or degenerative in nature. There is strong evidence for the inflammatory component, as detailed above, including clonally restricted, antigen-driven, infiltrating CD8-positive T cells; the strong genetic association with HLA genes; and the association with cN-1A antibodies and other autoimmune conditions such as systemic lupus erythematous and Sjögren’s syndrome. Unlike in inclusion body myositis, these findings are not encountered in other neurodegenerative disorders. Regarding the degenerative component, there is growing evidence that inflammation can cause secondary degenerative features. In inflammatory myopathies, including IBM, inflammatory cytokines can induce the expression of the immunoproteasome (usually only expressed in hematopoietic cells) in muscle, which strongly co-localizes with fibers expressing MHC-I [77]. Overexpression of MHC-I in mice can cause severe myopathy and induce ER stress and protein unfolding [78]. In myoblast cultures, overexpression of β-amyloid precursor protein and exposure to inflammatory cytokines can both induce cytoplasmic mislocalization of TDP-43 [79]. Furthermore, pro-inflammatory mediators can upregulate the production of β-amyloid proteins and the expression of inducible nitric oxide synthase (iNOS) in skeletal muscle [80, 81]. It has also been shown that the severity of the inflammation strongly correlates with β-amyloid production and mitochondrial dysfunction [80, 82]. As mentioned above, in vitro and in vivo passive immunization of mice with sera of patient’s with cN-1A antibodies can result in p62/SQSTM1 sarcoplasmic aggregates [21]. On the other hand, overexpression of β-APP activated nuclear factor kB in myoblast cultures [79]. Therefore, protein accumulation could theoretically trigger inflammation; however, further experimental studies in IBM patients are still needed. Nonetheless, one of the main arguments for a primarily degenerative component remains the lack of response to immunotherapy.
Genetics
Among immune- and neurodegenerative-related genes, the HLA region has the strongest association with IBM, especially HLA-DRB1 [83,84,85]. Furthermore, HLA-DRB1*03:01, DRB1*01:01, and DRB1*13:01 alleles can modify the phenotype and be associated with more severe muscle weakness [86]. Among neurodegenerative-related genes, there has not been any association between IBM and genes related to Alzheimer’s disease, or Parkinson’s disease. Three likely pathogenic or pathogenic rare missense variants in VCP and 4 in SQSTM1 were found in patients with IBM [87, 88]. None of the patients had developed inclusion body myopathy with Paget’s disease of bones, frontotemporal dementia, or amyotrophic lateral sclerosis, and none of the patients had family history of such disorders. All patients fulfilled clinical and pathological criteria for IBM. Although there is no clear association between apolipoprotein E and translocase of outer mitochondrial membrane 40 (TOMM40) genotypes with the risk of developing IBM, the presence of a very long polyT repeat allele in TOMM40 may delay onset of symptoms by about 5 years, especially when associated with apolipoprotein E genotype ε3/ε3. [89, 90] TOMM40 encodes an outer mitochondrial membrane protein involved in the transport of peptides into the mitochondria including amyloid-β [91]. Studying the proteomics of rimmed vacuoles, rare missense variants in FYCO1 were overrepresented in IBM patients (11.3%) compared with ALS (2.6%) patients and healthy controls (3.4%) [57]. FYCO1 is an autophagic adaptor protein [92].
Treatment
Better understanding of the pathogenesis and further characterization of the involved degenerative pathways resulted in casting the net wide searching for a treatment addressing inflammatory and degenerative pathways (summarized in Table 2). However, there continues to be no effective treatment in inclusion body myositis.
Targeting Inflammatory Pathways
Despite the clear inflammatory component, immunosuppressive therapy (such as corticosteroids, intravenous immunoglobulin (IVIG), methotrexate, and azathioprine) offers at best a mild and transient benefit [93,94,95,96,97,98]. In an open-label uncontrolled [94] and 2 placebo-controlled studies [95, 96], IVIG treatment showed overall marginal to no improvement. Despite reported improvement in swallowing and functionally significant improvement in strength in occasional patients [95, 99], IVIG does not seem to have a sustained benefit, nor does it alter the long-term disease course [100]. Therefore, IVIG treatment is not recommended in clinical practice, although on a case-by-case basis, it can be considered in patients with marked dysphagia.
Two randomized controlled studies of β-interferon-1a at standard [101] or high dose [102] showed no improvement in muscle strength in treated patients. Similarly, clinical trials with anti-T-lymphocyte globulin treatment [103], etanercept which is a tumor necrosis factor-alpha fusion protein [104], alemtuzumab which is a humanized monoclonal antibody that causes an immediate depletion or severe reduction of peripheral blood lymphocytes [105], anakinra which is an IL1 receptor antagonist [106], and simvastatin for its pleiotropic anti-inflammatory effect [107], showed no clinically meaningful benefit in IBM.
Targeting Degenerative Pathways
Based on the multiple unsuccessful attempts to treat IBM by acting on the immune system, and regardless whether the degenerative component is primary to the pathogenesis or not, degenerative pathways have become a novel potential therapeutic target. Arimoclomol prolongs the activation of heat shock factor 1 selectively in stressed cells and, subsequently, augments HSP levels [108]. HSP inducers [109] are under investigation for various disorders such as ALS [110], sphingolipidoses [111], and inclusion body myositis [79]. There is no good animal model for IBM; however, in mutant VCP mice, arimoclomol ameliorated muscle strength and disease pathology [79]. A proof-of-concept safety randomized controlled trial targeting drug safety in 24 IBM patients demonstrated arimoclomol to be safe and well tolerated. Three of the efficacy secondary outcomes demonstrated trends favoring arimoclomol at 8 months. A current phase II/III trial is underway (NCT02753530).
Therapeutic effect of myostatin inhibition has also been investigated. Bimagrumab, activin receptor II (ARII) inhibitory monoclonal antibody, was studied in a pilot trial in which 10 patients were randomized to bimagrumab and 4 to placebo [112]. Thigh muscle volume evaluated by MRI was increased by 6.5% on the right and 7.6% on the left in the treated group (p = 0.024 and 0.009, respectively); however, there was no statistically significant difference in muscle function. A follow-up randomized controlled trial did not reach its primary outcomes and the results remain unpublished.
Follistatin is a myostatin antagonist [113]. In a nonrandomized open-label study, 6 IBM patients were treated with follistatin gene therapy and showed improvement in the 6-min walk test (5-153 m) [114]. However, the treated group also received high-dose prednisone and a prescribed exercise program, which was not accounted for in the matched control group [115]. Therefore, further studies are needed to determine the efficacy of follistatin gene therapy in IBM. Increasing muscle mass was also attempted via treatment with oxandrolone, an anabolic steroid, which showed only borderline benefit improving whole-body strength, with more noticeable improvement in upper extremity strength [116].
Despite the conflicting evidence regarding the effect of rapamycin on VCP mice, a recent randomized, double blind, placebo-controlled clinical trial was conducted. The study did not reach the primary outcome defined as stabilization of maximal voluntary quadriceps isometric strength assessed with a dynamometer, although in the treated group, the 6MWT deteriorated less and the forced vital capacity improved [117]. The study is yet to be published. (NCT02481453).
Nonpharmacological Therapeutic Options
There is limited data regarding the role of exercise in idiopathic inflammatory myopathies in general and IBM in particular [118]. In 3 uncontrolled trials with limited number of patients (≤ 7 patients per trial), home exercise (resistance training with or without aerobic exercise) is at least not harmful and may indeed preserve or even improve muscle strength [119,120,121]. In a rat model with chloroquine-induced IBM, resistance training was noted to increase muscle strength and decrease p62 levels [122].
IBM patients with dysphagia may benefit from cricopharyngeal myotomy and pharyngoesophageal dilation which help in relaxing the upper esophageal sphincter [123]. In a retrospective study, 12 patients with IBM received botulinum toxin injection of the cricopharyngeus muscle, with subsequent improvement of their swallowing [124]. However, in a subset of patients in which dysphagia may be due to decreased hypolaryngeal excursion with normal upper esophageal sphincter relaxation, these interventions may not be helpful [125].
Future Therapeutic Options
Promising novel mechanistic approaches involve reducing endoplasmic reticulum stress, promoting autophagy, optimizing oxidative and mitochondrial dysfunction, and removal of toxic protein aggregates. There is marked patient excitement about the potential role of stem cells in IBM, but there is no current data to support the efficacy and safety of this approach.
In addition to muscle biopsies in IBM expressing large numbers of CD3+ cells that co-localized with Kv1.3, circulating PBMC had an increased number of Kv1.3+ cells in IBM as compared with healthy controls and other inflammatory myopathies [126]. Kv1.3 is frequently found on T effector memory cells, which have been implicated in T-cell-mediated autoimmune disorders, and targeting these cells in IBM may be a new promising strategy.
Prognosis
There is no clear evidence that IBM affects life expectancy. However, loss of ambulation and dysphagia remain the main source of disability. The use of a wheelchair is needed in about a third of patients 14 years from onset and nearly all patients 20 years from onset [127, 128]. During a 12-year follow-up study of 64 Dutch patients with IBM, 46 patients died during follow-up with a median age at death of 81 years [127]. Although the life expectancy was not different from an age-matched Dutch general population, death from respiratory disease, especially pneumonia, was markedly more common in the IBM group. Lastly, as mentioned above, there is preliminary evidence that patients with positive cN-1A antibodies may have mildly higher adjusted mortality risk [24].
Conclusion
IBM is an inexorably progressive muscle disorder characterized by distinctive clinical and histopathological features. Clinically, it is characterized by the predominant involvement of deep finger flexors and quadriceps muscles and, histopathologically, by the combination of inflammatory and degenerative changes. There remain many unanswered questions regarding IBM pathogenesis and, most importantly, the refractoriness to treatment. Perhaps, IBM is primarily an immune-mediated disorder, which unlike any other immune disorder, triggers downstream degenerative changes early on in the process. On the other hand, a primarily degenerative disorder with secondary inflammation is also a possibility. Regardless of the nature of the primary process, a successful treatment may necessitate addressing both the immune and degenerative components simultaneously. Alternatively, it may be that the therapeutic window of opportunity is confined, and requires intervention early on, prior to the development of the degenerative changes.
References
Callan A, Capkun G, Vasanthaprasad V, Freitas R, Needham M (2017) A systematic review and meta-analysis of prevalence studies of sporadic inclusion body myositis. J Neuromuscul Dis 4:127–137
Capkun G, Callan A, Tian H, Wei Z, Zhao C, Agashivala N, Barghout V (2017) Burden of illness and healthcare resource use in United States patients with sporadic inclusion body myositis. Muscle Nerve 56:861–867
Dimachkie MM, Barohn RJ (2014) Inclusion body myositis. Neurol Clin 32:629–646, vii
Lotz BP, Engel AG, Nishino H, Stevens JC, Litchy WJ (1989) Inclusion body myositis. Observations in 40 patients. Brain 112 (Pt 3):727–747
Oh TH, Brumfield KA, Hoskin TL, Stolp KA, Murray JA, Basford JR (2007) Dysphagia in inflammatory myopathy: clinical characteristics, treatment strategies, and outcome in 62 patients. Mayo Clin Proc 82:441–447
Ghosh PS, Milone M (2015) Camptocormia as presenting manifestation of a spectrum of myopathic disorders. Muscle Nerve 52:1008–1012
Ghosh PS, Laughlin RS, Engel AG (2014) Inclusion-body myositis presenting with facial diplegia. Muscle Nerve 49:287–289
Voermans NC, Vaneker M, Hengstman GJD, ter Laak HJ, Zimmerman C, Schelhaas HJ, Zwarts MJ (2004) Primary respiratory failure in inclusion body myositis. Neurology 63:2191–2192
Chahin N, Engel AG (2008) Correlation of muscle biopsy, clinical course, and outcome in PM and sporadic IBM. Neurology 70:418–424
Brady S, Squier W, Sewry C, Hanna M, Hilton-Jones D, Holton JL (2014) A retrospective cohort study identifying the principal pathological features useful in the diagnosis of inclusion body myositis. BMJ Open 4:e004552
Bernier FP, Boneh A, Dennett X, Chow CW, Cleary MA, Thorburn DR (2002) Diagnostic criteria for respiratory chain disorders in adults and children. Neurology 59:1406–1411
Griggs RC, Askanas V, DiMauro S, Engel A, Karpati G, Mendell JR, Rowland LP (1995) Inclusion body myositis and myopathies. Ann Neurol 38:705–713
Hilton-Jones D, Miller A, Parton M, Holton J, Sewry C, Hanna MG (2010) Inclusion body myositis: MRC Centre for Neuromuscular Diseases, IBM workshop, London, 13 June 2008. Neuromuscul Disord 20:142–147
Salajegheh M, Pinkus JL, Taylor JP, Amato AA, Nazareno R, Baloh RH, Greenberg SA (2009) Sarcoplasmic redistribution of nuclear TDP-43 in inclusion body myositis. Muscle Nerve 40:19–31
Nogalska A, Terracciano C, D’Agostino C, King Engel W, Askanas V (2009) p62/SQSTM1 is overexpressed and prominently accumulated in inclusions of sporadic inclusion-body myositis muscle fibers, and can help differentiating it from polymyositis and dermatomyositis. Acta Neuropathol 118:407–413
Dubourg O, Wanschitz J, Maisonobe T, Béhin A, Allenbach Y, Herson S, Benveniste O (2011) Diagnostic value of markers of muscle degeneration in sporadic inclusion body myositis. Acta Myol 30(2):103–8
Rose MR (2013) 188th ENMC International Workshop: inclusion body myositis, 2–4 December 2011, Naarden, The Netherlands. Neuromuscul Disord 23:1044–1055
Salajegheh M, Lam T, Greenberg SA (2011) Autoantibodies against a 43 kDa muscle protein in inclusion body myositis. PLoS One 6:e20266
Pluk H, van Hoeve BJA, van Dooren SHJ, et al (2013) Autoantibodies to cytosolic 5′-nucleotidase 1A in inclusion body myositis. Ann Neurol 73:397–407
Larman BH, Salajegheh M, Nazareno R, et al (2013) Cytosolic 5′-nucleotidase 1A autoimmunity in sporadic inclusion body myositis. Ann Neurol 73:408–418
Tawara N, Yamashita S, Zhang X, et al (2017) Pathomechanisms of anti-cytosolic 5′-nucleotidase 1A autoantibodies in sporadic inclusion body myositis. Ann Neurol 81:512–525
Lloyd TE, Christopher-Stine L, Pinal-Fernandez I, Tiniakou E, Petri M, Baer A, Danoff SK, Pak K, Casciola-Rosen LA, Mammen AL (2016) Cytosolic 5’-nucleotidase 1A as a target of circulating autoantibodies in autoimmune diseases Arthritis Care Res (Hoboken) 68:66–71
Goyal NA, Cash TM, Alam U, Enam S, Tierney P, Araujo N, Mozaffar FH, Pestronk A, Mozaffar T (2016) Seropositivity for NT5c1A antibody in sporadic inclusion body myositis predicts more severe motor, bulbar and respiratory involvement. J Neurol Neurosurg Psychiatry 87:373–378
Lilleker JB, Rietveld A, Pye SR, et al (2017) Cytosolic 5′-nucleotidase 1A autoantibody profile and clinical characteristics in inclusion body myositis. Ann Rheum Dis 76:862–868
Herbert MK, Stammen-Vogelzangs J, Verbeek MM, et al (2016) Disease specificity of autoantibodies to cytosolic 5′-nucleotidase 1A in sporadic inclusion body myositis versus known autoimmune diseases. Ann Rheum Dis 75:696–701
Kazamel M, Sorenson EJ, Milone M (2016) Clinical and electrophysiological findings in hereditary inclusion body myopathy compared with sporadic inclusion body myositis. J Clin Neuromuscul Dis 17:190–196
Badrising UA, Maat-Schieman M, van Duinen SG, et al (2000) Epidemiology of inclusion body myositis in the Netherlands: a nationwide study. Neurology 55:1385–1387
Hilton-Jones D, Miller A, Parton M, Holton J, Sewry C, Hanna MG (2010) Inclusion body myositis. Neuromuscul Disord 20:142–147
Lloyd TE, Mammen AL, Amato AA, Weiss MD, Needham M, Greenberg SA (2014) Evaluation and construction of diagnostic criteria for inclusion body myositis. Neurology 83:426–433
Jones KL, Sejersen T, Amato AA, Hilton-Jones D, Schmidt J, Wallace AC, Badrising UA, Rose MR, IBM Guideline Development Group (2016) A protocol to develop clinical guidelines for inclusion-body myositis. Muscle Nerve 53:503–507
Engel AG, Arahata K (1984) Monoclonal antibody analysis of mononuclear cells in myopathies. II: phenotypes of autoinvasive cells in polymyositis and inclusion body myositis. Ann Neurol 16:209–215
Amemiya K, Granger RP, Dalakas MC (2000) Clonal restriction of T-cell receptor expression by infiltrating lymphocytes in inclusion body myositis persists over time. Studies in repeated muscle biopsies. Brain 123 (Pt 10):2030–2039
Salajegheh M, Rakocevic G, Raju R, Shatunov A, Goldfarb LG, Dalakas MC (2007) T cell receptor profiling in muscle and blood lymphocytes in sporadic inclusion body myositis. Neurology 69:1672–1679
Tateyama M, Fujihara K, Misu T, Itoyama Y (2009) CCR7+ myeloid dendritic cells together with CCR7+ T cells and CCR7+ macrophages invade CCL19+ nonnecrotic muscle fibers in inclusion body myositis. J Neurol Sci 279:47–52
Greenberg SA, Pinkus GS, Amato AA, Pinkus JL (2007) Myeloid dendritic cells in inclusion-body myositis and polymyositis. Muscle Nerve 35:17–23
Greenberg SA, Bradshaw EM, Pinkus JL, Pinkus GS, Burleson T, Due B, Bregoli LS, O’Connor KC, Amato AA, Amato AA (2005) Plasma cells in muscle in inclusion body myositis and polymyositis. Neurology 65:1782–1787
Bradshaw EM, Orihuela A, McArdel SL, Salajegheh M, Amato AA, Hafler DA, Greenberg SA, O’Connor KC (2007) A local antigen-driven humoral response is present in the inflammatory myopathies. J Immunol 178:547–556
Uruha A, Noguchi S, Hayashi YK, Tsuburaya RS, Yonekawa T, Nonaka I, Nishino I (2016) Hepatitis C virus infection in inclusion body myositis. Neurology 86:211–217
Alverne ARSM, Marie SKN, Levy-Neto M, de Souza FHC, de Carvalho MS, Shinjo SK (2013) Inclusion body myositis: series of 30 cases from a Brazilian tertiary center. Acta Reumatol Port 38:179–185
Cupler EJ, Leon-Monzon M, Miller J, Semino-Mora C, Anderson TL, Dalakas MC (1996) Inclusion body myositis in HIV-1 and HTLV-1 infected patients. Brain 119 (Pt 6):1887–1893
Dalakas MC, Rakocevic G, Shatunov A, Goldfarb L, Raju R, Salajegheh M (2007) Inclusion body myositis with human immunodeficiency virus infection: four cases with clonal expansion of viral-specific T cells. Ann Neurol 61:466–475
Greenberg SA, Pinkus JL, Amato AA, Kristensen T, Dorfman DM (2016) Association of inclusion body myositis with T cell large granular lymphocytic leukaemia. Brain 139:1348–1360
Bareau B, Rey J, Hamidou M, et al (2010) Analysis of a French cohort of patients with large granular lymphocyte leukemia: a report on 229 cases. Haematologica 95:1534–41
Hohlfeld R, Schulze-Koops H (2016) Cytotoxic T cells go awry in inclusion body myositis. Brain 139:1312–1314
Lamy T, Moignet A, Loughran TP (2017) LGL leukemia: from pathogenesis to treatment. Blood 129:1082–1094
Zanusso G, Vattemi G, Ferrari S, et al (2001) Increased expression of the normal cellular isoform of prion protein in inclusion-body myositis, inflammatory myopathies and denervation atrophy. Brain Pathol 11:182–189
Askanas V, Engel WK, Alvarez RB, Glenner GG (1992) beta-Amyloid protein immunoreactivity in muscle of patients with inclusion-body myositis. Lancet (London, England) 339:560–561
Mendell JR, Sahenk Z, Gales T, Paul L (1991) Amyloid filaments in inclusion body myositis. Novel findings provide insight into nature of filaments. Arch Neurol 48:1229–1234
Catalán-García M, Garrabou G, Morén C, et al (2015) BACE-1, PS-1 and sAPPβ levels are increased in plasma from sporadic inclusion body myositis patients: surrogate biomarkers among inflammatory myopathies. Mol Med 21:1
Nogalska A, D’Agostino C, Engel WK, Klein WL, Askanas V (2010) Novel demonstration of amyloid-β oligomers in sporadic inclusion-body myositis muscle fibers. Acta Neuropathol 120:661–666
Sarkozi E, Askanas V, Johnson SA, Engel WK, Alvarez RB (1993) beta-Amyloid precursor protein mRNA is increased in inclusion-body myositis muscle. Neuroreport 4:815–818
Abdo WF, van Mierlo T, Hengstman GJ, Schelhaas HJ, van Engelen BG, Verbeek MM (2009) Increased plasma amyloid-beta42 protein in sporadic inclusion body myositis. Acta Neuropathol 118:429–431
Nogalska A, D’Agostino C, Engel WK, Cacciottolo M, Asada S, Mori K, Askanas V (2015) Activation of the unfolded protein response in sporadic inclusion-body myositis but not in hereditary GNE inclusion-body myopathy. J Neuropathol Exp Neurol 74:538–546
Finley D (2009) Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem 78:477–513
Fratta P, Engel WK, McFerrin J, Davies KJA, Lin SW, Askanas V (2005) Proteasome inhibition and aggresome formation in sporadic inclusion-body myositis and in amyloid-beta precursor protein-overexpressing cultured human muscle fibers. Am J Pathol 167:517–526
Fukuhara N, Kumamoto T, Tsubaki T (1980) Rimmed vacuoles. Acta Neuropathol 51:229–235
Güttsches A-K, Brady S, Krause K, et al (2017) Proteomics of rimmed vacuoles define new risk allele in inclusion body myositis. Ann Neurol 81:227–239
Ju J-S, Varadhachary AS, Miller SE, Weihl CC (2010) Quantitation of “autophagic flux” in mature skeletal muscle Autophagy 6:929–935
Ching JK, Elizabeth S V., Ju J-S, Lusk C, Pittman SK, Weihl CC (2013) mTOR dysfunction contributes to vacuolar pathology and weakness in valosin-containing protein associated inclusion body myopathy. Hum Mol Genet 22:1167–1179
Nalbandian A, Llewellyn KJ, Nguyen C, Yazdi PG, Kimonis VE (2015) Rapamycin and chloroquine: the in vitro and in vivo effects of autophagy-modifying drugs show promising results in valosin containing protein multisystem proteinopathy. PLoS One 10:e0122888
Nogalska A, D’Agostino C, Terracciano C, Engel WK, Askanas V (2010) Impaired autophagy in sporadic inclusion-body myositis and in endoplasmic reticulum stress-provoked cultured human muscle fibers. Am J Pathol 177:1377–1387
Lünemann JD, Schmidt J, Dalakas MC, Münz C (2007) Macroautophagy as a pathomechanism in sporadic inclusion body myositis. Autophagy 3:384–386
Kumamoto T, Ueyama H, Tsumura H, Toyoshima I, Tsuda T (2004) Expression of lysosome-related proteins and genes in the skeletal muscles of inclusion body myositis. Acta Neuropathol 107:59–65
Seibenhener ML, Babu JR, Geetha T, Wong HC, Krishna NR, Wooten MW (2004) Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol Cell Biol 24:8055–8068
Nakano S, Oki M, Kusaka H (2017) The role of p62/SQSTM1 in sporadic inclusion body myositis. Neuromuscul Disord 27:363–369
Kaufman RJ (1999) Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev 13:1211–1233
Brown IR (2007) Heat shock proteins and protection of the nervous system. Ann N Y Acad Sci 1113:147–158
Cacciottolo M, Nogalska A, D’Agostino C, Engel WK, Askanas V (2013) Chaperone-mediated autophagy components are upregulated in sporadic inclusion-body myositis muscle fibres. Neuropathol Appl Neurobiol 39:750–761
Nogalska A, Engel WK, McFerrin J, Kokame K, Komano H, Askanas V (2006) Homocysteine-induced endoplasmic reticulum protein (Herp) is up-regulated in sporadic inclusion-body myositis and in endoplasmic reticulum stress-induced cultured human muscle fibers. J Neurochem 96:1491–1499
Banwell BL, Engel AG (2000) AlphaB-crystallin immunolocalization yields new insights into inclusion body myositis. Neurology 54:1033–1041
Wójcik S, Engel WK, McFerrin J, Askanas V (2005) Myostatin is increased and complexes with amyloid-β within sporadic inclusion-body myositis muscle fibers. Acta Neuropathol 110:173–177
Nogalska A, Wojcik S, King Engel W, McFerrin J, Askanas V (2007) Endoplasmic reticulum stress induces myostatin precursor protein and NF-κB in cultured human muscle fibers: Relevance to inclusion body myositis. Exp Neurol 204:610–618
Sachdev R, Kappes-Horn K, Paulsen L, Duernberger Y, Pleschka C, Denner P, Kundu B, Reimann J, Vorberg I (2018) Endoplasmic reticulum stress induces myostatin high molecular weight aggregates and impairs mature myostatin secretion. Mol Neurobiol. doi: https://doi.org/10.1007/s12035-018-0997-9
Gonzalez-Cadavid NF, Bhasin S (2004) Role of myostatin in metabolism. Curr Opin Clin Nutr Metab Care 7:451–457
Catalan-Garcia M, Garrabou G, Moren C, et al (2016) Mitochondrial DNA disturbances and deregulated expression of oxidative phosphorylation and mitochondrial fusion proteins in sporadic inclusion body myositis. Clin Sci 130:1741–1751
Rygiel KA, Tuppen HA, Grady JP, Vincent A, Blakely EL, Reeve AK, Taylor RW, Picard M, Miller J, Turnbull DM (2016) Complex mitochondrial DNA rearrangements in individual cells from patients with sporadic inclusion body myositis. Nucleic Acids Res 44:5313–5329
Bhattarai S, Ghannam K, Krause S, et al (2016) The immunoproteasomes are key to regulate myokines and MHC class I expression in idiopathic inflammatory myopathies. J Autoimmun 75:118–129
Fréret M, Drouot L, Obry A, Ahmed-Lacheheb S, Dauly C, Adriouch S, Cosette P, Authier F-J, Boyer O (2013) Overexpression of MHC class I in muscle of lymphocyte-deficient mice causes a severe myopathy with induction of the unfolded protein response. Am J Pathol 183:893–904
Ahmed M, Machado PM, Miller A, et al (2016) Targeting protein homeostasis in sporadic inclusion body myositis. Sci Transl Med 8:331ra41–331ra41
Schmidt J, Barthel K, Wrede A, Salajegheh M, Bähr M, Dalakas MC (2008) Interrelation of inflammation and APP in sIBM: IL-1 beta induces accumulation of beta-amyloid in skeletal muscle. Brain 131:1228–1240
Adams V, Nehrhoff B, Späte U, Linke A, Schulze PC, Baur A, Gielen S, Hambrecht R, Schuler G (2002) Induction of iNOS expression in skeletal muscle by IL-1beta and NFkappaB activation: an in vitro and in vivo study. Cardiovasc Res 54:95–104
Rygiel KA, Miller J, Grady JP, Rocha MC, Taylor RW, Turnbull DM (2015) Mitochondrial and inflammatory changes in sporadic inclusion body myositis. Neuropathol Appl Neurobiol 41:288–303
Rojana-udomsart A, Bundell C, James I, Castley A, Martinez P, Christiansen F, Hollingsworth P, Mastaglia F (2012) Frequency of autoantibodies and correlation with HLA-DRB1 genotype in sporadic inclusion body myositis (s-IBM): a population control study. J Neuroimmunol 249:66–70
Rothwell S, Cooper RG, Lundberg IE, et al (2017) Immune-array analysis in sporadic inclusion body myositis reveals HLA-DRB1 amino acid heterogeneity across the myositis spectrum. Arthritis Rheumatol 69:1090–1099
Mastaglia FL, Needham M, Scott A, et al (2009) Sporadic inclusion body myositis: HLA-DRB1 allele interactions influence disease risk and clinical phenotype. Neuromuscul Disord 19:763–765
Rojana-udomsart A, James I, Castley A, et al (2012) High-resolution HLA-DRB1 genotyping in an Australian inclusion body myositis (s-IBM) cohort: an analysis of disease-associated alleles and diplotypes. J Neuroimmunol 250:77–82
Weihl CC, Baloh RH, Lee Y, Chou T-F, Pittman SK, Lopate G, Allred P, Jockel-Balsarotti J, Pestronk A, Harms MB (2015) Targeted sequencing and identification of genetic variants in sporadic inclusion body myositis. Neuromuscul Disord 25:289–296
Gang Q, Bettencourt C, Machado PM, et al (2016) Rare variants in SQSTM1 and VCP genes and risk of sporadic inclusion body myositis. Neurobiol Aging 47:218.e1–218.e9
Gang Q, Bettencourt C, Machado PM, et al (2015) The effects of an intronic polymorphism in TOMM40 and APOE genotypes in sporadic inclusion body myositis. Neurobiol Aging 36:1766.e1–1766.e3
Mastaglia FL, Rojana-udomsart A, James I, et al (2013) Polymorphism in the TOMM40 gene modifies the risk of developing sporadic inclusion body myositis and the age of onset of symptoms. Neuromuscul Disord 23:969–974
Hansson Petersen CA, Alikhani N, Behbahani H, et al (2008) The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc Natl Acad Sci U S A 105:13145–13150
Wild P, McEwan DG, Dikic I (2014) The LC3 interactome at a glance. J Cell Sci 127:3–9
Leff RL, Miller FW, Hicks J, Fraser DD, Plotz PH (1993) The treatment of inclusion body myositis: a retrospective review and a randomized, prospective trial of immunosuppressive therapy. Medicine (Baltimore) 72:225–235
Amato AA, Barohn RJ, Jackson CE, Pappert EJ, Sahenk Z, Kissel JT (1994) Inclusion body myositis: treatment with intravenous immunoglobulin. Neurology 44:1516–1518
Dalakas MC, Sonies B, Dambrosia J, Sekul E, Cupler E, Sivakumar K (1997) Treatment of inclusion-body myositis with IVIg: a double-blind, placebo-controlled study. Neurology 48:712–716
Walter MC, Lochmüller H, Toepfer M, Schlotter B, Reilich P, Schröder M, Müller-Felber W, Pongratz D (2000) High-dose immunoglobulin therapy in sporadic inclusion body myositis: a double-blind, placebo-controlled study. J Neurol 247:22–28
Badrising UA, Maat-Schieman MLC, Ferrari MD, et al (2002) Comparison of weakness progression in inclusion body myositis during treatment with methotrexate or placebo. Ann Neurol 51:369–372
Barohn RJ, Amato AA, Sahenk Z, Kissel JT, Mendell JR (1995) Inclusion body myositis: explanation for poor response to immunosuppressive therapy. Neurology 45:1302–1304
Cherin P, Pelletier S, Teixeira A, Laforet P, Simon A, Herson S, Eymard B (2002) Intravenous immunoglobulin for dysphagia of inclusion body myositis. Neurology 58:326
Dobloug C, Walle-Hansen R, Gran JT, Molberg Ø (2012) Long-term follow-up of sporadic inclusion body myositis treated with intravenous immunoglobulin: a retrospective study of 16 patients. Clin Exp Rheumatol 30:838–842
Muscle Study Group (2001) Randomized pilot trial of betaINF1a (Avonex) in patients with inclusion body myositis. Neurology 57:1566–1570
Muscle Study Group (2004) Randomized pilot trial of high-dose betaINF-1a in patients with inclusion body myositis. Neurology 63:718–720
Lindberg C, Trysberg E, Tarkowski A, Oldfors A (2003) Anti-T-lymphocyte globulin treatment in inclusion body myositis: a randomized pilot study. Neurology 61:260–262
Barohn RJ, Herbelin L, Kissel JT, King W, McVey AL, Saperstein DS, Mendell JR (2006) Pilot trial of etanercept in the treatment of inclusion-body myositis. Neurology 66:S123–S124
Dalakas MC, Rakocevic G, Schmidt J, et al (2009) Effect of Alemtuzumab (CAMPATH 1-H) in patients with inclusion-body myositis. Brain 132:1536–1544
Kosmidis ML, Alexopoulos H, Tzioufas AG, Dalakas MC (2013) The effect of anakinra, an IL1 receptor antagonist, in patients with sporadic inclusion body myositis (sIBM): a small pilot study. J Neurol Sci 334:123–125
Sancricca C, Mora M, Ricci E, Tonali PA, Mantegazza R, Mirabella M (2011) Pilot trial of simvastatin in the treatment of sporadic inclusion-body myositis. Neurol Sci 32:841–847
Hargitai J, Lewis H, Boros I, et al (2003) Bimoclomol, a heat shock protein co-inducer, acts by the prolonged activation of heat shock factor-1. Biochem Biophys Res Commun 307:689–695
Bíró K, Jednákovits A, Kukorelli T, Hegedüs E, Korányi L (1997) Bimoclomol (BRLP-42) ameliorates peripheral neuropathy in streptozotocin-induced diabetic rats. Brain Res Bull 44:259–263
Benatar M, Wuu J, Andersen PM, Atassi N, David W, Cudkowicz M, Schoenfeld D (2018) Randomized, double-blind, placebo-controlled trial of arimoclomol in rapidly progressive SOD1 ALS. Neurology 90:e565–e574
Kirkegaard T, Gray J, Priestman DA, et al (2016) Heat shock protein-based therapy as a potential candidate for treating the sphingolipidoses. Sci Transl Med 8:355ra118–355ra118
Amato AA, Sivakumar K, Goyal N, et al (2014) Treatment of sporadic inclusion body myositis with bimagrumab. Neurology 83:2239–2246
Lee S-J (2004) Regulation of muscle mass by myostatin. Annu Rev Cell Dev Biol 20:61–86
Mendell JR, Sahenk Z, Al-Zaidy S, et al (2017) Follistatin gene therapy for sporadic inclusion body myositis improves functional outcomes. Mol Ther 25:870–879
Greenberg SA (2017) Unfounded claims of improved functional outcomes attributed to follistatin gene therapy in inclusion body myositis. Mol Ther 25:2235–2237
Rutkove SB, Parker RA, Nardin RA, Connolly CE, Felice KJ, Raynor EM (2002) A pilot randomized trial of oxandrolone in inclusion body myositis. Neurology 58:1081–1087
Lilleker JB, Bukhari M, Chinoy H (2018) Rapamycin for inclusion body myositis: targeting non-inflammatory mechanisms. Rheumatology. doi: https://doi.org/10.1093/rheumatology/key043
Habers GEA, Takken T (2011) Safety and efficacy of exercise training in patients with an idiopathic inflammatory myopathy—a systematic review. Rheumatology (Oxford) 50:2113–2124
Arnardottir S, Alexanderson H, Lundberg IE, Borg K (2003) Sporadic inclusion body myositis: pilot study on the effects of a home exercise program on muscle function, histopathology and inflammatory reaction. J Rehabil Med 35:31–35
Johnson LG, Collier KE, Edwards DJ, Philippe DL, Eastwood PR, Walters SE, Thickbroom GW, Mastaglia FL (2009) Improvement in aerobic capacity after an exercise program in sporadic inclusion body myositis. J Clin Neuromuscul Dis 10:178–184
Spector SA, Lemmer JT, Koffman BM, Fleisher TA, Feuerstein IM, Hurley BF, Dalakas MC (1997) Safety and efficacy of strength training in patients with sporadic inclusion body myositis. Muscle Nerve 20:1242–1248
Kwon I, Lee Y, Cosio-Lima LM, Cho J-Y, Yeom D-C (2015) Effects of long-term resistance exercise training on autophagy in rat skeletal muscle of chloroquine-induced sporadic inclusion body myositis. J Exerc Nutr Biochem 19:225–234
Oh TH, Brumfield KA, Hoskin TL, Kasperbauer JL, Basford JR (2008) Dysphagia in inclusion body myositis. Am J Phys Med Rehabil 87:883–889
Schrey A, Airas L, Jokela M, Pulkkinen J (2017) Botulinum toxin alleviates dysphagia of patients with inclusion body myositis. J Neurol Sci 380:142–147
Claire Langdon P, Mulcahy K, Shepherd KL, Low VH, Mastaglia FL (2012) Pharyngeal dysphagia in inflammatory muscle diseases resulting from impaired suprahyoid musculature. Dysphagia 27:408–417
Karissa J. Munoz, Ali H. Mannaa, Jenna Kastenschmidt, Marie Wencel, Namita Goyal, S. Armando Villalta, Tahseen Mozaffar (2018) Circulatory Kv1.3+ cells in patients with sIBM. Neurology 90. http://n.neurology.org/content/90/15_Supplement/P3.437
Cox FM, Titulaer MJ, Sont JK, Wintzen AR, Verschuuren JJGM, Badrising UA (2011) A 12-year follow-up in sporadic inclusion body myositis: an end stage with major disabilities. Brain 134:3167–3175
Benveniste O, Guiguet M, Freebody J, et al (2011) Long-term observational study of sporadic inclusion body myositis. Brain 134:3176–3184
Acknowledgments
This work was supported by a Clinical and Translational Science Awards grant from National Center for Advancing Translational Sciences awarded to the University of Kansas Medical Center for Frontiers: The Heartland Institute for Clinical and Translational Research (# UL1TR000001). The contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health or National Center for Advancing Translational Sciences.
Author information
Authors and Affiliations
Corresponding author
Additional information
Invited review—theme: “Myopathies”
Rights and permissions
About this article

Cite this article
Naddaf, E., Barohn, R.J. & Dimachkie, M.M. Inclusion Body Myositis: Update on Pathogenesis and Treatment. Neurotherapeutics 15, 995–1005 (2018). https://doi.org/10.1007/s13311-018-0658-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-018-0658-8