
Abstract
Despite the unknown etiology and pathogenesis of sporadic inclusion body myositis (s-IBM), investigators have speculated that the lysosome system in muscle fiber plays a central role in rimmed vacuole formation, a hallmark of s-IBM. We explored the role of receptor-mediated intracellular transport and autophagy in the lysosomal system in the abnormal accumulation of rimmed vacuoles in s-IBM. Expressions of mannose 6-phosphate receptor (M6PR), clathrin and hApg5 and hApg12 were analyzed in muscle biopsy specimens from patients with s-IBM, amyotrophic lateral sclerosis (ALS) or peripheral neuropathy and in normal human muscle specimens by means of immunohistochemistry and reverse transcriptase-polymerase chain reaction (RT-PCR). Most muscle fibers in control specimens showed little or no immunoreactivity for clathrin and M6PR, which are involved in the receptor-mediated intracellular transport. Abnormal increases in both proteins were observed mainly in the sarcoplasm of atrophic fibers in all diseased specimens. In s-IBM muscles in particular, clathrin and M6PR were often observed inside rimmed vacuoles and in the sarcoplasm of vacuolated or non-vacuolated fibers. mRNA levels of hApg5 and hApg12, which are involved in autophagic vacuole formation, as well as of M6PR and clathrin were significantly increased in s-IBM muscles in comparison to levels in normal and ALS/peripheral neuropathy muscles. Our results suggest that the transport of newly synthesized lysosomal enzymes from the secretory pathway via the trans-Golgi network of the Golgi apparatus and autophagic vacuole formation (i.e., autophagy) in the lysosome system are activated in s-IBM muscles. Remarkable accumulation of rimmed vacuoles is thought to occur because of abnormal lysosome function, especially the formation or turnover of autolysosomes after the fusion of autophagic vacuoles with the early endosomes or because of the increase in the rate of muscle fiber breakdown.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Inclusion body myositis (IBM), which can be sporadic or autosomally inherited, is characterized pathologically by numerous rimmed vacuoles with the same properties as autophagic vacuoles and inflammatory cell infiltrates [7]. Rimmed vacuoles are considered not to be disease specific, but they are observed frequently in the skeletal muscles of patients with oculopharyngeal muscular dystrophy [4], distal myopathy with rimmed vacuoles (DMRV) [6, 13, 26] and tibial muscular dystrophy [33] as well as IBM. Ultrastructural and immunohistochemical studies have shown that rimmed vacuoles include many variably sized and shaped early endosomes, autophagic vacuoles and autolysosmoes [6, 13, 26]. Hence, authors have speculated that the lysosome system in the muscle fiber may play a central role in the formation of these vacuoles.
Our previous report showed upregulation of lysosome-related transport proteins such as clathrin, α- and γ-adaptins and Golgi-zone protein in DMRV muscles, suggesting that the lysosome system, especially formation of the early endosome (i.e., primary lysosome) and autophagic vacuoles, is activated in this myopathy [18]. However, the etiology and pathomechanism of abnormal accumulation of rimmed vacuoles including autophagic vacuoles and autolysosomes in DMRV muscles are still unknown.
In general, autophagy mediates the transport of newly synthesized lysosomal enzymes from the exocytotic pathway via the trans-Golgi network of the Golgi apparatus to the early endosome [20]. Many novel proteins and genes involved in these systems have been discovered [2, 12, 19, 29]. Mannose-6-phosphate receptor (M6PR), clathrin, adaptins or coatomer proteins in particular may be involved in the receptor-mediated intracellular transport pathways and formation of early endosomes and autolysosomes [12, 19, 29]. In particular, M6PR plays an important role in the exocytotic pathway via the trans-Golgi network to the early endosome [12, 19]. However, the function of the trans-Golgi network/early endosome pathway has not been studied in relation to IBM. Autophagy-defective mutants (apg ant mut) were discovered in the yeast Saccharomyces cerevisiae [32], and most of them were thought to have defects in autophagic vacuole formation [11]. Because scientists have been able to take advantage of yeast genetics, more than 14 Apg mutants, including Apg12p and Apg5p, have been isolated [32]. In particular, the Apg12p gene is covalently attached to Apg5p via an isopeptide bond. The Apg12p-conjugated Apg5p complex functions in autophagic vacuole formation or completion [22, 23, 24]. Ohsumi and colleagues found a new protein conjugation system in which the human Apg 12 (hApg12) homologue is conjugated to the human Apg 5 homologue (hApg5) [22]. This conjugation is found in various human tissues, including skeletal muscle. Ohsumi et al. suggest that the Apg12 system is well conserved and that it functions in autophagy in the human cell [22]. However, there is no direct evidence that the system is related to autophagy in humans. Expression of hApg12-conjugated hApg5 has never been studied in skeletal muscle, especially under pathologic conditions.
To understand the role of the receptor-mediated intracellular transport pathways and autophagy in the abnormal accumulation of rimmed vacuoles, we performed histochemical, immunohistochemical and semiquantitatve reverse transcriptase-polymerase chain reaction (RT-PCR) studies on muscle biopsy specimens obtained from patients with sporadic (s-) IBM or other neuromuscular disorders.
Materials and methods
Patients
Biopsy specimens were obtained from the biceps, rectus femoris or tibialis anterior muscles of 21 patients: eight with IBM (three women and five men, 50–75 years of age), four with amyotrophic lateral sclerosis (ALS; 45–68 years of age) and four with peripheral neuropathy (two with hereditary motor sensory neuropathy type 2 and 1 with chronic inflammatory demyelinating polyneuropathy; 41–78 years of age). All patients with IBM fulfilled the clinical, neurophysiologic and myopathologic criteria for IBM (Table 1) [7]. None had a family history of IBM; they represented sporadic cases. Mean age at onset was 57 years (range, 50–75 years), and the mean duration of illness was 11 years (range, 3–8 years). Serum creatine kinase levels were mildly to moderately elevated (mean, 518 U/l; range, 50–1,474 U/l). Electromyography revealed primarily myopathic changes and some neuropathic features in all IBM patients. Biopsies were performed on the biceps (six patients), rectus femoris (one patient) or tibialis anterior (one patient) with local anesthesia. Control specimens were obtained from the biceps or rectus femoris muscles of five patients (33–76 years of age) with orthopedic disorders and no neurological deficits at the time of biopsy. Biopsy specimens were frozen rapidly in isopentane cooled in liquid nitrogen and kept at −80°C until use.
Histochemical and immunohistochemical studies
Serial 10-μm-thick, transverse sections were cut with a cryostat. Frozen sections were stained by routine histochemical methods [4]. For immunohistochemical study, unstained frozen sections were fixed in acetone for 10 min. Each section was incubated for 15 min in phosphate-buffered saline (PBS) or Tris-buffered saline (TBS) containing 10% (w/v) nonfat dry milk and then washed. Specimens were incubated with each of the optimally diluted primary antibodies for 1 h at room temperature. Mouse monoclonal antibodies for bovine M6PR (Affinity BioReagents, Galden, Colo.) and bovine clathrin (Progen Biotechnik GmbH, Heidelberg) were diluted 1:100 in PBS and 1:20 in TBS. After a wash, biotinylated goat anti-mouse IgG was added for 30 min, or alkaline phosphatase-conjugated goat anti-mouse IgM was added for 1 h. Respective specimens were then incubated for 30 min with the use of a Vectastain ABC Elite kit or an Alkaline Phosphatase Substrate kit I (Vector Labs, Burlingame, Calif.). Antibody binding sites were visualized by staining with 0.02% 3, 3'-diaminobenzidine tetrahydrochloride (DAB) plus 0.01% H2O2 for 8 min when the Vectastain ABC kit was used. Immunostaining was specific; no staining was obtained when sections were allowed to react without first-layer antibodies or with normal mouse serum substituted for first-layer antibodies.
For quantitative analysis, the number of strongly stained M6PR- or clathrin-positive muscle fibers was determined from 400 muscle fibers per muscle with a Nikon Cosmozon ISA image analyzer (Nikon, Tokyo) attached to a Macintosh computer (Apple Computer, Cupertino, Calif.).
Semiquantitative RT-PCR
Total RNA was isolated from the patient and control muscle specimens with acid guanidininum thocyanate buffer (Nippon Gene, Tokyo) according to the manufacturer's instructions. Complementary DNA (cDNA) was synthesized from 1 μg of total RNA with the use of 200 units of Moloney murine leukemia virus reverse transcriptase (Gibco BRL, Gaithersburg, Md.) and 1 μg of oligo-(dT)12–18 primer (Invitrogen, Tokyo).
The PCR primers were constructed on the basis of published nucleotide sequences of the mouse M6PR gene [21] (5'-GCT GCT GGA CTG AAC-3' (sense)); 5'-ATC GAG CCC ACA CTG AGG-3' (antisense)), the human clathrin gene [25] (5'-AAT GAC CCA AGT AAT CCA-3' (sense)); 5'-TCC GTA AGA GCA ACC-3' (antisense)), the human hApg5 gene [8] (5'-GCT TCG AGA TGT GTG GTT-3' (sense)); 5'-AAT GTA CTG TGA TGT TCC-3' (antisense)), the human hApg12 gene [22] (5'-GCT GCA GTT TCC CCG GGA-3' (sense)); 5'-TTC TGA GCC ACA AAG TTT-3' (antisense)) and the rat glycerol aldehyde 3-phosphate dehydrogenase (GAPDH) gene [31] (5'-GCC AAG TTC AAT GGC ACA GT-3' (sense)); 5'- AAG GTG GAG GAA TGG GAG TT-3' (antisense)). The PCR reactions were performed in a final volume of 50 μl in a DNA thermal cycler (ASTEC Co., Tokyo). Each PCR reaction mixture contained 1 μl of cDNA, 10 X PCR buffer (200 mM Tris-HCl, pH 8.4, and 500 mM KCl), 2.5 mM dNTP, 25 mM MgCl2, 0.5 μM of each set of primers and 2.5 units of Taq DNA polymerase (Gibco BRL). The PCR mixture was incubated at 94°C for 2 min, and 30 or 35 cycles of amplification followed. Each cycle consisted of 1 min of denaturation at 94°C, 1 min of annealing at 50 or 55°C, and 2 min of extension at 72°C. After 30 or 35 cycles, a final extension step at 72°C for 5 min was carried out. A negative control was included with an RT-PCR reaction that was performed without reverse transcriptase contamination with genomic DNA that would be detected in this reaction. The efficiency of reverse transcription was controlled by PCR with GAPDH-specific primers. Target sequences of these primers were located on different exons. Only a proper RT reaction resulted in an amplification of a PCR product of the correct fragment size. After 2% agarose gel electrophoresis, scanning densitometry (Epson GT-8000 scanner, Epson; Tokyo) was used to determine the peak areas and assess the relative amounts of mRNA in ethiclium bromide-stained bands. The intensity of each band was quantified with NIH image (Version 1.61) on a Macintosh computer. M6PR, Apg5, Apg12 and clathrin mRNA levels were normalized in relation to GAPDH mRNA levels.
Statistical analysis
Differences between control specimens and disease specimens were evaluated with the unpaired t test. A P value of <0.05 was considered statistically significant.
Results
Histologic, histochemical and immunohistochemical studies
Our histologic and histochemical findings in s-IBM muscles corresponded to those of previous reports [7]. All muscles from patients with IBM showed mild to moderate accumulation of rimmed vacuoles, predominantly in the atrophic fibers. Rimmed vacuoles were observed in 1.9 to 18.1% of the 400 muscle fibers in each s-IBM specimen with evidence of inflammation such as mononuclear cell infiltration or perivascular cuffing. Muscles from patients with ALS or peripheral neuropathy showed disease-specific pathologic changes, but no rimmed vacuoles. The control specimens had a normal appearance and no rimmed vacuoles (Table 1).
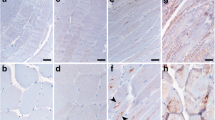
Immunohistochemical findings for clathrin in control and diseased muscles were similar to those described elsewhere [16, 18]. While most muscle fibers in control specimens showed little or no reaction for clathrin, some showed fine granular sarcoplasmic staining (Fig. 1a). In specimens from patients with ALS, peripheral neuropathy or s-IBM, numerous clathrin-positive granules were often scattered throughout the cytoplasm and, to a lesser extent, in the subsarcolemmal regions (Fig. 1b–c). Reactivity for clathrin in s-IBM muscles was stronger than that in the control, ALS or peripheral muscles.

Immunostaining for anti-clathrin antibody in frozen muscle sections obtained from control subjects and patients with neuromuscular disorders. Most muscle fibers in normal control specimens show little or no clathrin reactiviting (a), whereas clathrin-positive fibers are more frequent in muscles from patients with inclusion-body myositis (IBM) (b) and amyotrophic lateral sclerosis/peripheral neuropathy (c). Clathrin-positive granules are scattered through the cytoplasm and are found to a lesser extent in subsarcolemmal regions. These granules are often observed inside or on the rim of rimmed vacuoles (b). Reactivity for clathrin in DMRV specimens is stronger than that in control and other diseased specimens. Bar =50 μm
Most muscle fibers in control specimens showed no reaction for M6PR; some showed fine granular sarcolemmal staining, but sarcoplasmic staining was quite rare (Fig. 2a). M6PR was detected in the plasma membrane and often in the cytoplasm of s-IBM, ALS and peripheral neuropathy specimens (Fig. 2b–d). These granules were more abundant in s-IBM and ALS/peripheral neuropathy muscles than in control muscles and were more frequent in s-IBM than in ALS and peripheral neuropathy muscles. In ALS and peripheral neuropathy muscles, M6PR-positive granules appeared mainly in the subsarcolemmal regions or diffusely in the cytoplasm of atrophic fibers. In s-IBM muscles, M6PR-positive granules were often observed in fibers that appeared normal as well as in atrophic and vacuolated fibers (Fig. 2b). M6PR was sometimes positive within the vacuoles, but the rim of the rimmed vacuoles stained only rarely for M6PR (Fig. 2c). Quantitative immunohistochemistry analysis showed the following percentages of strongly M6PR- and clathrin-positive fibers: 47.6±13.3% and 48.4±12.6% in IBM; 18.5±15.9% and 11.1±9.2% in ALS/peripheral neuropathy; and 8.4±9.5% and 7.5±4.7% in control muscles (Table 1). The percentages were significantly higher in IBM specimens than in controls (P<0.0001) and ALS/peripheral neuropathy specimens (M6PR, P=0.002; clathrin, P<0.0001). The frequency of these fibers tended to be higher in ALS/peripheral neuropathy specimens than in controls, but the difference was not statistically significant.

Immunostaining for anti-mannose 6-phosphate receptor (M6PR) antibody in muscles from control subjects and patients with neuromuscular disorders. Immunoreactivity for M6PR is markedly stronger in muscles from sporadic inclusion body myositis (s-IBM) patients (b, c) than in muscles from control subjects (a) or patients with amyotrophic lateral sclerosis (ALS) and peripheral neuropathy (d). M6PR-positive granules in s-IBM (b) are often observed in the cytoplasm, in addition to sarcolemma, of muscle fibers that appeared normal as well as atrophic and vacuolated fibers, whereas in ALS muscles these granules are especially seen in small angular fibers (d). These granules are occasionally observed inside of rimmed vacuoles (c). Bar =50 μm
Semiquatitative RT-PCR
To test for possibile contamination of chromosomal DNA during RNA extraction, we performed PCR amplification without reverse transcription using the extracted RNA as a template. No bands appeared with 35 cycles of PCR, indicating the absence of contaminating DNA.
In the present study, we found M6PR, clathrin and hApg5 and hApg12 mRNAs to be constitutively expressed in the IBM and control muscles. The mRNA levels of these genes are given in Fig. 3a. Scanning densitometry showed increased expression of hApg5 and hApg12 mRNAs in s-IBM and ALS/peripheral neuropathy muscles in comparison to expression in normal muscles; the respective levels in s-IBM and ALS/peripheral neuropathy muscles were 1.5 and 1.8 times the control muscle levels. There was a significant increase in the s-IBM muscles relative to the control muscles (hApg5, P=0.001; hApg12, P=0.042) and ALS/peripheral neuropathy (hApg5, P=0.008; hApg12, NS). Expressions of M6PR and clathrin mRNA in s-IBM muscles were higher than expressions in ALS/peripheral neuropathy (M6PR, P=0.017; clathrin, P=0.043) and control muscles (M6PR, P=0.049; clathrin, P=0.013; Fig. 3b); the expression of M6PR and clathrin mRNA in s-IBM muscles was 1.5 and 1.6 times that of the controls, respectively.

Semiquantitative analysis of relative expression of mannose 6-phosphate receptor (M6PR), clathrin, hApg5, hApg12 and GAPDH m RNA in the biopsied muscles from controls and patients with neuromuscular disorders based on reverse transcriptase (RT-PCR). Ten-microgram PCR products were subjected to 3% agarose gel electrophoresis, stained with ethidium bromide and visualized by illumination with ultraviolet light. Lanes 2 to 9, sporadic inclusion body myositis (s-IBM); lanes 10 and 11, amyotrophic lateral sclerosis (ALS); lane 12, peripheral neuropathy; lanes 13 to 15, controls. b Densitometric quantification of M6PR, clathrin, and hApg5 and hApg12 mRNA expressions in control and diseased muscles. Scanning densitometry shows an increased expression of these genes' mRNA in s-IBM as compared with ALS/peripheral neuropathy and controls (*P<0.05, **P<0.01). PN peripheral neuropathy
Discussion
Despite the cloning of hApg5 and hApg12 genes from human tissues including skeletal muscle, expression of these gene mRNAs has not been yet observed in any diseased human muscle. In the present study, we showed expression of both hApg12 and hApg5 genes in normal and diseased human muscles. This suggests that the hApg12-conjugated hApg5 system may play an important role in physiological and pathological conditions in human muscles. Previous reports have indicated that the Apg12-conjugated Apg5 system may be involved in the function of autophagic vacuoles or completion in yeast cells [22, 23, 24]. Our study showed hApg12 and hApg5 mRNA levels to be significantly increased in s-IBM muscles, which have numerous rimmed vacuoles within muscle fibers, compared to levels in normal and ALS/peripheral neuropathy muscles without rimmed vacuoles. As described previously, rimmed vacuoles may include many autophagic vacuoles in addition to autolysosmes [6, 13, 15]. The hApg12-conjugated hApg5 gene, therefore, may be well conserved and may be involved in autophagic vacuole formation in human cells as well as in yeast cells [22, 23, 24]. This may reflect an increase in autophagy activity in s-IBM muscles, resulting in increased autophagic vacuole formation.
We also found that clathrin protein and its mRNA levels increased in s-IBM muscles. We reported that muscles from DMRV patients showed more numerous strongly clathrin-positive granules in the cytoplasm of muscle fibers than did muscles from patients with Duchenne and Becker muscular dystrophies, ALS, or peripheral neuropathies or muscles from control subjects, suggesting a relatively disease-specific upregulation of clathrin [18, 25]. These results suggest that the number of clathrin-coated vesicles increases in s-IBM-affected muscles as well as in DMRV-affected muscles. Clathrin levels were also elevated in denervated muscles from chloroquine-affected rats, which have numerous autophagic vacuoles in their muscle fibers [17]. Clathrin-coated vesicles are involved in receptor-mediated intracellular transport pathways, i.e., the export of aggregated materials from the trans-Golgi network of the Golgi apparatus for regulated secretion, transfer of lysosomal enzymes from the trans-Golgi network to lysosomes and receptor-mediated endocytosis at the plasma membrane [27, 28]. Hence, increased clathrin in s-IBM muscle may reflect an increase in secretory activity involving transport from the trans-Golgi network or in endocytotic activity at the plasma membrane in these muscles [16, 28].
The lysosome system including the trans-Golgi network/early endosome pathway and autophagic vacuole/autolysosome pathway is responsible for protein degradation or turnover of intracellular protein in skeletal muscle. Especially in the lysosome system, M6PRs mediate the transport of newly synthesized lysosomal enzymes from the secretory pathway via the trans-Golgi network to the early endosome [2, 12, 19, 20, 29]. M6PRs are found in the Golgi apparatus, the trans-Golgi network, prelysosomal compartments and the endosome and on the plasma membrane [21]. Most of the control muscle fibers in the present study did not react positively for M6PR, but some M6PR-positve granules were observed exclusively on the plasma membrane. The M6PR-positive granules were often present in the cytoplasm as well as on the plasma membrane of muscle fibers in s-IBM muscles and to some extent in ALS/neuropathy muscles. In s-IBM muscles, these granules were observed in the cytoplasm of muscle fibers with or without rimmed vacuoles, and occasionally within the vacuoles, whereas in ALS or peripheral neuropathy muscles, these granules were observed mainly in atrophic fibers. M6PR was significantly more numerous in s-IBM muscle fibers than in the control muscle fibers or ALS and peripheral neuropathy muscle fibers. The levels of M6PR mRNA also increased in s-IBM muscles in comparison to normal and ALS/peripheral neuropathy muscles. This may reflect an increase in the amount of M6PR, suggesting an eventual increase in the transport of lysosome enzyme from the trans-Golgi network to the early endosome and a relatively specific upregulation of M6PR as well as clathrin in s-IBM muscles.
In the present study, mRNA or protein levels of major lysosome-related proteins such as M6PR, clathrin and hApg5 and hApg12 increased in s-IBM muscles as well as in DMRV muscles [18]. This suggests that the lysosome system, especially the transport of newly synthesized lysosomal enzymes from the secretory pathway via the trans-Golgi network and autophagic vacuole formation, is activated in s-IBM muscles. However, etiology and pathomechanism of abnormal accumulation of rimmed vacuoles consisting many autophagic vacuoles and autolysosmes are still unknown. Recently, the UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase (GNE) gene mutation has been detected in cases of familial IBM and DMRV [5, 10]. The same gene mutation has been detected in cases of Salla disease with sialuria [5]. In this disease, ultrastructural analysis of blood lymphocytes, the skin and the liver reveals abnormal lysosomal morphology [1], suggesting autophagic vacuoles or autolysosome. Accumulation of autophagic vacuoles/autolysosome has appeared in both diseases with a GNE gene abnormality, suggesting that a certain lysosome system abnormality may be responsible for sporadic IBM development.
References
Aula P, Autio S, Raivio KO, Aula P, Autio S, Raivio KO, Rapola J, Thoden CJ, Koskela SL, Yamashina I (1979) "Salla disease": a new lysosomal storage disorder. Arch Neurol 36:88–94
Bruder G, Wiedenmann B (1986) Identification of a distinct 9S form of soluble clathrin in cultured cells and tissues. Exp Cell Res 164:449–462
De Duve C, Wattiaux R (1986) Functions of lysosomes. Annu Rev Physiol 28:435–92
Dubowitz V, Brooke MH (1973) Muscle biopsy−a modern approach. WB Saunders, Philadelphia
Eisenberg I, Avidan N, Potikha T, Hochner H, Chen M, Olender T, Barash M, Shemesh M, Sadeh M, Grabov-Nardini G, Shmilevich I, Friedmann A, Karpati G, Bradley WG, Baumbach L, Lancet D, Asher EB, Beckmann JS, Argov Z, Mitrani-Rosenbaum S (2001) The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nat Genet 29:83–87
Fukuhara N, Kumamoto T, Tsubaki T (1980) Rimmed vacuoles. Acta Neuropathol (Berl) 51:229–235
Griggs RC, Askanas V, DiMauro S, Engel A, Karpati G, Mendell JR, Rowland LP (1995) Inclusion body myositis and myopathies. Ann Neurol 38:705–713
Hammond EM, Brunet CL, Johnson GD, Parkhill J, Milner AE, Brady G, Gregory CD, Grand RJ (1998) Homology between a human apoptosis specific protein and the product of APG5, a gene involved in autophagy in yeast. FEBS Lett 425:391–395
Ii K, Hizawa K, Nonaka I, Sugita H, Kominami E, Katunuma N (1986) Abnormal increases of lysosomal cysteinine proteinases in rimmed vacuoles in the skeletal muscle. Am J Pathol 122:193–198
Kayashima T, Matsuo H, Satoh A, Ohta T, Yoshiura K, Matsumoto N, Nakane Y, Niikawa N, Kishino T (2002) Nonaka myopathy is caused by mutations in the UDP-N-acetylglucosamine-2-epimerase/N-acetylmannosamine kinase gene (GNE). J Hum Genet 47:77–79
Klionsky DJ, Ohsumi Y (1999) Vacuolar import of proteins and organelles from the cytoplasm. Annu Rev Cell Dev Biol 15:1–32
Klumperman J, Kuliawat R, Griffith JM, Geuze HJ, Arvan P (1998) Mannose 6-phosphate receptors are sorted from immature secretory granules via adaptor protein AP-1, clathrin, and syntaxin 6-positive vesicles. J Cell Biol 141:359–371
Kumamoto T, Fukuhara N, Nagashima M, Kanda T, Wakabayashi M (1982) Distal myopathy: histochemical and ultrastructural studies. Arch Neurol 39:367–371
Kumamoto T, Ueyama H, Watanabe S, Murakami T, Araki S (1993) Effect of denervation on overdevelopment of chloroquine-induced autophagic vacuoles in skeletal muscles. Muscle Nerve 16:819–826
Kumamoto T, Ueyama H, Watanabe S, Kominami E, Ando M (1994) Muscle fiber degradation in distal myopathy with rimmed vacuoles. Acta Neuropathol (Berl) 87:143–148.
Kumamoto T, Abe T, Nagao S, Ueyama H, Tsuda T (1998) Immunohistochemical study of clathrin in distal myopathy with rimmed vacuoles. Acta Neuropathol (Berl) 95:571–575
Kumamoto T, Nagao SI, Sugihara R, Abe T, Ueyama H, Tsuda T (1998) Effect of chloroquine-induced myopathy on rat soleus muscle sarcoplasm and expression of clathrin. Muscle Nerve 21:665–668.
Kumamoto T, Ito T, Horinouchi H, Ueyama H, Toyoshima I, Tsuda T (2000) Increased lysosome-related proteins in the skeletal muscles of distal myopathy with rimmed vacuoles. Muscle Nerve 23:1686–1693
Le Borgne R, Hoflack B (1997) Mannose 6-phosphate receptors regulate the formation of clathrin-coated vesicles in the TGN. J Cell Biol 137:335–345
Lemansky P, Hasilik A, von Figura K, Helmy S, Fishman J, Fine RE, Kedersha NL, Rome LH (1987) Lysosomal enzyme precursors in coated vesicles derived from the exocytic and endocytic pathways. J Cell Biol 104:1743–1748
Ma ZM, Grubb JH, Sly WS (1991) Cloning, sequencing, and functional characterization of the murine 46-kDa mannose 6-phosphate receptor. J Biol Chem 266:10589–10595
Mizushima N, Sugita H, Yoshimori T, Ohsumi Y (1998) A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J Biol Chem 273:33889–33892
Mizushima N, Noda T, Ohsumi Y (1999) Apg16p is required for the function of the Apg12p-Apg5p conjugate in the yeast autophagy pathway. EMBO J 18:3888–3896
Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y, Yoshimori T (2001) Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol 152:657–668
Nomura N, Miyajima N, Sazuka T, Tanaka A, Kawarabayasi Y, Sato S, Nagase T, Seki N, Ishikawa K, Tabata S (1994) Prediction of the coding sequences of unidentified human genes. I. The coding sequences of 40 new genes (KIAA0001-KIAA0040) deduced by analysis of randomly sampled cDNA clones from human immature myeloid cell line KG-1. DNA Res 1:27–35
Nonaka I, Sunohara N, Ishiura S, Satoyoshi E (1981) Familial distal myopathy with rimmed vacuole and lamellar (myeloid) body formation. J Neurol Sci 51:141–155
Pearse BM, Robinson MS (1990) Clathrin, adaptors, and sorting. Annu Rev Cell Biol 6:151–171.
Pley U, Parham P (1993) Clathrin: its role in receptor-mediated vesicular transport and specialized functions in neurons. Crit Rev Biochem Mol Biol 28:431–464
Schekman R, Orci L (1996) Coat proteins and vesicle budding. Science 271:1526–1533
Tajsharghi H, Thornell LE, Darin N, Martinsson T, Kyllerman M, Wahlstrom J, Oldfors A (2002) Myosin heavy chain IIa gene mutation E706 K is pathogenic and its expression increases with age. Neurology 58:780–786
Tso JY, Sun XH, Kao TH, Reece KS, Wu R (1985) Isolation and characterization of rat and human glyceraldehyde-3-phosphate dehydrogenase cDNAs: genomic complexity and molecular evolution of the gene. Nucleic Acids Res 13:2485–2502
Tsukada M, Ohsumi Y (1993) Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett 333:169–174
Udd B, Partanen J, Halonen P, Falck B, Hakamies L, Heikkila H, Ingo S, Kalimo H, Kaariainen H, Laulumaa V, Paljarvi L, Rapola J, Reunanen M, Sonninen V, Somer H (1993) Tibial muscular dystrophy. Late adult-onset distal myopathy in 66 Finnish patients. Arch Neurol 50:604–608
Yan C, Ikezoe K, Nonaka I (2001) Apoptotic muscle fiber degeneration in distal myopathy with rimmed vacuoles. Acta Neuropathol (Berl) 101:9–16
Acknowledgements
We are grateful to Ms. M. Ono and Ms. K. Hirano for their technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kumamoto, T., Ueyama, H., Tsumura, H. et al. Expression of lysosome-related proteins and genes in the skeletal muscles of inclusion body myositis. Acta Neuropathol 107, 59–65 (2004). https://doi.org/10.1007/s00401-003-0774-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-003-0774-2