
Abstract
Diabetes mellitus (DM) significantly impairs patients’ quality of life, primarily because of its complications, which are the leading cause of mortality among individuals with the disease. Autophagy has emerged as a key process closely associated with DM, including its complications such as diabetic nephropathy (DN). DN is a major complication of DM, contributing significantly to chronic kidney disease and renal failure. The intricate connection between autophagy and DM, including DN, highlights the potential for new therapeutic targets. This review examines the interplay between autophagy and these conditions, aiming to uncover novel approaches to treatment and enhance our understanding of their underlying pathophysiology. It also explores the role of autophagy in maintaining renal homeostasis and its involvement in the development and progression of DM and DN. Furthermore, the review discusses natural compounds that may alleviate these conditions by modulating autophagy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
This review explains how autophagy is associated with type 1 and type 2 diabetes mellitus, detailing mechanisms that cause a decrease in autophagy in these conditions |
The mechanisms of kidney damage in diabetic nephropathy, particularly the role of oxidative stress and its link to autophagy, are described in the review |
The role of autophagy in maintaining kidney health is discussed, and key molecules that are targeted by autophagy in diabetic nephropathy are identified |
This review also lists natural compounds that could potentially reduce the impact of diabetes mellitus and diabetic nephropathy by influencing autophagy |
Introduction
Diabetes mellitus (DM) results from a complex interaction of genetic, environmental, and other factors. Characterized by chronic hyperglycemia, DM can damage islet β-cells and cause insulin resistance (IR). These conditions disrupt glucose metabolism and can trigger chronic low-grade inflammation.
DM complications severely affect quality of life and are a leading cause of death, often due to sustained hyperglycemia and ongoing metabolic issues. Diabetic nephropathy (DN), a key microvascular complication of DM, not only contributes significantly to chronic kidney disease, potentially leading to renal failure and death, but also elevates the risk of cardiovascular disease and cancer [1, 2]. Yet, the underlying causes of DN are not fully understood, and effective treatments for its progression are scarce [3].
Autophagy, a fundamental metabolic process, allows cells to break down and recycle abnormal proteins and damaged organelles for energy. Recent research indicates that modulating autophagy is a promising therapeutic approach for DM and its complications. Additionally, some established drugs used to treat DM and DN have been found to induce autophagy [4,5,6]. Consequently, the modulation of autophagy-related signaling pathways has garnered increasing interest. Understanding autophagy’s impact on DM and DN, and its molecular mechanisms, may lead to innovative treatments.
Autophagy is essential for pancreatic β-cells, maintaining their function by clearing damaged proteins and organelles. Dysregulated autophagy can cause β-cell dysfunction or death, impacting insulin production and release [7,8,9]. Autophagy also regulates the cell’s response to nutrients in DM by controlling growth and metabolism via the mammalian target of rapamycin (mTOR, a serine/threonine protein kinase that negatively regulates autophagy) pathway [10,11,12,13,14]. The activation of adenosine 5ʹ-monophosphate-activated protein kinase (AMPK) can lead to the inhibition of mTOR. This pathway has been proven to be effective in alleviating IR [4, 5]. Hyperglycemia and lipid metabolic disorders (glucolipotoxicity) that appear in the prediabetic stage are major risk factors for β-cell functional damage. Studies have found that glucolipotoxicity causes excessive activation of autophagy in β-cells, leading to β-cell damage [8, 15, 16].
Modulating the AMPK/mTOR/sirtuin 1 (SIRT1) pathway affects both β-cell homeostasis and kidney health by influencing autophagy. In DN, the activation of the AMPK/mTOR/SIRT1 pathway can enhance autophagy activity and reduce kidney damage [17, 18]. On the other hand, the phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt)/mTOR pathway negatively regulates DN by inhibiting autophagy, causing kidney damage and fibrosis [19, 20]. In DN, autophagy is linked to inflammatory responses, with high glucose (HG) conditions correlating with reduced autophagy and increased kidney damage [21,22,23,24,25,26,27]. Mitochondrial autophagy, which clears damaged mitochondria, is vital for maintaining cellular homeostasis [28]. In the context of DN or a HG environment, mitochondrial autophagy becomes disrupted in renal glomerular mesangial cells (MCs) and proximal tubular epithelial cells (PTECs), which is associated with the AMPK/ UNC-51-like kinase 1 (ULK1)/phosphatase and tensin homolog deleted on chromosome ten (PTEN)-induced putative kinase 1 (PINK1)/parkin pathway. This disruption can lead to increased oxidative stress and inflammatory responses, thereby accelerating the progression of DN [17, 29, 30].
This review synthesizes findings across disciplines to offer a multidisciplinary perspective on targeting autophagy for treating DM and DN. Additionally, the review lists and discusses a variety of natural compounds that may alleviate DM and DN by modulating autophagy. It not only reviews preclinical studies but also explores how these findings can be translated into clinical applications through the limited existing clinical trials and case reports, including improvements to current treatment methods and the development of new drugs. The review particularly emphasizes the importance of assessing safety and efficacy when considering autophagy modulators, a critical factor in drug development. Finally, the review concludes by proposing future research directions and challenges, aiming to foster further advancements.
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Autophagy and DM
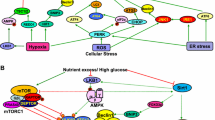
The primary pathological changes in DM include islet β-cell apoptosis in type 1 diabetes mellitus (T1DM) and IR in type 2 diabetes mellitus (T2DM). Autophagy plays a key role in both T1DM and T2DM processes [3] (Fig. 1). Consequently, the interaction between autophagy and DM has been extensively studied in both in vivo and in vitro settings.

Mechanisms of autophagy inhibition in T1DM and T2DM. This figure was created using Figdraw tools (www.figdraw.com). Abbreviations: 1-DSL 1-deoxysphingolipids, CUL3 cullin 3, HG high glucose, IRS-1 insulin receptor substrate-1, KLHL9/13 Kelch-like protein 9/13, ROS reactive oxygen species, T1DM type 1 diabetes mellitus, T2DM type 2 diabetes mellitus
Apoptosis and Regeneration of Islet β-Cells
In T1DM, functional islet β-cell mass is drastically reduced to < 10% of normal levels because of autoimmune-mediated apoptosis, impairing insulin secretion [9]. Early in T1DM, autophagy is paradoxically overactivated by HG-induced oxidative stress, leading to lysosome depletion and autophagy deficiency [7, 8]. This deficiency worsens mitochondrial damage and oxidative stress, further inducing lysosomal damage and β-cell apoptosis. For example, the study by Canet et al. [7] found that T1DM can impair mitochondrial function and promote oxidative stress and autophagy in leukocytes. Research shows that intermittent hypoxia can stimulate β-cell autophagy but may also induce apoptosis and worsen dysfunction [31]. This adverse effect can be mitigated by rapamycin, which enhances autophagy induction. In addition to hypoxia, the combination of glucose and palmitic acid (PA), a saturated fatty acid, can induce β-cell apoptosis and inhibit autophagy. Mitogen-activated protein kinase phosphatase-5 (MKP-5), a regulator in the extracellular signal-related kinase (MAPK) pathway, plays a role in lipid metabolism and reducing lipid toxicity. Insufficient levels of MKP-5 can lead to increased lipid toxicity. Song et al. [32] found that overexpression of MKP-5 reversed the impairment of insulin secretion and abnormal decrease in the expression of islet function genes induced by glucose and PA, thereby maintaining normal insulin secretory function, while the absence of MKP-5 exacerbated islet cell dysfunction. Additionally, MKP-5 overexpression also reduced the production of reactive oxygen species (ROS) in response to PA and the increase in inflammation-related genes. On the other hand, Zhao et al. [33] confirmed that MKP-5 overexpression improved the inhibition of autophagic signaling in pancreatic β-cells by enhancing the c-Jun N-terminal kinase (JNK) and p38MAPK pathways, thereby suppressing the apoptosis, dysfunction, inflammatory response, and oxidative stress induced by PA. On a molecular level, quercetin has been shown to significantly increase the expression of autophagy-related markers and decrease the expression of ER stress indicators as well as several micro-ribonucleic acids (RNAs) such as eukaryotic elongation factor 2, JNK, X-box binding protein 1, C/EBP homologous protein, heavy-chain binding protein, and activating transcription factor 6 (ATF-6). These micro-RNAs regulate the signaling pathways of both autophagy and ER stress, potentially enhancing insulin secretion [21]. Furthermore, Bai et al. [34] discovered that rno-circRNA-008565 can inhibit JNK expression and promote autophagy in rat islet β-cells through the MAPK signaling pathway. This finding suggests that β-cell autophagy is modulated at the genetic level and could be a target for therapeutic intervention in T1DM.
Autophagy is recognized for its crucial role in promoting β-cell regeneration, which can have profound implications for DM therapy. In a study by Hu et al. [11], it was discovered that the overexpression of the wild-type hCDC14A gene in murine β-TC3 cells enhances their viability, proliferation, and autophagy. This enhancement is believed to be associated with increased islet secretion and a reduction in cell apoptosis, potentially through the inhibition of the mTOR pathway and activation of the AMPK pathway. These pathways are pivotal in cellular metabolism and growth, and their modulation by hCDC14A overexpression suggests a mechanism by which β-cell function and mass could be preserved or even expanded. Intermittent fasting can counteract the inhibitory effects of a high-fat diet (HFD) on autophagy in β-cells, supporting cell survival [12]. This restoration is mediated through the AMPK/mTOR pathway, which is a key regulator of cellular energy homeostasis. By activating autophagy, intermittent fasting helps maintain the quality of cellular organelles through the autophagy-lysosome pathway, which is essential for β-cell health and function. As a result, these fasting-induced changes can enhance β-cell survival and improve glucose tolerance, offering a potential dietary strategy for managing DM and promoting β-cell regeneration.
IR
IR, a key mechanism in T2DM, is influenced by autophagy, which regulates β-cells and insulin-responsive tissues, such as the liver, skeletal muscle, and adipose tissue [3]. Mice lacking the autophagy-related 16 like 1 (Atg16L1) gene show increased expression of Kelch-like protein (KLHL) 9 and KLHL13, which interact with Cullin 3, to degrade insulin receptor substrate-1 (IRS-1, a key mediator of insulin signaling), contributing to IR [35]. Studies by Yang et al. [36] indicate that inducing autophagy can alleviate IR in HFD-fed rats by stabilizing IRS-1. Furthermore, Yang et al. [4] and Nowell et al. [5] have identified that glucagon-like-peptide-1 (GLP-1) receptor agonists, such as liraglutide and exenatide, which are widely used in T2DM treatment, can induce autophagy. This induction of autophagy by GLP-1 receptor agonists is suggested to alleviate IR through the AMPK/mTOR pathway. By modulating this pathway, GLP-1 receptor agonists may improve insulin sensitivity and provide therapeutic benefits for patients with T2DM.
Inflammation in the liver can lead to steatosis, a sign of liver IR, impairing lysosomal function and autophagy. The inducible nitric oxide synthase (iNOS), an inflammatory marker, becomes activated under conditions of obesity and generates nitric oxide. This overproduction can impair lysosomal function, a critical component of autophagy, thereby disrupting the cell's ability to recycle and remove damaged cellular components. Qian et al. [14] discovered that the co-overexpression of mTOR and SLC38A9 can exacerbate lysosomal nitric oxide production, inhibiting autophagy. Conversely, the absence of iNOS can activate the transcription factor EB (TFEB), a master regulator of lysosomal biogenesis. This activation enhances lysosomal function and autophagy, improving hepatocyte insulin sensitivity. However, this beneficial effect can be attenuated by the inhibition of TFEB and Atg7, a key autophagy-related gene. Sitagliptin, a widely prescribed dipeptidyl-peptidase 4 (DPP-4) inhibitor for T2DM, offers another perspective on the interplay between inflammation, autophagy, and IR. It has been shown to suppress inflammatory responses and stimulate autophagy via the AMPK/mTOR pathway, leading to a significant alleviation of liver steatosis and IR [6]. Further research by Du et al. [10] indicates that HFD can excessively activate mammalian target of rapamycin complex 1 (mTORC1), which in turn inhibits the phosphorylation of IRS-1, promoting IR in vivo. This inhibition can result in decreased glucose uptake and impaired glycogen synthesis. The study suggests that enhancing autophagy could be a viable strategy to reduce liver steatosis and IR, offering potential therapeutic benefits in the context of T2DM and metabolic syndrome.
Studies on human skeletal muscle reveal how insulin signaling and mTOR interact, with fasting triggering autophagy for cellular repair. During the fed state, insulin signaling activates mTOR, promoting anabolic processes. In stark contrast, fasting results in insulin signaling having the opposite effect, leading to mTOR inactivation. This inactivation triggers autophagy, a cellular housekeeping mechanism that facilitates the clearance of damaged cellular components and supports cellular repair [13]. Furthermore, a substantial body of evidence indicates that enhancing autophagy activity can effectively improve IR and the balance of glucolipid metabolism in key tissues such as the hypothalamus, skeletal muscle, liver, and adipose tissue. Moreover, autophagy can also increase the number and functionality of β-cells in the pancreas. This suggests that the activation of autophagy represents an innovative approach to the treatment of T2DM [13, 37]. Exercise, like fasting, strongly induces autophagy and can mitigate IR and muscle atrophy through various pathways. The AMPK/ULK1 and mTOR pathways are key players in these processes, with the former activating autophagy and the latter being negatively regulated by it. Interestingly, Yang et al. [38] found that exercise can improve IR by adjusting the gut microbiota, reversing the decrease in total intestinal and plasma short-chain fatty acids and strengthening autophagy in skeletal muscle cells through their interaction with G protein-coupled receptor 43. Studies by Xiang et al. [39] and Cui et al. [40] found that exercise can promote autophagy and enhance insulin sensitivity via the AMPK/mTOR/ULK1 pathway, and it can show superior effects on reducing muscle atrophy than metformin. The aforementioned studies offer a complex but effective mechanism by which exercise can enhance metabolic health.
Adipose tissue serves as the central hub for fat storage, energy expenditure, glucose and insulin metabolism, and hormonal regulation, playing a crucial role in maintaining systemic energy metabolism homeostasis. Obesity triggers inflammation in white adipose tissue, increasing the levels of pro-inflammatory mediators [2]. Furthermore, in individuals with IR, excessive lipolysis and impaired lipid formation in adipose tissue lead to the release of cytokines and lipid metabolites [41]. This suggests that the substantial accumulation of adipose tissue could potentially induce IR. Shi et al. [42] discovered that the deletion of Forkhead box O1 (FoxO1) in adipocytes, a transcription factor that regulates autophagy, suppresses B-cell lymphoma/leukemia 2 (Bcl2) interacting protein 3 and activates the FUN14 domain containing 11/dynamin-related protein 1/optic atrophy 1 cascade. They also found upregulation of Atg7 and cathepsin L, which are involved in autophagy. However, the suppression of autophagy via cathepsin L induced by HFD can negate these benefits. This implies that the autophagic clearance of mitochondria is essential for the healthy browning of adipose tissue. Song et al. [43] reported that the absence of phosphatidylinositol 3-kinase catalytic subunit type 3 (PIK3C3) inhibits adipocyte autophagy and impedes the differentiation, survival, and function of adipocytes. This leads to the loss of white adipose tissues and an impaired “browning” process of white adipose tissues, ultimately contributing to dyslipidemia, IR, and T2DM. Notably, this study did not find any changes in glucose tolerance in adipocyte-specific PIK3C3-deficient mice. Xiong et al. [44] also found that apigenin can promote browning by activating autophagy through the inhibition of the PI3K/ Akt/mTOR pathway. While evidence suggests a link between autophagy and IR in adipose tissue, further research is needed to clarify this relationship.
Autophagy and DN
Oxidative Stress and DN
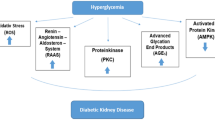
Oxidative stress, central to kidney injury in DN, is closely linked to autophagy. Oxidative stress in DN arises from pathways including the polyol pathway, advanced glycation end product (AGE)-receptor interactions, protein kinase C (PKC) activation, and hexosamine pathway overactivation. Each of these mechanisms plays a significant role in the injury and dysfunction of renal cells in DN (Fig. 2). Yet, clinical studies show that targeting a single mechanism is inadequate to stop DN progression. This suggests that these pathways do not operate in isolation but are interrelated and contribute collectively to the pathogenesis of DN [28, 45]. Therefore, understanding the complex interplay among these mechanisms is crucial for gaining a comprehensive understanding of the overall pathophysiology of DN.

Mechanisms of oxidative stress, autophagy, and DN. This figure was created using Figdraw tools (www.figdraw.com). Abbreviations: AGEs advanced glycation end products, Akt protein kinase B, AMPK adenosine 5ʹ-monophosphate-activated protein kinase, DNA deoxyribonucleic acid, eNOS endothelial nitric oxide synthase, GAPD glyceraldehyde-3-phosphate dehydrogenase, GlcNAc N-acetylglucosamine, HSP72 heat shock protein 72, mTOR mammalian target of rapamycin, P53 tumor protein p53, PAI-1 plasminogen activator inhibitor-1, PINK1 phosphatase and tensin homolog deleted on chromosome ten-induced putative kinase 1, ROS reactive oxygen species, SOD superoxide dismutase, UDP uridine diphosphate
Hyperglycemia activates the polyol pathway, reducing superoxide dismutase (SOD) production and increasing oxidative stress. This decrease in SOD activity results in an insufficient capacity to break down the superoxide radicals generated within the cell, thereby causing an escalation in oxidative stress [46]. Furthermore, Morresi et al. [47, 48] have discovered that high glucose levels can activate the p53 protein and suppress the activity of various antioxidant enzymes, including glutathione peroxidase. This suppression disrupts the delicate balance between cellular oxidation and antioxidation processes, leading to an increase in intracellular oxidative stress. Consequently, this results in elevated levels of reactive ROS and the formation of AGEs. Given that all four aforementioned mechanisms stem from intermediates of glucose metabolism, it is plausible that oxidative stress induced by hyperglycemia acts as an upstream signaling pathway that triggers these pathogenic mechanisms. This highlights the central role of oxidative stress in the etiology of diabetic complications and suggests that interventions targeting oxidative stress may have broad therapeutic benefits in mitigating the renal and other tissue injuries associated with DM.
Hyperglycemia-induced glucose metabolism disorder is a primary cause of increased superoxide production and heightened oxidative stress in the kidney. To mitigate cellular damage from oxidative stress, the body activates an antioxidant defense system designed to neutralize free radicals. Regulating the nicotinamide adenine dinucleotide phosphate, reduced form (NADPH)/nicotinamide adenine dinucleotide phosphate, oxidized form (NADP+) ratio is key to the antioxidant defense system, facilitating glutathione and cysteine interconversion. This dynamic process supports the continuous cycling of these antioxidants, allowing them to be activated or deactivated as needed. Reduced glutathione plays a crucial role as an electron donor, participating in the reduction of hydrogen peroxide (H2O2) to water, thereby preserving intracellular oxidation balance. However, hyperglycemia and oxidative stress can inhibit glyceraldehyde-3-phosphate dehydrogenase, an enzyme in the glycolysis pathway, leading to the activation of the polyol pathway. This activation results in NADPH depletion and a diminished production of reduced glutathione. Consequently, the cell's ability to promptly detoxify excessive superoxide is compromised, exacerbating oxidative damage within the cell [49]. Oxidative damage not only leads to the accumulation of superoxide but can also result in protein carbonylation, inflicting irreversible harm on proteins and serving as a marker of severe, chronic oxidative damage. To preserve intracellular protein stability, cells employ proteasomal and autophagic systems to degrade damaged proteins, both small and large. These mechanisms are essential for minimizing the buildup of oxidatively damaged proteins, especially during periods of mild oxidative stress [50].
Mitochondria initiate oxidative stress and are the main targets for oxidative damage. To maintain mitochondrial homeostasis, the cell employs mitochondrial repair, regeneration, and autophagy degradation. The PINK1/parkin pathway, activated by mitochondrial damage, initiates mitophagy to degrade damaged mitochondria. Through this process, impaired organelles and biomacromolecules are broken down into nucleotide fragments and fatty acids. This not only preserves protein stability within the cell but also releases nutrients into the cytoplasm, which can be repurposed for cellular metabolism and the synthesis of new biomolecules [28]. In the context of DN, renal autophagy can be impaired because of deoxyribonucleic acid (DNA) damage inflicted by excessive oxidative stress, the downregulation of the AMPK signaling pathway by HG or the upregulation of mTOR, while metformin can activate mitochondrial autophagy through the AMPK pathway and alleviate renal oxidative stress and tubulointerstitial fibrosis in diabetic mice [51]. The accumulation of superoxide can lead to damage in mitochondrial DNA, causing an increased accumulation of dysfunctional mitochondria. This results in a reduced number of normal mitochondria, decreased glycolysis rate, and insufficient adenosine triphosphate (ATP) supply. These factors further exacerbate the inhibitory effects of oxidative stress on renal structure and function [51, 52]. Li et al. [52] found that by activating DNA damage repair, the levels of nicotinamide adenine dinucleotide (NAD+) can be reduced and mitochondrial function can be improved in diabetic rats, thereby reducing the production of superoxide under HG conditions. Moreover, the inhibition of autophagy due to oxidative stress can lead to a further downregulation of autophagy levels, creating a vicious cycle that perpetuates cellular damage and contributes to the progression of DN.
The activation of PKC and intensification of oxidative stress are key regulatory factors that can modulate the expression of various genes implicated in the pathogenesis of DN. Specifically, these factors can induce the production of collagen fibers and pro-inflammatory agents through the action of transcription factors such as β and the plasminogen activator inhibitor-1 (PAI-1), as well as the nuclear factor kappa B (NF-κB). This induction is instrumental in accelerating the process of renal fibrosis, a hallmark of DN. Jiang et al. [53] have reported findings in diabetic mice that suggest a significant role for the PKC-α/P66SHC pathway in DN-mediated podocyte injury. In their study, the inhibition of Klotho protein expression, a protein known for its anti-aging effects and protective role in the kidney, led to the upregulation of PKC-α and p66SHC, along with an increase in ROS generation. These changes were associated with podocyte injury and the onset of proteinuria, a critical indicator of renal dysfunction. The PKCα/P66SHC pathway, therefore, appears to play a pivotal role in the development and progression of DN, particularly in the context of podocyte injury. Understanding the mechanisms by which PKCα and P66SHC contribute to renal pathology may offer new therapeutic targets for the treatment of DN and the preservation of renal function.
The overactivation of the hexosamine pathway, a biosynthetic route involved in the utilization of glucose, culminates in the production of uridine diphosphate and N-acetylglucosamine (UDP-GlcNAc). Excess UDP-GlcNAc can inhibit the Akt/endothelial nitric oxide synthase (eNOS) and heat shock protein 72 phosphorylation through a kinase-like effect on Ser/Thr phosphorylation sites. This inhibition disrupts the normal signaling pathways that are protective against cellular stress. The dysregulation of these pathways results in the upregulation of profibrotic factors such as transforming growth factor-β (TGF-β) and plasminogen activator inhibitor-1 (PAI-1). TGF-β, in particular, is a potent inducer of fibrosis, and PAI-1 is known to contribute to the pathogenesis of DN by affecting fibrinolysis and wound healing. The increased expression of these factors drives DN-related oxidative stress and renal fibrosis, leading to the progressive loss of kidney function characteristic of the disease [54]. In conclusion, understanding the interplay among oxidative stress pathways is crucial for developing comprehensive DN treatments.
Autophagy and Renal Homeostasis
Podocytes are specialized, terminally differentiated cells within the glomerulus that are essential for preserving the integrity of the glomerular filtration barrier. Their damage and loss are primary contributors to the development of glomerulosclerosis and significant proteinuria in patients with DN. Podocytes require robust autophagy for physiological function, which is often inhibited early in DN, contributing to podocyte damage. This inhibition accelerates podocyte and endothelial damage, leading to the breakdown of the glomerular filtration barrier, glomerulosclerosis, and ultimately the progression of DN and severe proteinuria [1]. Zhou et al. [55] have identified that the progranulin (PGRN) signal enhances mitochondrial biogenesis and autophagy, mitigating the mitochondrial dysfunction in podocytes induced by HG through the PGRN/SIRT1/peroxisome proliferator-activated receptor γ coactivator 1-α (PGC-1α)/FoxO1 pathway. The inhibition of mitophagy, a specific form of autophagy targeting damaged mitochondria, can negate these benefits, underscoring the involvement of mitophagy in podocyte injury. Hou et al. [56] observed that the activation of AGEs results in decreased lysosomal activity and the inactivation of the transcription factor EB (TFEB), leading to the inhibition of autophagy and damage to podocytes. The induction of autophagy through the use of rapamycin can ameliorate lysosomal dysfunction, reduce proteinuria, and alleviate podocyte damage in DN. These findings collectively highlight the critical role of autophagy in maintaining podocyte homeostasis and the potential therapeutic benefits of targeting autophagy pathways in the treatment of DN.
Phenotypic alterations in PETCs mark one of the earliest indicators of DN, with the severity of tubulointerstitial damage being a strong predictor of patient prognosis. Interestingly, despite only low levels of autophagy being observed in renal PTECs, mice with proximal tubular-specific deletion of Atg5 or Atg7, key autophagy-related genes, exhibit severe mitochondrial dysfunction, albumin accumulation, and deteriorated renal histology, ultimately leading to exacerbated renal function impairment and premature renal failure [57, 58]. Lee et al. [30] also found that autophagy shows a protective effect in resisting the death of PTECs induced by HG. These findings suggests that even a low level of autophagy is critical for the maintenance of renal health. Furthermore, the inhibition of renal tubular glucose reabsorption through the use of sodium-glucose cotransporter 2 (SGLT-2) inhibitors has been shown to increase microtubule-associated protein 1 light chain 3B, lipidated form levels in PTECs, which can mitigate HG-induced oxidative stress and apoptosis [59], suggesting that that the level of glucose uptake may directly influence the autophagic activity within PTECs. Liu et al. [60] demonstrated that tumor necrosis factor α (TNF-α)-induced protein 8-like 1 can disrupt the PINK1/Parkin pathway, which is essential for mitochondrial autophagy and mitochondrial homeostasis in PTECs. Li et al. [22] observed that HG stimulation and AGE overload in PTECs can inhibit autophagy. The impaired degradation of AGEs due to autophagy deficiency can trigger inflammatory responses and exacerbate DN, suggesting that autophagy deficiency in PTECs can contribute to HG-induced inflammation in DN. Fujimura et al. [61] discovered that the absence of mitochondrial autophagy in PTECs results in mitochondrial dysfunction, increased ROS levels, and decreased ATP levels, leading to cellular damage and senescence of renal tubular cells. The induction of mitophagy has been shown to ameliorate renal tubular injury, improve renal function, and effectively inhibit hyperphosphatemia caused by renal injury.
MCs play a vital role in preserving the structural integrity of the glomerular microvascular bed and maintaining the homeostasis of the mesangial matrix. Early pathological changes in DN, such as HG-induced proliferation of MCs, expansion of the mesangial matrix, and thickening of the basement membrane, are indicative of the disease’s progression. Mao et al. [62] found that HG-induced MC proliferation not only increases the level of serum TGFβ1 but also inhibits autophagy. This leads to apoptosis, cytoskeletal rearrangement, and ultimately podocyte injury. Additional research has indicated that AGEs and HG can suppress the autophagy levels in MCs through the receptor of AGEs/signal transducer and activator of transcription 5 pathway and by inhibiting the expression of F-box and WD repeat domain containing 7, contributing to accelerated cellular senescence and exacerbating renal inflammation and fibrosis in DN [63,64,65]. Meng et al. [63] found that the application of insulin in conjunction with improved autophagy led to further improvements in various aspects of mice with DN, including blood glucose, glycated hemoglobin (HbA1c), glucose tolerance, renal function, oxidative stress, and AGEs. E et al. [29] and Wen et al. [66] discovered that activating autophagy in MCs can mitigate AGE-induced MC injury by scavenging ROS and inhibit apoptosis and oxidative stress. These findings highlight the protective role of autophagy in MCs and suggest that inducing autophagic processes could offer a promising therapeutic strategy for the repair of MCs injury and slow the progression of DN. Furthermore, the studies provide a foundation for further investigation into the therapeutic potential of modulating autophagy in MCs as a treatment for DN.
Injury to glomerular endothelial cells (GECs) is a key early factor in DN development, with autophagy playing a crucial role in maintaining GEC integrity. Conditions such as HG levels, ROS accumulation, and inhibition of autophagy can trigger GECs dysfunction and apoptosis, which in turn can lead to proteinuria and early renal injury. The damage to GECs has a cascading effect, impacting mesangial cells, glomerular epithelial cells, and podocytes, ultimately exacerbating the progression of DN [67]. Takagaki et al. [24] provided evidence that the knockdown of Atg5, a critical gene involved in autophagy, can intensify GECs damage and worsen DN. This underscores the importance of autophagy in maintaining the integrity of GECs. Furthermore, Tian et al. [68] discovered that inducing autophagy in GECs through the activation of the AMPK/nuclear factor-erythroid 2-related factor 2 (Nrf2) pathway can reduce kidney injury markers and lower the levels of serum creatinine, urinary protein, and urea nitrogen in mice with DN. The collective findings from these studies suggest that autophagy in GECs plays a crucial role in DN treatment and management. Enhancing autophagy may offer a valuable strategy to protect GECs, ameliorate renal injury, and possibly slow the progression of DN. In summary, modulating autophagy in renal cells is a promising therapeutic strategy for DN treatment, protecting renal health and function.
DN, characterized by pro-inflammatory factor overexpression and inflammatory cell infiltration, involves macrophage autophagy in polarization and fibrosis. In DN, there is an increase in macrophage infiltration and phenotypic changes in the kidneys of both mice and patients, which are linked to proteinuria and renal fibrosis. Macrophage autophagy plays a critical role in polarization, chronic inflammation, and organ fibrosis. Zhao et al. [26] found that HG stimulates macrophage exosome secretion, inhibiting PTEC autophagy and affecting renal cell function by targeting Atg9b. This finding suggests that HG-induced changes in macrophages can affect renal cell function through modulation of autophagy. Yan et al. [25] proposed that the activation of the TFEB and induction of autophagy can promote the M2 polarization of macrophages. This shift towards an anti-inflammatory phenotype can help alleviate inflammation and renal injury in DN, indicating the potential of macrophage autophagy as a therapeutic target. Furthermore, in the glomerular tissue of patients with DN, there are significant changes in the number of various immune cells and multiple immune-related signaling pathways. The presence of specific phenotypes of T cells, B cells, dendritic cells, and natural killer cells can either increase or decrease the risk of DN [69,70,71]. Wang et al. [72] conducted in vivo research showing that knockdown of SGLT-2 can significantly alleviate the Th17/Treg imbalance in db/db mice and inhibit the progression of DN. Given the association between SGLT-2 and autophagy [59], it is plausible that other immune cells may also be involved in the progression of inflammation and fibrosis in DN through autophagy-related mechanisms. However, more evidence is needed to fully understand and support this hypothesis.
Autophagic-related Molecules and DN
Autophagy is a complex process that involves a multitude of molecules and signaling pathways, some of which have been proven to be associated with DN (Table 1).
mTOR, a central regulator of autophagy, exists in two distinct complexes, mTORC1 and mTORC2. These complexes are influenced by a variety of upstream signaling pathways and are sensitive to intracellular nutrient levels and redox states. mTOR directly or indirectly regulates the expression of genes or protein modifications associated with autophagy. Previous research has established a link between the overexpression of mTOR, resultant inhibition of autophagy, and IR. This inhibition has been shown to promote the synthesis and accumulation of AGEs, which are implicated in the pathogenesis of diabetic complications [10, 73]. Further studies have demonstrated that inhibiting mTOR expression, thereby inducing autophagy, can have significant therapeutic effects in DN. Enhancement of autophagy through mTOR inhibition can substantially alleviate key pathological features of DN, including mesangial cell proliferation, podocyte apoptosis, glomerular injury, renal fibrosis, and proteinuria [74,75,76].
SIRT1, an NAD+-dependent deacetylase, plays a key role in cellular repair processes that mitigate DN pathogenesis. These processes include antioxidant defense, autophagy modulation, and anti-senescence. SIRT1 promotes autophagy in different cell types by regulating multiple autophagy-related pathways. Under fasting conditions, it interacts with PGC-1α, FoxO1, and CREB-regulated transcription coactivator 2 (CRTC2) to enhance gluconeogenesis and inhibit glycolysis [77,78,79]. Furthermore, SIRT1 regulates PGC-1α and peroxisome proliferator-activated receptor α (PPAR-α) to promote fatty acid oxidation and gluconeogenesis, as well as liver glucose output, thereby maintaining energy supply and ensuring stable blood glucose levels [80]. The expression of SIRT1 is tightly controlled by the intracellular concentration of NAD+, which is often decreased in the diabetic state [81]. Consequently, SIRT1 expression is significantly reduced in the kidneys of both T1DM and patients with T2DM. Studies on diabetic animals have shown that activating SIRT1 can reduce inflammation and oxidative stress, decrease MCs proliferation, improve renal autophagy activity, promote mitochondrial regeneration, improve blood glucose homeostasis, and alleviate IR by deacetylating transcription factors involved in the pathogenesis of the disease, such as NF-κB, FoxO, and p53. This contributes to the renoprotective role in DN, making SIRT1 a promising target for the treatment of DN [17, 82].
AMPK, an energy sensor expressed in renal cells, inhibits the mTOR pathway to induce autophagy and plays a protective role in DN. By enhancing autophagy activity, AMPK can ameliorate damage to the glomerular filtration barrier, a key pathological feature of DN [83]. Research has indicated that AMPK expression is significantly suppressed in DN model mice. Boosting AMPK expression has been shown to effectively mitigate DN-associated damage, including podocyte and glomerular endothelial cell injury, thickening of the glomerular basement membrane, and inflammatory cell infiltration. These effects result in improved laboratory indicators of renal function [23, 24]. Li et al. [22] discovered that vitamin D and its receptors can activate AMPK, which in turn alleviates the autophagy deficiency observed in renal cells of mice with DN. This activation leads to a reduction in renal tubular injury, inflammation, and renal fibrosis, further highlighting the potential of AMPK as a therapeutic target in DN. In summary, targeting autophagy-related pathways like mTOR, SIRT1, and AMPK offers promising therapeutic strategies for DN treatment.
Natural Substances to Alleviate DM and DN via Autophagy
The United Kingdom Prospective Diabetes Study highlights the need for a multifaceted approach to control blood glucose, often combining oral antidiabetic drugs with insulin. Limitations of traditional oral diabetes medications are driving the search for new treatments. Emerging treatments like DPP-4 inhibitors, SGLT-2 inhibitors, and GLP-1 or GLP-1 receptor agonists broaden therapeutic options for DM [84] (Table 1). While inducing autophagy in DN has therapeutic potential, conventional inducers like rapamycin can have significant side effects [85]. This has led to interest in natural substances that induce autophagy with lower toxicity, offering a safer therapeutic alternative (Table 2).
DPP-4 Inhibitors
Quercetin, a flavanol compound extracted from plants like Portulaca oleracea L. and Abelmoschus esculentus. Studies show quercetin has hypoglycemic effects and enhances insulin sensitivity by targeting the DPP-4 enzyme [86, 87]. Beyond its effects on glucose levels and insulin sensitivity, quercetin has also shown promise as an anti-aging agent. Its potential as an anti-aging agent may be due to effects on autophagy and apoptosis pathways [88]. Notably, Khater et al. [21] found that quercetin can significantly reduce blood glucose levels, oxidative, and inflammatory markers in the pancreas of T1DM model rats by alleviating the deficiency in insulin secretion through the induction of β-cell autophagy. This discovery reveals the therapeutic value of quercetin for T1DM. Additionally, Zhu et al. [89] have shown that quercetin can significantly improve blood glucose and kidney function indicators in db/db mice by activating autophagy, which in turn helps to alleviate podocyte dedifferentiation via the Notch pathway. Despite these promising findings, the clinical application of quercetin is limited by its low bioavailability. To overcome this challenge, there is a need to develop innovative dosage forms, such as liposomes, designed to enhance the bioavailability of quercetin [21]. Moreover, Abelmoschus esculentus, from which quercetin is extracted, contains a variety of bioactive components, including carbohydrates and polysaccharides. Huang et al. [87] have observed that these components can reduce the activity of DPP-4 and its downstream signals associated with IR in T2DM.
Dihydromyricetin, derived from Ampelopsis grossedentata, is a natural dihydroflavanol compound [90]. Wu et al. [91] showed dihydromyricetin stimulates cyclic adenosine monophosphate (cAMP) and inhibits DPP-4, lowering blood glucose levels. Wen et al. [92] have reported that dihydromyricetin can effectively reduce blood glucose levels through the activation of the PI3K/Akt/MAPK signaling pathway, which is a well-established mediator of autophagy. This suggests that dihydromyricetin may enhance cellular autophagy, a process critical for maintaining cellular health and function. Yang et al. [36] also found that dihydromyricetin can promote the formation of autophagic lysosomes and induce autophagy through the AMPK/PGC-1α and PPAR-α pathways, thereby alleviating IR in HFD-fed rats. Furthermore, Guo et al. [19] discovered that dihydromyricetin has the potential to mitigate renal interstitial fibrosis in DN by promoting autophagy. This protective effect is mediated through the PI3K/Akt/mTOR pathway and the micro RNA-155-5p (miR-155-5p)/PTEN pathway, both of which are implicated in the regulation of autophagy and cellular responses to stress.
Ginsenoside Rg1 is an active component extracted from Panax ginseng C. A. Mey. Jiang et al. [93] discovered that ginsenoside Rg1 functions as a DPP-4 inhibitor, which helps modulate glucolipid metabolism. The anti-diabetic and IR alleviation effects of ginsenoside Rg1 are likely mediated through the activation of the Akt/glycogen synthase kinase 3β (GSK-3β) signaling pathway, a key regulator of glucose homeostasis. Building on these findings, Zong et al. [94] and Chen et al. [95] demonstrated the protective effects of ginsenoside Rg1 in mice with T1DM. These effects may be attributed to its ability to increase serum insulin levels and tissue autophagy markers, which can help alleviate islet injury and tissue inflammation. This suggests that ginsenoside Rg1 has therapeutic and preventive potential for treating pancreatic injury in patients with DM. Furthermore, Shi et al. [96] found that ginsenoside Rg1 can induce autophagy and reduce the levels of fasting blood glucose (FBG) and HbA1c in diabetic mouse models through the Akt/GSK3β/β-Catenin signaling pathway and also mitigate the epithelial-mesenchymal transition of podocytes. This finding indicates that ginsenoside Rg1 may have therapeutic potential for the treatment of DN by preserving podocyte integrity and function.
Berberine, an isoquinoline alkaloid from the root of Berberis aristate, has significant antidiabetic effects. It acts as a DPP-4 inhibitor, enhancing insulin sensitivity and managing blood glucose levels [97, 98]. Geng et al. [99] have observed that berberine can upregulate the phosphorylation of the insulin receptor and its downstream signaling molecules. This upregulation is involved in enhancing cell viability and autophagy in diabetic rats, leading to improved mesenteric vasodilation and vascular insulin sensitivity, and reduced blood glucose levels. Furthermore, Li et al. [100] discovered that berberine can protect islet β-cells from injury induced by PA, potentially through the regulation of mitophagy, a process critical for maintaining mitochondrial health and cellular function. A clinical trial by Zhang et al. [101] demonstrated that berberine significantly reduced fasting and postload plasma glucose levels, as well as HbA1c levels in patients with T2DM, with the side effects being extremely mild. Shao et al. [102] reported a case in which a 12-year-old Chinese boy with T2DM experienced significant alleviation of symptoms and maintained normal FBG levels through an integrated treatment approach that included berberine hydrochloride tablets, physical exercise, and dietary control. Berberine also shows renal protective effects in DN. First, it exerts renal protection through AMPK and Toll-like receptor 4 (TLR4) signaling pathways. Second, it activates podocyte autophagy by inhibiting the mTOR/70 kDa ribosomal protein S6 kinase (p70S6K)/eukaryotic translation initiation factor 4E binding protein 1 (EBP1) signaling pathway, which can help alleviate podocyte apoptosis. Third, berberine protects against diabetic kidney dysfunction by inhibiting the expansion of the glomerular mesangial matrix and activating autophagy, thereby preserving renal health [103,104,105].
SGLT-2 Inhibitors
Phlorizin, a natural dihydrochalcone, is a well-known SGLT-2 inhibitor [106]. Research by Kumar et al. [107] shows that phlorizin enhances adipocyte function. It achieves this by reducing inflammatory cytokine levels and increasing adiponectin, both in vivo and in vitro. These effects contribute to a decrease in the hyperglycemic index and hepatic IR. Madrakhimov et al. [108] also showed that phlorizin induces autophagy by inhibiting mTOR. This suggests that the therapeutic value of phlorizin may be associated with its capacity to stimulate autophagy, a cellular process crucial for maintaining cellular health. Additionally, dapagliflozin, a novel oral hypoglycemic agent derived from phlorizin, has proven therapeutic value for treating DM and DN. Its beneficial effects are mediated through the AMPK/mTOR/SIRT1 and PI3K/Akt pathways [18, 20]. In addition, Borkar et al. [106] have identified diglucuronide metabolites, resulting from the mono-glucuronidation of phlorizin, as novel SGLT-2 inhibitors. These metabolites exhibit better binding affinity and hypoglycemic effects compared to phlorizin itself. This discovery expands the potential therapeutic applications of phlorizin-derived compounds in managing glucose homeostasis.
Mangiferin, a phenolic compound derived from various parts of the Mangifera indica L, including its leaves, bark, and fruit, has potent antioxidant properties and inhibits SGLT-2 [109, 110]. Wang et al. [111] found that mangiferin activates autophagy via the AMPK/mTOR/ULK1 pathway, which plays a crucial role in protecting podocytes under diabetic conditions and slowing the progression of DN. Foudah et al. [109] developed mangiferin-loaded solid lipid nanoparticles with enhanced and sustained hypoglycemic and lipid-lowering effects, indicating their potential as an efficient oral treatment for DM. Furthermore, Olusola et al. [110] identified neomangiferin, a mangiferin congener from natural resources, which may offer better stabilization of SGLT-2 compared to dapagliflozin, a well-established SGLT-2 inhibitor. This suggests that neomangiferin could be a promising candidate for the development of new SGLT-2 inhibitors. Mangiferin also acts as a natural DPP-4 inhibitor, offering dual mechanisms for blood glucose control. It has the capacity to reduce high-sensitivity C-reactive protein levels and improve β-cell function in diabetic rats with metabolic syndrome, thereby achieving blood glucose control through both insulin sensitization and stimulation of insulin release from pancreatic cells [97].
GLP-1 and GLP-1 Receptor Agonists
Garlic (Allium sativum.) is known for its health benefits, potentially reducing cancer, hypertension, and DM incidence [112]. Allicin, the main bioactive component of garlic, helps maintain glucose homeostasis in DM. This is achieved through the activation of the transient receptor potential ankyrin 1 (TRPA1) channel, which stimulates the secretion of GLP-1 [113]. Qian et al. [114] showed that allicin induces autophagy and inhibits apoptosis, reducing β cell injury from streptozotocin (STZ). This effect is mediated through the AMPK/mTOR pathway, which is a critical regulator of cellular metabolism and survival. The induction of autophagy by allicin leads to significant improvements in islet structure and insulin expression, as well as a reduction in blood glucose levels. These findings suggest that allicin possesses therapeutic potential for patients with T1DM. Additionally, Arellano Buendia et al. [115] have confirmed in rat experiments that allicin can improve oxidative stress caused by metabolic syndrome, impaired glucose tolerance, and reduce kidney injury markers through the Nrf2 pathway. Although this study was not entirely focused on kidney damage caused by DN, it also demonstrates the potential of allicin in treating DN.
Glycyrrhizic acid (GA), a triterpenoid saponin found in licorice roots, regulates metabolism [116]. Wang et al. [116] found that GA enhances GLP-1 secretion and lowers plasma glucose via the G-protein-coupled bile acid receptor 1 (TGR5) receptor, a pathway known to contribute to its anti-diabetic effects. Zhao et al. [117] also showed that GA mitigates high glucose-induced fibrosis and inflammation in podocytes. This protective effect is mediated through the sucrose non-fermenting AMP-activated protein kinase (SNARK)/AMPK pathway, which is associated with the regulation of autophagy, a cellular process critical for maintaining cellular health. Fei et al. [118] reported significant blood glucose control in a 73-year-old female with reactive perforating collagenosis and T2DM using glycyrrhizin tablets. While these findings suggest that GA may exert therapeutic benefits in DM and DN through its impact on autophagy, additional research is necessary to confirm this mechanism and fully understand the extent of GA's therapeutic potential.
Oxymatrine is a quinolizidine alkaloid from Styphnolobium Schott [119]. Guo et al. [120] found that oxymatrine increases insulin and GLP-1 levels in diabetic rats. This suggests that oxymatrine may alleviate hyperlipidemia and hyperglycemia in HFD and STZ-induced diabetic rats by enhancing insulin secretion and sensitivity. Zuo et al. [121] also observed that oxymatrine can modulate the expression of key proteins in the liver, such as KH-type splicing regulatory protein (KSRP), PETN, and Akt, which may help to reduce blood glucose levels and protect rats with T2DM from IR. Moreover, Huan et al. [119] have summarized the pharmacological mechanisms by which oxymatrine may combat DM and highlighted the need for appropriate administration methods and concentrations. Further clinical studies are needed to confirm oxymatrine’s potential benefits in diabetes treatment.
Other Polyphenols and Saponins
Resveratrol, a polyphenol found in the roots of Veratrum gandiflorum and discovered in 1939, has been extensively studied for its potential to promote autophagy. This is achieved by activating SIRT1 and SIRT3 and through the upregulation of miR-18a-5p, as well as the downregulation of miR-142-3p. These actions may alleviate IR, improve lipid metabolism, and protect podocytes from injury in DN, ultimately leading to improved renal function [122,123,124,125,126]. Liu et al. [127] highlighted that podocytes exposed to AGEs are susceptible to lysosomal membrane permeabilization, which can trigger autophagy inhibition and cell damage. Resveratrol has been shown to ameliorate the oxidative state and cytoskeleton disorder under diabetic conditions. When combined with vitamin E, which also has oxidative state-alleviating properties, the therapeutic effect on DN may be further enhanced. Despite some studies suggesting that resveratrol has the potential to treat DM, for example, Abhale et al. [128] found that resveratrol combined with stem cell conditioned media can regenerate damaged pancreatic β-cells in mice with T1DM and exert an anti-DM effect; Wahab et al. [129] and Heydarpour et al. [130] pointed out that resveratrol can regulate carbohydrate digestion through the TGF-β signaling pathway and alleviate IR. Most research is concentrated on its therapeutic value in diabetic complications. Moreover, despite an extensive array of clinical trials and a wealth of literature on resveratrol, the definitive outcomes and impacts of resveratrol in humans remain unclear. This uncertainty stems from factors such as inconsistent dosages, varied experimental designs, insufficient statistical power, and a complex interplay of biological and other confounding elements [122]. Notably, Yadegar et al. [131] found that in elderly patients with DM, resveratrol was not more effective than a placebo. This finding indicates that the hypoglycemic effect of resveratrol requires further investigation.
Curcumin, a natural polyphenol antioxidant derived from turmeric, possesses anti-aging and DM management properties via autophagy induction [88, 132]. Wahab et al. [129] found that encapsulating resveratrol and curcumin in a starch matrix reduces starch digestion. This finding suggests that the development of novel functional foods incorporating these compounds could assist in the treatment of T2DM. Khater et al. [133] observed that curcumin can alleviate β-cell damage and reduce FBG levels through the regulation of autophagy pathways, specifically by modulating miR-137 and miR-29b. These micro RNAs are believed to mitigate ER stress in β-cells, and liposomal curcumin has shown a improved pharmacokinetics and effects compared to the native compound. Further studies indicate that curcumin, when administered at appropriate doses, can promote autophagy by inhibiting the mTOR pathway. It also exhibits anti-inflammatory, antioxidant, anti-fibrosis, and anti-apoptotic effects in DN [27, 103, 134]. Further research by Zhang et al. [134] found that curcumin induced autophagy and significantly slowed the progression of DN in mice by regulating Beclin1/UV radiation resistance associated gene (UVRAG)/Bcl-2 pathway, which inhibited podocyte apoptosis. This study further confirmed the significant protective role of curcumin in DN. However, Rainey et al. [135] noted that high doses of curcumin can enhance the permeability of the lysosomal membrane, potentially leading to cell death and exacerbation of DN. This observation underscores the importance of careful dosage consideration when applying curcumin therapeutically.
Kaempferol, a flavonoid compound found in a variety of natural plants, has been studied for its effects on AMPK and mTOR. Varshney et al. [136] discovered that kaempferol activates AMPK and inhibits mTOR, regulating cellular energy balance and autophagy. The autophagy induced by kaempferol was found to be abrogated by AMPK inhibitors, resulting in β-cell apoptosis under PA-stressed conditions. Furthermore, studies by Zhang et al. [137] and Sheng et al. [138] have indicated that kaempferol possesses protective properties against mitochondrial damage. This damage is often caused by the activation of AGEs and the accumulation of ROS. By mitigating these harmful effects, kaempferol contributes to the alleviation of podocyte damage, glomerular basement membrane thickening, and mesangial matrix expansion in DN.
Puerarin is an isoflavone derivative extracted from Pueraria lobata [139]. Li et al. [139] discovered that puerarin protected the kidneys and improved podocyte injury in STZ-induced diabetic mice by activating SIRT1 and its antioxidant effects, which led to an improved urinary albumin-to-creatinine ratio and renal damage in mice with STZ-induced DN. Xu et al. [140] found that puerarin elevated the level of autophagy in STZ-induced DN mice, particularly under conditions of endoplasmic reticulum stress (ERS), through the activation of the RNA-dependent protein kinase-like endoplasmic reticulum kinase (PERK)/eukaryotic initiation factor 2 subunit α (eIF2α)/ATF4 signaling pathway. This improved FBG, renal function, and podocyte protein expression in DN mice. This finding suggests that PERK could be a potential therapeutic target for DN. In addition to its renal protective effects, Heydarpour et al. [130] have highlighted that puerarin can also regulate autophagy via the TGF-β pathway. This regulation can lead to the mitigation of DM and its complications. The findings suggest that puerarin may offer a promising alternative to current treatments for DM, providing a new therapeutic option for managing the disease and its associated complications.
Astragaloside IV is a saponin compound and the main active ingredient derived from Astragalus membranaceus [141]. Research has indicated that astragaloside IV can modulate the PI3K/Akt/mTOR and SIRT/NF-κB pathways, which are critical in promoting autophagy. This modulation contributes to the alleviation of podocyte injury, proteinuria, glomerular sclerosis, and renal fibrosis in DN [77, 103]. Further research has found that astragaloside IV inhibits the activation of MCs under high glucose conditions and enhances autophagy by increasing the expression of SIRT1 and reducing the acetylation of NF-κB p65. The autophagy inhibitor 3-methyladenine abolished the improving effects of astragaloside IV on MCs, while the autophagy activator rapamycin restored the activation of MCs induced by HG. Additionally, astragaloside IV also improved the renal function and fibrosis in mice with DN in in vivo experiments [77]. Furthermore, Zhou et al. [142] discovered that astragaloside IV significantly mitigated hyperglycemia, glucose intolerance, IR, placental oxidative stress, and the production of inflammatory cytokines in gestational DM via the TLR4/NF-κB pathway. This effect may be associated with the promotion of autophagy [143]. Further research is needed to determine astragaloside IV’s effects on non-pregnant individuals with diabetes [141].
Discussion
Inhibition of autophagy in islet β-cells is observed in patients with T1DM and T2DM. Studies agree that inducing autophagy may improve β cell dysfunction in DM [3]. However, Sakai et al. [8] found that in the early stages of HG treatment, the level of autophagy in PTECs increased, but was later suppressed because of lysosomal stress caused by excessive autophagy, and insulin could inhibit HG-induced autophagy. At the same time, the process of autophagy suppression in mice with T1DM did not involve the activation of the mTOR pathway observed in mice with T2DM. This finding suggests that there are differences in the mechanisms of autophagy suppression between T1DM and T2DM. In T1DM, autophagy suppression is due to lysosomal depletion caused by excessive initial activation of autophagy, while in T2DM, it is due to reduced lysosomal activity. The study then found that inhibiting autophagy in mice with T1DM could exacerbate their proteinuria, but this phenomenon was not observed in mice with T2DM. At the same time, using rapamycin to induce autophagy in mice with T1DM at an early stage of the disease actually exacerbated their kidney damage, but it could alleviate kidney damage in mice with T2DM. This indicates that in the early stages of T1DM, to prevent kidney damage, autophagy is over-activated, and further induction of autophagy in this state exacerbates lysosomal stress and accelerates disease progression. Tran et al. [15] found 1-deoxysphingolipids (1-DSL) upregulated in patients with T2DM or those at risk. 1-DSL not only induces myoblast autophagy but also significantly inhibits insulin-stimulated glucose uptake in myotubes, potentially leading to skeletal muscle cell functional impairment and IR. The possible mechanism behind this phenomenon may be due to the early activation of autophagy leading to excessive energy intake by cells and impaired signaling for energy deficiency recognition, ultimately resulting in the inhibition of autophagy. However, this hypothesis requires further research for confirmation. These findings suggest that, despite the ultimate inhibition of autophagy, both T1DM and T2DM may initially exhibit increased autophagy levels, with different mechanisms leading to inhibition (Fig. 1). This insight underscores the need for tailored treatment strategies based on the type and stage of DM and its complications. Wu et al. [144] reported that islet tissue replacement may offer a promising strategy for patients with DM, with autophagy shown to be beneficial for the prognosis of patients undergoing stem cell or organ transplantation [145, 146]. In addition, some studies have found that certain treatments for DM and DN can alter the composition of the gut microbiota, and these changes have been proven to be related to therapeutic benefits, with the mechanism possibly related to the impact of changes in gut bacterial metabolites on autophagy [38, 91, 147]. Future research can continue to focus on the therapeutic value of the gut microbiota and autophagy for DM and DN. Despite the potential of autophagy as an important target in DM treatment, most research has focused on static autophagy-related proteins. Given that autophagy is a dynamic process, further research is needed to determine whether it is a key factor in the progression of DM and DN [3]. Additionally, the species-specific nature of many drugs, such as those effective in mice but not in humans, and vice versa, complicates direct translation to human therapy. Even when effective in humans, autophagy's impact on IR can vary across different organs [148]. Furthermore, factors like age, sex, and the presence of other diseases may also influence drug efficacy [3]. Therefore, future research is needed to further investigate how autophagy interacts with cellular environmental disorders such as oxidative stress and ERS as well as the specific mechanisms by which key molecules and signaling pathways involved in the autophagy process respond to metabolic disorders related to DM. It is also necessary to clarify the definitive impact of autophagy as a dynamic process on DM and DN to verify the application effects of autophagy modulation in clinical practice and to explore additional strategies.
Currently, there is no consensus among scholars regarding the precise mechanisms of kidney injury in DN. It is widely accepted that no single factor can cause kidney injury in DN. Instead, various pathogenic factors interact, collectively contributing to the disease’s onset and progression [149]. Autophagy has been implicated in many of the proposed mechanisms of kidney injury in DN. HG levels have been consistently shown to inhibit autophagy and are commonly used to study the effects of autophagy inhibition in DM and DN. In addition, autophagy regulatory mechanisms identified in other diabetes complications, such as circadian rhythm disruptions and pyroptosis, may similarly influence DN [150, 151]. These findings suggest that inducing autophagy could be a promising therapeutic strategy for DN. However, while most studies concur that inducing autophagy can alleviate DN, the different mechanisms of autophagy inhibition in T1DM and T2DM necessitate a nuanced approach. For instance, the use of rapamycin to induce autophagy has been shown to alleviate DN in T2DM but exacerbate it in T1DM [8]. Furthermore, Feng et al. [152] discovered that excessive autophagy activation can lead to autophagic cell death and myocardial damage in diabetic cardiomyopathy, a finding with implications for DN treatment strategies targeting autophagy. Chen et al. [153] found that astilbin, a flavonoid compound from the rhizome of Smilax China L., can inhibit excessive autophagy and apoptosis in PTECs in DN via the PI3K/Akt pathway, potentially improving clinical management of DN. Tang et al. [154] also noted that excessive autophagy levels may worsen muscle atrophy induced by DN. Despite suggestions that autophagy may act as a bridge in the pathogenesis of DN [28], conclusive evidence supporting this theory is lacking [68]. Future research should focus on determining the optimal dosage for autophagy induction therapy and thoroughly evaluating its side effects [1]. Additionally, autophagy is a process involving various immune cells and is closely related to the body’s immune system. Therefore, future research can further explore how autophagy participates in immune responses in DN and how to improve immune-mediated tissue damage by regulating autophagy. Moreover, diabetes complications are not limited to DN alone, and there may be connections between various complications. For example, the reduced kidney filtration function and water-sodium retention caused by DN lead to increased cardiac burden and ultimately develop into cardiorenal syndrome [155, 156]. Future in-depth research to explore the connections between diabetes complications and the role of autophagy in them may help to break through existing treatment strategies for DN [8, 28, 149].
Natural substances, with their wide availability, low cost, and significant therapeutic effects, have fewer toxic and side effects. These benefits have led to their widespread use in treating various diseases. This review highlights a variety of natural substances that target autophagy, demonstrating promising potential in the treatment of DM and DN. Many of these substances have also shown good therapeutic potential in other diabetic complications [157]. However, clinical data supporting their use are lacking, limiting research to basic science. The pharmacological mechanisms and pharmacokinetic properties of these substances require further elucidation [158]. Most of the current basic research also focuses only on the impact of natural substances on basic indicators such as blood glucose and kidney function. Rarely are data on HbA1c mentioned in the results of animal experiments, which better reflects the level of blood glucose control in DM. Additionally, excessive dosages may cause cell damage and potentially worsen the disease [135]. Therefore, a large number of randomized, double-blind, and placebo-controlled clinical trials are necessary to investigate the efficacy, optimal dosage, long-term effects, side effects, synergistic interactions, and potential incompatibilities of these natural substances [131]. At the same time, due to the current lack of research on natural substances for the treatment of cardiorenal syndrome, subsequent research on natural substances for the treatment of DN should also comprehensively observe the impact on cardiorenal function. In addition, Palmer et al. [159] compared SGLT-2 inhibitors with GLP-1 receptor agonists, finding that both drug classes reduced all-cause mortality and cardiovascular, myocardial, and renal injuries. However, they also noted that SGLT-2 inhibitors and GLP-1 receptor agonists may increase the risk of genital infections and severe gastrointestinal events, respectively. This underscores the importance of selecting appropriate drugs based on individual patient conditions. Furthermore, there is significant research potential in developing new formulations and exploring structural modifications of existing natural substances. These efforts could enhance their therapeutic effects and lead to the discovery of new treatments for DM and DN [18, 20, 21, 105, 106, 109, 110, 129, 133]. Beyond natural substances, the development of new formulations or structural modifications of existing chemical drugs, or the use of variants of autophagy-related genes to treat or prevent the progression of DM or DN, could be explored. The development and evaluation of these new drugs and therapies can further confirm the application value of autophagy as a therapeutic target for DM and DN.
In summary, targeting autophagy is a promising strategy for DM and DN treatment and research. However, to advance its clinical application, there is a need for more comprehensive and in-depth studies. These investigations should aim to fully understand the mechanisms involved, optimize treatment approaches and evaluate the safety and efficacy of autophagy modulation in the context of diabetes and its complications.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
References
Liu T, Jin Q, Yang L, et al. Regulation of autophagy by natural polyphenols in the treatment of diabetic kidney disease: therapeutic potential and mechanism. Front Endocrinol (Lausanne). 2023;14:1142276.
Szablewski L. Insulin resistance: the increased risk of cancers. Curr Oncol. 2024;31:998–1027.
Wang LH, Wang YY, Liu L, Gong Q. From diabetes to diabetic complications: role of autophagy. Curr Med Sci. 2023;43:434–44.
Yang X, Feng P, Ji R, Ren Y, Wei W, Holscher C. Therapeutic application of GLP-1 and GIP receptor agonists in Parkinson’s disease. Expert Opin Ther Targets. 2022;26:445–60.
Nowell J, Blunt E, Gupta D, Edison P. Antidiabetic agents as a novel treatment for Alzheimer’s and Parkinson’s disease. Ageing Res Rev. 2023;89: 101979.
Zheng W, Zhou J, Song S, et al. Dipeptidyl-peptidase 4 inhibitor sitagliptin ameliorates hepatic insulin resistance by modulating inflammation and autophagy in ob/ob mice. Int J Endocrinol. 2018;2018:8309723.
Canet F, Diaz-Pozo P, Luna-Marco C, et al. Mitochondrial redox impairment and enhanced autophagy in peripheral blood mononuclear cells from type 1 diabetic patients. Redox Biol. 2022;58: 102551.
Sakai S, Yamamoto T, Takabatake Y, et al. Proximal tubule autophagy differs in type 1 and 2 diabetes. J Am Soc Nephrol. 2019;30:929–45.
Wu M, Chen W, Zhang S, et al. Rotenone protects against β-cell apoptosis and attenuates type 1 diabetes mellitus. Apoptosis. 2019;24:879–91.
Du X, Di Malta C, Fang Z, et al. Nuciferine protects against high-fat diet-induced hepatic steatosis and insulin resistance via activating TFEB-mediated autophagy-lysosomal pathway. Acta Pharm Sin B. 2022;12:2869–86.
Hu H, Yin JH, Shao DD, et al. The phosphorylation of hCDC14A modulated by ZIPK regulates autophagy of murine pancreatic islet β-TC3 cells upon glucose stimulation. Eur Rev Med Pharmacol Sci. 2020;24:10028–35.
Liu H, Javaheri A, Godar RJ, et al. Intermittent fasting preserves beta-cell mass in obesity-induced diabetes via the autophagy-lysosome pathway. Autophagy. 2017;13:1952–68.
Parikh HM, Elgzyri T, Alibegovic A, et al. Relationship between insulin sensitivity and gene expression in human skeletal muscle. BMC Endocr Disord. 2021;21:32.
Qian Q, Zhang Z, Li M, et al. Hepatic lysosomal iNOS activity impairs autophagy in obesity. Cell Mol Gastroenterol Hepatol. 2019;8:95–110.
Tran D, Myers S, McGowan C, et al. 1-deoxysphingolipids, early predictors of type 2 diabetes, compromise the functionality of skeletal myoblasts. Front Endocrinol (Lausanne). 2021;12: 772925.
Wu T, Shao Y, Li X, et al. NR3C1/glucocorticoid receptor activation promotes pancreatic β-cell autophagy overload in response to glucolipotoxicity. Autophagy. 2023;19:2538–57.
Ji J, Tao P, Wang Q, Li L, Xu Y. SIRT1: mechanism and protective effect in diabetic nephropathy. Endocr Metab Immune Disord Drug Targets. 2021;21:835–42.
Jaikumkao K, Thongnak L, Htun KT, et al. Dapagliflozin and metformin in combination ameliorates diabetic nephropathy by suppressing oxidative stress, inflammation, and apoptosis and activating autophagy in diabetic rats. Biochim Biophys Acta Mol Basis Dis. 2024;1870: 166912.
Guo L, Tan K, Luo Q, Bai X. Dihydromyricetin promotes autophagy and attenuates renal interstitial fibrosis by regulating miR-155-5p/PTEN signaling in diabetic nephropathy. Bosn J Basic Med Sci. 2020;20:372–80.
Wu J, Chen Y, Shi S, et al. Exploration of pharmacological mechanisms of dapagliflozin against type 2 diabetes mellitus through PI3K-Akt signaling pathway based on network pharmacology analysis and deep learning technology. Curr Comput Aided Drug Des. 2024. https://doi.org/10.2174/0115734099274407231207070451. (Online ahead of print).
Khater SI, El-Emam MMA, Abdellatif H, et al. Lipid nanoparticles of quercetin (QU-Lip) alleviated pancreatic microenvironment in diabetic male rats: the interplay between oxidative stress—unfolded protein response (UPR)—autophagy, and their regulatory miRNA. Life Sci. 2024;344: 122546.
Li A, Yi B, Han H, et al. Vitamin D-VDR (vitamin D receptor) regulates defective autophagy in renal tubular epithelial cell in streptozotocin-induced diabetic mice via the AMPK pathway. Autophagy. 2022;18:877–90.
Li F, Chen Y, Li Y, Huang M, Zhao W. Geniposide alleviates diabetic nephropathy of mice through AMPK/SIRT1/NF-κB pathway. Eur J Pharmacol. 2020;886: 173449.
Takagaki Y, Lee SM, Dongqing Z, Kitada M, Kanasaki K, Koya D. Endothelial autophagy deficiency induces IL6—dependent endothelial mesenchymal transition and organ fibrosis. Autophagy. 2020;16:1905–14.
Yan J, Li X, Liu N, He JC, Zhong Y. Relationship between macrophages and tissue microenvironments in diabetic kidneys. Biomedicines. 2023;11:1889.
Zhao J, Chen J, Zhu W, Qi XM, Wu YG. Exosomal miR-7002-5p derived from highglucose-induced macrophages suppresses autophagy in tubular epithelial cells by targeting Atg9b. FASEB J. 2022;36: e22501.
Zhu X, Xu X, Du C, et al. An examination of the protective effects and molecular mechanisms of curcumin, a polyphenol curcuminoid in diabetic nephropathy. Biomed Pharmacother. 2022;153: 113438.
Yu J, Liu Y, Li H, Zhang P. Pathophysiology of diabetic kidney disease and autophagy: a review. Medicine (Baltimore). 2023;102: e33965.
E Y, Lin Y, Yan G, et al. Exogenous H(2)S alleviates senescence of glomerular mesangial cells through up-regulating mitophagy by activation of AMPK-ULK1-PINK1-parkin pathway in mice. Biochim Biophys Acta Mol Cell Res. 2023;1870: 119568.
Lee WC, Chiu CH, Chen JB, Chen CH, Chang HW. Mitochondrial fission increases apoptosis and decreases autophagy in renal proximal tubular epithelial cells treated with high glucose. DNA Cell Biol. 2016;35:657–65.
Song S, Tan J, Miao Y, Sun Z, Zhang Q. Intermittent-hypoxia-induced autophagy activation through the ER-stress-related PERK/eIF2α/ATF4 pathway is a protective response to pancreatic β-Cell apoptosis. Cell Physiol Biochem. 2018;51:2955–71.
Song Z, Ma J, Lu Y, et al. The protective role of the MKP-5-JNK/P38 pathway in glucolipotoxicity-induced islet β-cell dysfunction and apoptosis. Exp Cell Res. 2019;382: 111467.
Zhao T, Ma J, Li L, et al. MKP-5 relieves lipotoxicity-induced islet β-cell dysfunction and apoptosis via regulation of autophagy. Int J Mol Sci. 2020;21:7161.
Bai C, Yang W, Lu Y, Wei W, Li Z, Zhang L. Identification of circular RNAs regulating islet β-cell autophagy in type 2 diabetes mellitus. Biomed Res Int. 2019;2019:4128315.
Frendo-Cumbo S, Jaldin-Fincati JR, Coyaud E, et al. Deficiency of the autophagy gene ATG16L1 induces insulin resistance through KLHL9/KLHL13/CUL3-mediated IRS1 degradation. J Biol Chem. 2019;294:16172–85.
Yang Y, Qiu W, Xiao J, Sun J, Ren X, Jiang L. Dihydromyricetin ameliorates hepatic steatosis and insulin resistance via AMPK/PGC-1α and PPARα-mediated autophagy pathway. J Transl Med. 2024;22:309.
Yang X, Ding W, Chen Z, Lai K, Liu Y. The role of autophagy in insulin resistance and glucolipid metabolism and potential use of autophagy modulating natural products in the treatment of type 2 diabetes mellitus. Diabetes Metab Res Rev. 2024;40: e3762.
Yang L, Lin H, Lin W, Xu X. Exercise ameliorates insulin resistance of type 2 diabetes through motivating short-chain fatty acid-mediated skeletal muscle cell autophagy. Biology (Basel). 2020;9:203.
Xiang M, Yuan X, Zhang N, et al. Effects of exercise, metformin, and combination treatments on type 2 diabetic mellitus-induced muscle atrophy in db/db mice: crosstalk between autophagy and the proteasome. J Physiol Biochem. 2024;80:235–47.
Cui D, Drake JC, Wilson RJ, et al. A novel voluntary weightlifting model in mice promotes muscle adaptation and insulin sensitivity with simultaneous enhancement of autophagy and mTOR pathway. FASEB J. 2020;34:7330–44.
Bodis K, Roden M. Energy metabolism of white adipose tissue and insulin resistance in humans. Eur J Clin Invest. 2018;48: e13017.
Shi L, Yang J, Tao Z, et al. Loss of FoxO1 activates an alternate mechanism of mitochondrial quality control for healthy adipose browning. Clin Sci (Lond). 2024;138:371–85.
Song W, Postoak JL, Yang G, et al. Lipid kinase PIK3C3 maintains healthy brown and white adipose tissues to prevent metabolic diseases. Proc Natl Acad Sci USA. 2023;120: e2214874120.
Xiong S, Yu S, Wang K, et al. Dietary apigenin relieves body weight and glycolipid metabolic disturbance via pro-browning of white adipose mediated by autophagy inhibition. Mol Nutr Food Res. 2023;67: e2200763.
Roohi TF, Faizan S, Parray ZA, et al. Beyond glucose: the dual assault of oxidative and ER stress in diabetic disorders. High Blood Press Cardiovasc Prev. 2023;30:513–31.
Samsu N. Diabetic nephropathy: challenges in pathogenesis, diagnosis, and treatment. Biomed Res Int. 2021;2021:1497449.
Morresi C, Vasarri M, Bellachioma L, Ferretti G, Degl Innocenti D, Bacchetti T. Glucose uptake and oxidative stress in caco-2 cells: health benefits from Posidonia oceanica (L.) delile. Mar Drugs. 2022;20:457.
Morresi C, Cianfruglia L, Sartini D, et al. Effect of high glucose-induced oxidative stress on paraoxonase 2 expression and activity in caco-2 cells. Cells. 2019;8:1616.
Li Y, Li Y, Zheng S. Inhibition of NADPH oxidase 5 (NOX5) suppresses high glucose-induced oxidative stress, inflammation and extracellular matrix accumulation in human glomerular mesangial cells. Med Sci Monit. 2020;26: e919399.
Chiaradia E, Renzone G, Scaloni A, et al. Protein carbonylation in dopaminergic cells exposed to rotenone. Toxicol Lett. 2019;309:20–32.
Han YC, Tang SQ, Liu YT, et al. AMPK agonist alleviate renal tubulointerstitial fibrosis via activating mitophagy in high fat and streptozotocin induced diabetic mice. Cell Death Dis. 2021;12:925.
Li S, Deng J, Sun D, et al. FBXW7 alleviates hyperglycemia-induced endothelial oxidative stress injury via ROS and PARP inhibition. Redox Biol. 2022;58: 102530.
Jiang W, Xiao T, Han W, et al. Klotho inhibits PKCα/p66SHC-mediated podocyte injury in diabetic nephropathy. Mol Cell Endocrinol. 2019;494: 110490.
Ma X, Ma J, Leng T, et al. Advances in oxidative stress in pathogenesis of diabetic kidney disease and efficacy of TCM intervention. Ren Fail. 2023;45:2146512.
Zhou D, Zhou M, Wang Z, et al. PGRN acts as a novel regulator of mitochondrial homeostasis by facilitating mitophagy and mitochondrial biogenesis to prevent podocyte injury in diabetic nephropathy. Cell Death Dis. 2019;10:524.
Hou B, Li Y, Li X, et al. HGF protected against diabetic nephropathy via autophagy-lysosome pathway in podocyte by modulating PI3K/Akt-GSK3β-TFEB axis. Cell Signal. 2020;75: 109744.
Minami S, Sakai S, Yamamoto T, et al. FGF21 and autophagy coordinately counteract kidney disease progression during aging and obesity. Autophagy. 2024;20:489–504.
Uchida Y, Torisu K, Ueki K, Tsuruya K, Nakano T, Kitazono T. Autophagy gene ATG7 regulates albumin transcytosis in renal tubule epithelial cells. Am J Physiol Renal Physiol. 2021;321:F572–86.
Liu H, Chen W, Wan S, et al. Canagliflozin ameliorates high glucose-induced apoptosis in NRK-52E cells via inhibiting oxidative stress and activating AMPK/mTOR-mediated autophagy. Mol Biol Rep. 2023;50:10325–37.
Liu L, Bai F, Song H, et al. Upregulation of TIPE1 in tubular epithelial cell aggravates diabetic nephropathy by disrupting PHB2 mediated mitophagy. Redox Biol. 2022;50: 102260.
Fujimura R, Yamamoto T, Takabatake Y, et al. Autophagy protects kidney from phosphate-induced mitochondrial injury. Biochem Biophys Res Commun. 2020;524:636–42.
Mao X, Xu Z, Xu X, et al. TGF-β1 inhibits the autophagy of podocytes by activating mTORC1 in IgA nephropathy. Exp Cell Res. 2019;385: 111670.
Meng F, Cao Y, Khoso MH, et al. Therapeutic effect and mechanism of combined use of FGF21 and insulin on diabetic nephropathy. Arch Biochem Biophys. 2021;713: 109063.
Shi M, Yang S, Zhu X, et al. The RAGE/STAT5/autophagy axis regulates senescence in mesangial cells. Cell Signal. 2019;62: 109334.
Gao C, Fan F, Chen J, et al. FBW7 regulates the autophagy signal in mesangial cells induced by high glucose. Biomed Res Int. 2019;2019:6061594.
Wen D, Tan RZ, Zhao CY, et al. Astragalus Mongholicus bunge and Panax notoginseng (Burkill) F.H. Chen formula for renal injury in diabetic nephropathy-in vivo and in vitro evidence for autophagy regulation. Front Pharmacol. 2020;11:732.
Casalena GA, Yu L, Gil R, et al. The diabetic microenvironment causes mitochondrial oxidative stress in glomerular endothelial cells and pathological crosstalk with podocytes. Cell Commun Signal. 2020;18:105.
Tian H, Zheng X, Wang H. Isorhapontigenin ameliorates high glucose-induced podocyte and vascular endothelial cell injuries via mitigating oxidative stress and autophagy through the AMPK/Nrf2 pathway. Int Urol Nephrol. 2023;55:423–36.
Zhou W, Liu Y, Hu Q, Zhou J, Lin H. The landscape of immune cell infiltration in the glomerulus of diabetic nephropathy: evidence based on bioinformatics. BMC Nephrol. 2022;23:303.
Ren Y, Zhang H. The causal effect of inflammatory proteins and immune cell populations on diabetic nephropathy: evidence from Mendelian randomization. Int Urol Nephrol. 2024. https://doi.org/10.1007/s11255-024-04017-5. (Online ahead of print).
Kim H, Kim M, Lee HY, Park HY, Jhun H, Kim S. Role of dendritic cell in diabetic nephropathy. Int J Mol Sci. 2021;22:7554.
Wang D, Zhang Q, Dong W, et al. SGLT2 knockdown restores the Th17/Treg balance and suppresses diabetic nephropathy in db/db mice by regulating SGK1 via Na. Mol Cell Endocrinol. 2024;584: 112156.
Ramasubbu K, Devi RV. Impairment of insulin signaling pathway PI3K/Akt/mTOR and insulin resistance induced AGEs on diabetes mellitus and neurodegenerative diseases: a perspective review. Mol Cell Biochem. 2023;478:1307–24.
Yang F, Qu Q, Zhao C, et al. Paecilomyces cicadae-fermented Radix astragali activates podocyte autophagy by attenuating PI3K/AKT/mTOR pathways to protect against diabetic nephropathy in mice. Biomed Pharmacother. 2020;129: 110479.
Jin J, Shi Y, Gong J, et al. Exosome secreted from adipose-derived stem cells attenuates diabetic nephropathy by promoting autophagy flux and inhibiting apoptosis in podocyte. Stem Cell Res Ther. 2019;10:95.
Chen K, Yu B, Liao J. LncRNA SOX2OT alleviates mesangial cell proliferation and fibrosis in diabetic nephropathy via Akt/mTOR-mediated autophagy. Mol Med. 2021;27:71.
Wang X, Gao Y, Tian N, et al. Astragaloside IV represses high glucose-induced mesangial cells activation by enhancing autophagy via SIRT1 deacetylation of NF-κB p65 subunit. Drug Des Devel Ther. 2018;12:2971–80.
Wang W, Sun W, Cheng Y, Xu Z, Cai L. Role of sirtuin-1 in diabetic nephropathy. J Mol Med (Berl). 2019;97:291–309.
Lu Z, Liu H, Song N, et al. METTL14 aggravates podocyte injury and glomerulopathy progression through N(6)-methyladenosine-dependent downregulating of Sirt1. Cell Death Dis. 2021;12:881.
Yap KH, Yee GS, Candasamy M, et al. Catalpol ameliorates insulin sensitivity and mitochondrial respiration in skeletal muscle of type-2 diabetic mice through insulin signaling pathway and AMPK/SIRT1/PGC-1α/PPAR-γ activation. Biomolecules. 2020;10:1360.
Cui Z, Zhao X, Amevor FK, et al. Therapeutic application of quercetin in aging-related diseases: SIRT1 as a potential mechanism. Front Immunol. 2022;13: 943321.
Qiu D, Song S, Wang Y, et al. NAD(P)H: quinone oxidoreductase 1 attenuates oxidative stress and apoptosis by regulating Sirt1 in diabetic nephropathy. J Transl Med. 2022;20:44.
Szrejder M, Piwkowska A. AMPK signalling: implications for podocyte biology in diabetic nephropathy. Biol Cell. 2019;111:109–20.
Tsang MW. The management of type 2 diabetic patients with hypoglycaemic agents. ISRN Endocrinol. 2012;2012: 478120.
Barlow AD, Nicholson ML, Herbert TP. Evidence for rapamycin toxicity in pancreatic β-cells and a review of the underlying molecular mechanisms. Diabetes. 2013;62:2674–82.
Zhang L, Pan MY, Li T, et al. Study on optimal extraction and hypoglycemic effect of quercetin. Evid Based Complement Alternat Med. 2023;2023:8886503.
Huang CN, Lin CL, Li HH, Tsou SH, Peng CH. Abelmoschus esculentus (Okra) prevents insulin resistance and restores neuron autophagy by regulating dipeptidyl peptidase-4 and thus improving hippocampal function. J Med Food. 2023;26:462–9.
Luo M, Mai M, Song W, et al. The antiaging activities of phytochemicals in dark-colored plant foods: involvement of the autophagy- and apoptosis-associated pathways. Int J Mol Sci. 2022;23:11038.
Zhu X, Zhang C, Liu L, Xu L, Yao L. Senolytic combination of dasatinib and quercetin protects against diabetic kidney disease by activating autophagy to alleviate podocyte dedifferentiation via the Notch pathway. Int J Mol Med. 2024;53:26.
Sun L, Xiao Y, San W, Chen Y, Meng G. Dihydromyricetin regulates RIPK3-CaMKII to prevent necroptosis in high glucose-stimulated cardiomyocytes. Heliyon. 2024;10: e28921.
Wu L, Zhou M, Xie Y, et al. Dihydromyricetin enhances exercise-induced GLP-1 elevation through stimulating cAMP and inhibiting DPP-4. Nutrients. 2022;14:4583.
Wen X, Lv C, Zhou R, Wang Y, Zhou X, Qin S. The molecular mechanism underlying the therapeutic effect of dihydromyricetin on type 2 diabetes mellitus based on network pharmacology, molecular docking, and transcriptomics. Foods. 2024;13:344.
Jiang S, Wu X, Wang Y, Zou J, Zhao X. The potential DPP-4 inhibitors from Xiao-Ke-An improve the glucolipid metabolism via the activation of AKT/GSK-3β pathway. Eur J Pharmacol. 2020;882: 173272.
Zong Y, Yu W, Hong H, et al. Ginsenoside Rg1 improves inflammation andautophagy of the pancreas and spleen in streptozotocin-induced type 1 diabetic mice. Int J Endocrinol. 2023;2023:3595992.
Chen J, Zhu G, Xiao W, Huang X, Wang K, Zong Y. Ginsenoside Rg1 ameliorates pancreatic injuries via the AMPK/mTOR pathway in vivo and in vitro. Diabetes Metab Syndr Obes. 2023;16:779–94.
Shi Y, Gao Y, Wang T, et al. Ginsenoside Rg1 alleviates podocyte EMT passage by regulating AKT/GSK3 β/β-Catenin pathway by restoring autophagic activity. Evid Based Complement Alternat Med. 2020;2020:1903627.
Suman RK, Borde MK, Mohanty IR, Singh HK. Mechanism of action of natural dipeptidyl peptidase-IV inhibitors (berberine and mangiferin) in experimentally induced diabetes with metabolic syndrome. Int J Appl Basic Med Res. 2023;13:133–42.
Naik S, Deora N, Pal SK, et al. Purification, biochemical characterization, and DPP-IV and α-amylase inhibitory activity of Berberine from Cardiospermum halicacabum. J Mol Recognit. 2022;35: e2983.
Geng FH, Li GH, Zhang X, et al. Berberine improves mesenteric artery insulin sensitivity through up-regulating insulin receptor-mediated signalling in diabetic rats. Br J Pharmacol. 2016;173:1569–79.
Li M, She J, Ma L, Ma L, Ma X, Zhai J. Berberine protects against palmitate induced beta cell injury via promoting mitophagy. Genes Genomics. 2022;44:867–78.
Zhang Y, Li X, Zou D, et al. Treatment of type 2 diabetes and dyslipidemia with the natural plant alkaloid berberine. J Clin Endocrinol Metab. 2008;93:2559–65.
Shao B, Hou S, Chan Y, Shao C, Lao L. Remission of new-onset type 2 diabetes mellitus in an adolescent using an integrative medicine approach: a case report. J Integr Med. 2021;19:85–8.
Gong L, Wang R, Wang X, et al. Research progress of natural active compounds on improving podocyte function to reduce proteinuria in diabetic kidney disease. Ren Fail. 2023;45:2290930.
Li C, Guan XM, Wang RY, et al. Berberine mitigates high glucose-induced podocyte apoptosis by modulating autophagy via the mTOR/P70S6K/4EBP1 pathway. Life Sci. 2020;243: 117277.
Zhang M, Zhang Y, Xiao D, et al. Highly bioavailable berberine formulation ameliorates diabetic nephropathy through the inhibition of glomerular mesangial matrix expansion and the activation of autophagy. Eur J Pharmacol. 2020;873: 172955.
Borkar RM, Kanwal A, Raju B, et al. A pharmacokinetic study to correlate the hypoglycemic effect of phlorizin in rats: identification of metabolites as inhibitors of sodium/glucose cotransporters. J Mass Spectrom. 2023;58: e4964.
Kumar S, Chhimwal J, Kumar S, et al. Phloretin and phlorizin mitigates inflammatory stress and alleviate adipose and hepatic insulin resistance by abrogating PPARγ S273-Cdk5 interaction in type 2 diabetic mice. Life Sci. 2023;322: 121668.
Madrakhimov SB, Yang JY, Kim JH, Han JW, Park TK. mTOR-dependent dysregulation of autophagy contributes to the retinal ganglion cell loss in streptozotocin-induced diabetic retinopathy. Cell Commun Signal. 2021;19:29.
Foudah AI, Ayman Salkini M, Alqarni MH, Alam A. Preparation and evaluation of antidiabetic activity of mangiferin-loaded solid lipid nanoparticles. Saudi J Biol Sci. 2024;31: 103946.
Olusola AJ, Famuyiwa SO, Faloye KO, et al. Neomangiferin, a naturally occurring mangiferin congener, inhibits sodium-glucose co-transporter-2: an in silico approach. Bioinform Biol Insights. 2024;18:11779322231223852.
Wang X, Gao L, Lin H, et al. Mangiferin prevents diabetic nephropathy progression and protects podocyte function via autophagy in diabetic rat glomeruli. Eur J Pharmacol. 2018;824:170–8.
Torres-Narvaez JC, Perez-Torres I, Del Valle-Mondragon L, et al. Garlic prevents the oxidizing and inflammatory effects of sepsis induced by bacterial lipopolysaccharide at the systemic and aortic level in the rat. Role of trpv1. Heliyon. 2023;9: e21230.
Khare P, Mahajan N, Singh DP, et al. Allicin, a dietary trpa1 agonist, prevents high fat diet-induced dysregulation of gut hormones and associated complications. Food Funct. 2021;12:11526–36.
Qian R, Chen H, Lin H, et al. The protective roles of allicin on type 1 diabetes mellitus through AMPK/mTOR mediated autophagy pathway. Front Pharmacol. 2023;14:1108730.
Arellano Buendia AS, Juarez Rojas JG, Garcia-Arroyo F, et al. Antioxidant and anti-inflammatory effects of allicin in the kidney of an experimental model of metabolic syndrome. PeerJ. 2023;11: e16132.
Wang LY, Cheng KC, Li Y, Niu CS, Cheng JT, Niu HS. Glycyrrhizic acid increases glucagon like peptide-1 secretion via TGR5 activation in type 1-like diabetic rats. Biomed Pharmacother. 2017;95:599–604.
Zhao TQ, Li Y, Zhang M, Zhao MC, Cao X, Hou SZ. Glycyrrhizic acid protects glomerular podocytes induced by high glucose by modulating SNARK/AMPK signaling pathway. Curr Med Sci. 2023;43:696–707.
Fei C, Wang Y, Gong Y, Xu H, Yu Q, Shi Y. Acquired reactive perforating collagenosis: a report of a typical case. Medicine (Baltimore). 2016;95: e4305.
Huan DQ, Hop NQ, Son NT. Oxymatrine: a current overview of its health benefits. Fitoterapia. 2023;168: 105565.
Guo S, Yan T, Shi L, et al. Matrine, as a CaSR agonist promotes intestinal GLP-1 secretion and improves insulin resistance in diabetes mellitus. Phytomedicine. 2021;84: 153507.
Zuo ML, Wang AP, Tian Y, Mao L, Song GL, Yang ZB. Oxymatrine ameliorates insulin resistance in rats with type 2 diabetes by regulating the expression of KSRP, PETN, and AKT in the liver. J Cell Biochem. 2019;120:16185–94.
Springer M, Moco S. Resveratrol and its human metabolites-effects on metabolic health and obesity. Nutrients. 2019;11:143.
Zhao YH, Fan YJ. Resveratrol improves lipid metabolism in diabetic nephropathy rats. Front Biosci (Landmark Ed). 2020;25:1913–24.
Salami M, Salami R, Mafi A, Aarabi MH, Vakili O, Asemi Z. Therapeutic potential of resveratrol in diabetic nephropathy according to molecular signaling. Curr Mol Pharmacol. 2022;15:716–35.
Xu XH, Ding DF, Yong HJ, et al. Resveratrol transcriptionally regulates miRNA-18a-5p expression ameliorating diabetic nephropathy via increasing autophagy. Eur Rev Med Pharmacol Sci. 2017;21:4952–65.
Sha W, Liu M, Sun D, et al. Resveratrol improves Gly-LDL-induced vascular endothelial cell apoptosis, inflammatory factor secretion and oxidative stress by regulating miR-142-3p and regulating SPRED2-mediated autophagy. Aging (Albany NY). 2021;13:6878–89.
Liu WJ, Gan Y, Huang WF, et al. Lysosome restoration to activate podocyte autophagy: a new therapeutic strategy for diabetic kidney disease. Cell Death Dis. 2019;10:806.
Abhale K, Veeranjaneyulu A, Desai S, Sanap A, Bhonde R. Effects of mesenchymal stem cell-conditioned media with natural immunomodulatory agent resveratrol on type 1 diabetes. Curr Drug Discov Technol. 2024. https://doi.org/10.2174/0115701638276524240305054259. (Online ahead of print).
Wahab M, Janaswamy S. Porous corn starch granules as effective host matrices for encapsulation and sustained release of curcumin and resveratrol. Carbohydr Polym. 2024;333: 121967.
Heydarpour F, Sajadimajd S, Mirzarazi E, et al. Involvement of TGF-β and autophagy pathways in pathogenesis of diabetes: a comprehensive review on biological and pharmacological insights. Front Pharmacol. 2020;11: 498758.
Yadegar S, Mohammadi F, Yadegar A, et al. Effects and safety of resveratrol supplementation in older adults: a comprehensive systematic review. Phytother Res. 2024;38:2448–61.
Xiaoqin S, Yi T, Xiaoyu L, Ya B, Jingwen S, Yin L. Research progress of traditional Chinese medicine monomer in treating diabetic peripheral neuropathy: a review. Medicine (Baltimore). 2024;103: e37767.
Khater SI, Dowidar MF, Abdel-Aziz AE, et al. β-Cell autophagy pathway and endoplasmic reticulum stress regulating-role of liposomal curcumin in experimental diabetes mellitus: a molecular and morphometric study. Antioxidants (Basel). 2022;11:2400.
Zhang P, Fang J, Zhang J, Ding S, Gan D. Curcumin inhibited podocyte cell apoptosis and accelerated cell autophagy in diabetic nephropathy via regulating Beclin1/UVRAG/Bcl2. Diabetes Metab Syndr Obes. 2020;13:641–52.
Rainey N, Motte L, Aggarwal BB, Petit PX. Curcumin hormesis mediates a cross-talk between autophagy and cell death. Cell Death Dis. 2015;6: e2003.
Varshney R, Gupta S, Roy P. Cytoprotective effect of kaempferol against palmitic acid-induced pancreatic β-cell death through modulation of autophagy via AMPK/mTOR signaling pathway. Mol Cell Endocrinol. 2017;448:1–20.
Zhang N, Zhao S, Hong J, Li W, Wang X. Protective effects of kaempferol on D-ribose-induced mesangial cell injury. Oxid Med Cell Longev. 2019;2019:7564207.
Sheng H, Zhang D, Zhang J, et al. Kaempferol attenuated diabetic nephropathy by reducing apoptosis and promoting autophagy through AMPK/mTOR pathways. Front Med (Lausanne). 2022;9: 986825.
Li X, Zhu Q, Zheng R, et al. Puerarin attenuates diabetic nephropathy by promoting autophagy in podocytes. Front Physiol. 2020;11:73.
Xu X, Chen B, Huang Q, Wu Y, Liang T. The effects of puerarin on autophagy through regulating of the PERK/eIF2α/ATF4 signaling pathway influences renal function in diabetic nephropathy. Diabetes Metab Syndr Obes. 2020;13:2583–92.
Qu C, Tan X, Hu Q, et al. A systematic review of astragaloside IV effects on animal models of diabetes mellitus and its complications. Heliyon. 2024;10: e26863.
Zhou L, Zhang R, Yang S, Zhang Y, Shi D. Astragaloside IV alleviates placental oxidative stress and inflammation in GDM mice. Endocr Connect. 2020;9:939–45.
Awad AM, Elshaer SL, Gangaraju R, Abdelaziz RR, Nader MA. Ameliorative effect of montelukast against STZ induced diabetic nephropathy: targeting HMGB1, TLR4, NF-κB, NLRP3 inflammasome, and autophagy pathways. Inflammopharmacology. 2024;32:495–508.
Wu J, Li T, Guo M, et al. Treating a type 2 diabetic patient with impaired pancreatic islet function by personalized endoderm stem cell-derived islet tissue. Cell Discov. 2024;10:45.
Liu Y, Zhang Z, Ma C, Song J, Hu J, Liu Y. Transplanted MSCs promote alveolar bone repair via hypoxia-induced extracellular vesicle secretion. Oral Dis. 2024. https://doi.org/10.1111/odi.14982. (Online ahead of print).
Kaminska D, Skrzycki M. Lipid droplets, autophagy, and ER stress as key (survival) pathways during ischemia-reperfusion of transplanted grafts. Cell Biol Int. 2024;48:253–79.
Zhao N, Chen X, Chen QG, et al. NLRP3-mediated autophagy dysfunction links gut microbiota dysbiosis to tau pathology in chronic sleep deprivation. Zool Res. 2024;45:857–74.
Li X, Gong H, Yang S, Yang L, Fan Y, Zhou Y. Pectic bee pollen polysaccharide from Rosa rugosa alleviates diet-induced hepatic steatosis and insulin resistance via induction of AMPK/mTOR-mediated autophagy. Molecules. 2017;22:699.
Forbes JM, Cooper ME. Mechanisms of diabetic complications. Physiol Rev. 2013;93:137–88.
Liu C, Yao Q, Hu T, et al. Cathepsin B deteriorates diabetic cardiomyopathy induced by streptozotocin via promoting NLRP3-mediated pyroptosis. Mol Ther Nucleic Acids. 2022;30:198–207.
Qi X, Mitter SK, Yan Y, Busik JV, Grant MB, Boulton ME. Diurnal rhythmicity of autophagy is impaired in the diabetic retina. Cells. 2020;9:905.
Feng Y, Xu W, Zhang W, Wang W, Liu T, Zhou X. LncRNA DCRF regulates cardiomyocyte autophagy by targeting miR-551b-5p in diabetic cardiomyopathy. Theranostics. 2019;9:4558–66.
Chen F, Sun Z, Zhu X, Ma Y. Astilbin inhibits high glucose-induced autophagy and apoptosis through the PI3K/Akt pathway in human proximal tubular epithelial cells. Biomed Pharmacother. 2018;106:1175–81.
Tang G, Du Y, Guan H, et al. Butyrate ameliorates skeletal muscle atrophy in diabetic nephropathy by enhancing gut barrier function and FFA2-mediated PI3K/Akt/mTOR signals. Br J Pharmacol. 2022;179:159–78.
Pan HC, Chen JY, Chen HY, et al. GLP-1 receptor agonists’ impact on cardio-renal outcomes and mortality in T2D with acute kidney disease. Nat Commun. 2024;15:5912.
Ronco C, Haapio M, House AA, Anavekar N, Bellomo R. Cardiorenal syndrome. J Am Coll Cardiol. 2008;52:1527–39.
Yang K, Cao F, Wang W, Tian Z, Yang L. The relationship between HMGB1 and autophagy in the pathogenesis of diabetes and its complications. Front Endocrinol (Lausanne). 2023;14:1141516.
Xuan X, Zhang S. Targeting the programmed cell death (PCD) signaling mechanism with natural substances for the treatment of diabetic cardiomyopathy (DCM). Phytother Res. 2023;37:5495–508.
Palmer SC, Tendal B, Mustafa RA, et al. Sodium-glucose cotransporter protein-2 (SGLT-2) inhibitors and glucagon-like peptide-1 (GLP-1) receptor agonists for type 2 diabetes: systematic review and network meta-analysis of randomised controlled trials. BMJ. 2021;372: m4573.
Acknowledgements
The signal mechanism diagrams (Figs. 1 and 2) in this manuscript were drawn using www.Figdraw.com.
Funding
The review was funded by: 1. Traditional Chinese and Western Medicine clinical medicine brand construction project of Jiangsu Higher Education Institutions (Phase II); no: 2020PPZXL261. 2. Research project of Inner Mongolia Medical University Affiliated Hospital; no: 2023NYFYGG004. 3. The open fund project of the key laboratory of Chinese and Mongolian Medicine in Inner Mongolia Autonomous Region; no: MYX2022-K07. The journal’s Rapid Service Fee was funded by the authors.
Author information
Authors and Affiliations
Contributions
LI Qirui: investigation and writing. XU Huiying: writing—review and editing. MA Ruiting: writing—review and editing. MA Yuanyuan: writing—review and editing and funding acquisition. CHEN Meijuan: writing—review and editing and funding acquisition.
Corresponding authors
Ethics declarations
Conflict of Interest
All authors (Qi-Rui LI, Hui-Ying XU, Rui-Ting MA, Yuan-Yuan MA, Mei-Juan CHEN) disclosed no relevant relationships.
Ethical Approval
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Li, QR., Xu, HY., Ma, RT. et al. Targeting Autophagy: A Promising Therapeutic Strategy for Diabetes Mellitus and Diabetic Nephropathy. Diabetes Ther 15, 2153–2182 (2024). https://doi.org/10.1007/s13300-024-01641-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13300-024-01641-3