
Abstract
Acquired evidence indicated that microRNAs (miRNAs) played essential roles in cancer development, including hepatocellular carcinoma (HCC). Functions and mechanisms of miRNAs involved in HCC remain largely unknown. Here, we found that miR-384 was significantly downregulated in HCC cells and tissues by RT-PCR. Gain and loss of function studies revealed that miR-384 significantly suppressed HCC cell proliferation. Insulin receptor substrate 1(IRS1) was identified as a direct and functional target of miR-384. Moreover, miR-384 decreased IRS1 expression, subsequently downregulating cyclin D1 and upregulating p21 and p-Rb expression. In addition, promotion of cell proliferation caused by miR-384-in was counteracted by silencing IRS1 expression with siRNAs. Taken together, our data provided convincing evidence that miR-384 exerted suppressive effect on HCC cell proliferation through the direct inhibition of IRS1 expression, suggesting miR-384 may serve as a potential therapeutic target for HCC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hepatocellular carcinoma (HCC) is the second leading cause of cancer death worldwide in males [1]. Despite the improvement of current treatment, the disease of HCC remains a high mortality rate; thus, there is an urgency need to find the underlying molecular mechanisms and find new biomarkers that can be used for early screening and diagnosis of HCC [2].
Lots of reports have indicated that microRNAs (miRNAs) are small noncoding RNA oligonucleotides (18–25 nt) that play essential roles in the regulation of multiple biological processes, such as cell proliferation, apoptosis, invasion, and migration [3–6]. Accumulating evidence has shown that miRNAs can act as tumor suppressors or oncogenes, functioning by targeting their specific target gene [7–9]. MiR-1180 was reported to promote apoptotic resistance of HCC by activating NF-kB signaling pathway [10]. MiR-361-5p was found to inhibit cancer cell growth by targeting CXCR6 in HCC [11]. Tu K et al. demonstrated that miR-519a promoted tumor growth by targeting PTEN/PI3K/AKT signaling in HCC [12]. Furthermore, loss of miR-532-5p influenced CXCL2 expression and then promoted cell proliferation and metastasis in HCC [13]. But the biological function of miR-384 and its mechanism of HCC remain unclear. This study explored the relationship between miR-384 and insulin receptor substrate 1 (IRS1) in HCC cells and investigates its function in cell proliferation. Here, we found that miR-384 was markedly downregulated in HCC cells and clinical tissues. Further experiment suggested that ectopic expression of miR-384 decreased HCC cell proliferation by regulating IRS1 expression.
Materials and methods
Clinical specimens
Eight human HCC tissues and adjacent normal tissues (ANT) were obtained from HCC patients at the Department of General Surgery, Guangzhou First People’s Hospital, Guangzhou Medical University (Guangzhou, People’s Republic of China). The study was approved by the ethics committee of Guangzhou First People’s Hospital, Guangzhou Medical University (Guangzhou, People’s Republic of China). Written informed consent was obtained from all patients. Tissue samples were collected at surgery, immediately frozen in liquid nitrogen and stored until total RNAs or proteins were extracted.
Cell culture
Human HCC cell lines MHCC97H, BEL-7402, MHCC97L, HepG2, Huh7, Hep3B, HCCC-9810, and QGY-7703 were obtained from the American Type Culture Collection (ATCC, Manassas, VA), and human hepatic cell lines THLE3 were purchased from the China Center for Type Culture Collection. All cells were grown in Dulbecco’s Modified Eagle Medium (Gibco BRL, Rockville, MD) McCoy’s 5A modified (Invitrogen; Life Technologies, Carlsbad, CA) medium supplemented with 10 % fetal bovine serum (FBS, Sigma, USA), 100 units/ml of penicillin-streptomycin (Invitrogen, Carlsbad, CA) in a humidified incubator in 5 % CO2 at 37 °C.
Plasmids, small interfering RNA, and transfection
MiR-384, miR-384-inhibitor (miR-384-in), miR-384-mut, and relative control miRNAs were synthesized by RiboBio Corporation (Guangzhou, China), and transfection was performed using Lipofectamine 2000 (Life Technologies, Carlsbad, CA, USA) as recommended by the manufacturer’s instructions.
Two shRNA lentiviruses against IRS1 were purchased from Genecopoeia (Genecopoeia Co. Ltd.) and transfection of siRNAs were performed using lipofectamine 2000 (Invitrogen), according to the manufacturer’s protocol.
The IRS1 ORF was amplified from the HepG2 cell cDNA and subcloned into pEGFP-N3 (Invitrogen). The wild-type 3′UTR of IRS1 was synthesized and subcloned into the firefly luciferase reporter (RiboBio, Guangzhou, China) and then transfected into HCC cells using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s instructions.
RNA extraction and real-time quantitative PCR
Total RNA was extracted using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA). The expression of miR-384 was detected by TaqMan® MicroRNA Assays (Applied Biosystems) according to the manufacturer’s instructions. The cyclin D1 and p21 levels were quantified using qRT-PCR with the TaqMan®. We assessed the RNA expression according to relative quantification using the 2−ΔΔCt method to determine the fold change in the expression.
MTT assays and colony formation
Cell proliferation assay was carried out using the MTT assay. Twenty microliters of 5 mg/ml MTT solution (Sigma-Aldrich) was added to each well and incubated at 37 °C for 4 h, and the resulting MTT formazan was solubilized in 150 μl of DMSO. The absorbance at 490 nm was measured in a Thermo Scientific Multiskan (Thermo Fisher Scientific, USA).
For colony formation assays, 1000 HepG2 cells were seeded into each well of a six-well plate. On 2 weeks after seeding, the cells were stained with 0.1 % crystal violet for 30 s and fixation with 10 % formaldehyde for 10 min. Images were taken and the colonies were counted.
Anchorage-independent growth assay
One thousand HepG2 cells were trypsinized and resuspended in 2 ml complete medium plus 0.3 % agar (Sigma). The agar-cell mixture was plated on top of a bottom layer comprising 0.66 % complete medium agar mixture. After 10 days, colony sizes were measured with an ocular micrometer and colonies greater than 0.1 mm in diameter were counted. The experiment was performed for independently three times for each cell line.
Luciferase assays
Cells were seeded in triplicate in 24-well plates at a density of 50000 cells per well. After 24 h, cells were co-transfected with miR-384 or miR-384-in or miR-384-mut and pGL3-IRS1-3′-UTR. Forty-eight hours after transfection, the cells were lysed and assayed for the firefly luciferase and the Renilla luciferase activities using the Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA).
Western blotting
The cultured cells were lysed, equal quantities of protein (30 μg) were separated on 10 % SDS-PAGE gels, and gels were transferred onto nitrocellulose membrane. The membranes were incubated with anti-IRS1 (#2382; 1:1000; Cell Signaling Technology), anti-cyclin D1 (#2922; 1:1000; Cell Signaling Technology), anti-p21 (#2947; 1:1000; Cell Signaling Technology), anti-phosphorylated Rb (p-Rb; #8516; 1:1000; Cell Signaling Technology), and anti-Rb (#9313; 1:1000; Cell Signaling Technology). And as the control sample loading, the membranes were stripped and re-probed with an anti-α-tubulin antibody (SAB4500087; Sigma-Aldrich). The membranes were then washed and incubated with the respective goat anti-rabbit secondary antibodies (Beyotime Biotechnology, China). Immunocomplexes were visualized using the chemiluminesence (GE, USA) according to the manufacturer’s protocol.
Statistical analysis
All results in this study were statistically presented as mean ± standard error of the mean by using the SPSS 19.0 (IBM). A P < 0.05 was considered to be statistically significant by Student’s t test.
Result
MiR-384 expression was downregulated in HCC cell lines and HCC tissues
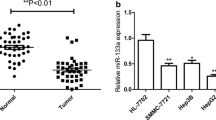
In order to understand the role of miR-384 in HCC development, we were interested in the identification of the miR-384 expression in HCC cell lines and primary HCC tissue samples by quantitative real-time PCR analyses. MiR-384 expression was significantly lower in eight HCC cell lines compared with human hepatic cell lines THLE3 (Fig. 1a). Similarly, miR-384 expression was remarkably downregulated in HCC tissues compared with the corresponding adjacent non-tumor tissues (Fig. 1b). Together, these results suggested that miR-384 was significantly increased in HCC.

Expression of miR-384 in human hepatocellular carcinoma (HCC) tissues and cell lines. a Real-time PCR analysis of miR-384 expression in human hepatic cell lines THLE3 cells and HCC cell lines, including MHCC97H, BEL-7402, MHCC97L, HepG2, Huh7, Hep3B, HCCC-9810, and QGY-7703. b Relative miR-384 mRNA expression levels in 8 paired primary HCC tissues (T) and the matched adjacent normal tissues (ANT) from the same patient were detected by PCR analysis. Experiments were repeated at least three times (a, b). Each bar represents the mean of three independent experiments. *P < 0.05
MiR-384 suppressed HCC cell proliferation
To investigate the biological functions of miR-384 in HCC development, we transfected with miR-384, miR-384-in, and their relative negative controls in HepG2 cells, and the expression of miR-384 was confirmed by real-time qPCR (Figs. 2a and 3a). Result of MTT assay and colony formation assay demonstrated that overexpression of miR-384 resulted in significant reduction in cell proliferation and colony formation capability of HepG2 cells (Fig. 2b, c). Consistently, ectopic expression of miR-384 dramatically attenuated the anchorage-independent cell growth ability than the relative control cells (Fig. 2d). Moreover, HepG2 cells after transfected with miR-384-in resulted in significant increasing of cell proliferation and colony formation capabilities and increased the anchorage-independent cell growth ability (Fig. 3b–d). These results suggested suppressive effects of miR-384 on HCC cell proliferation.

miR-384 upregulation inhibited HCC cell proliferation. a Validation of miR-384 expression levels after transfection by PCR analysis. b MTT assays revealed that upregulation of miR-384 suppressed growth of HepG2 cells. c Representative micrographs (left) and quantification (right) of crystal violet-stained cell colonies. d Upregulation of miR-384 promoted the anchorage-independent growth of HepG2 cells (×100). Representative micrographs (left) and quantification of colonies that were >0.1 mm (right). Each bar represents the mean of three independent experiments. *P < 0.05

Inhibition of miR-384 promoted HCC cell proliferation. a Validation of miR-384 expression levels after transfection by PCR analysis. b MTT assays revealed that miR-384-in increased growth of HepG2 cells. c Representative micrographs (left) and quantification (right) of crystal violet-stained cell colonies. d Inhibition of miR-384 inhibited the anchorage-independent growth of HepG2 cells (×100). Representative micrographs (left) and quantification of colonies that were >0.1 mm (right). Each bar represents the mean of three independent experiments. *P < 0.05
MiR-384 directly targets IRS1 by binding to its 3′-UTR and altered levels of proteins related to cell proliferation in HepG2 cells
It is reported that miRNAs exert their functions by regulating their downstream target genes expression. We found that miR-384 might be a potential regulator of IRS1 by TargetScan 6.2 (Fig. 4a).

MiR-384 suppresses IRS1 expression by directly targeting the IRS1 3′-UTR and altered levels of proteins related to proliferation in HCC cells. a Predicted miR-384 target sequence in the 3′-UTR of IRS1 (IRS1-3′-UTR) and positions of three mutated nucleotides (red) in the 3′-UTR of miR-384-mut. b IRS1 protein expression in HepG2 and Hep3B cells transfected with miR-384 or the miR-384-in were detected by Western blotting analysis. α-Tubulin served as the loading control. c Luciferase reporter assay of HepG2 cells transfected with the pGL3-IRS1-3′-UTR reporter and miR-384 or miR-384-in or miR-384-mut with increasing amounts (10 and 50 nM) oligonucleotides. d Real-time PCR analysis of expression of cyclin D1 and p21 in HepG2 cells. e Western blotting analysis of protein expression of cyclin D1, p21, p-Rb, and Rb in HepG2 cells. α-Tubulin was used to serve as the loading control. *P < 0.05
To determine whether IRS1 can be directly targeted by miR-384, IRS1 expressions were detected in HepG2 cells after transfected with miR-384, miR-384-in or the respective controls. Results of Western blot showed that miR-384 significantly suppressed IRS1 protein levels, while miR-384-in clearly increased IRS1 protein expression in HepG2 and Hep3B cells (Fig. 4b). Using the luciferase reporter, as showed in Fig. 4c, we found that overexpression of miR-384 in HepG2 cells suppressed the activity of reporter gene, whereas miR-384-in increased wild-type IRS1 luciferase activity. Meanwhile, miR-384-mut showed no changes in the reporter gene activity in HepG2 cells (Fig. 4c).
It has been shown that IRS1-induced increase translation of β-catenin leads to upregulation of its downstream targets related to cancer cell proliferation, such as c-Myc and cyclin D1 [14]. In this study, we evaluated that the mRNA of the IRS1 downstream genes (cyclin D1, p21, Rb and p-Rb). Quantitative PCR results revealed a significant reduction in the mRNA levels of cyclin D1 and p21 (a protein product of CDKN1A gene) in miR-384 transferred-HepG2 cells, while miR-384-in showed the opposite effect (Fig. 4d). The results of Western blot analysis showed that the levels of cyclin D1 were decreased while the levels of p21 and p-Rb were increased in the miR-384 group compared with the miR-NC group, while miR-384-in showed the opposite function (Fig. 4e). Take together, our results demonstrated that miR-384 functionally modulated IRS1 and then regulated cellular proliferation regulators, cyclin D1, p21, and p-Rb, thus relevant to cell proliferation. All these results demonstrated that miR-384 could combine with IRS1 3′UTR and play a role in suppressing the expression of IRS1 gene.
IRS1 downregulation counteracted the promotion of cell proliferation by miR-384-in
Given that IRS1 acted as a direct target of miR-384 and possibly involve in HCC tumorigenesis, we investigated the role of IRS1 on HCC cell proliferation. Downregulation of IRS1 in miR-384-in-transfected HepG2 cells by transfecting with IRS1-siRNAs and was confirmed by Western blot analysis (Fig. 5a). Result of MTT, colony formation and anchorage-independent growth assays indicated that the decreased expression of IRS1 significantly decreased cell proliferation and cell growth, and colony formation capabilities of HepG2 cells after transfected with miR-384-in (Fig. 5b–d). Taken together, these results suggested that the tumor-suppressive effect of miR-384 is mediated by inhibition of IRS1 in HCC.

IRS1 downregulation counteracted the promotion of cell proliferation by miR-384-in. a Western blot analysis verified that silencing IRS1 effectively decreased the expression of IRS1 in miR-384-in-transfected HepG2 cells. b miR-384-in-transfected HepG2 cells after transfection with IRS1-siRNAs decreased cell growth. c miR-384-in-transfected HepG2 cells after transfection with IRS1-siRNAs decreased cell colonies formation. d miR-384-in-transfected HepG2 cells after transfection with IRS1-siRNAs decreased the anchorage-independent growth. Representative quantification of colonies that were >0.1 mm. Each bar represents the mean of three independent experiments. *P < 0.05
Discussion
In this present study, we investigated the biological role of miR-384 in the progression of HCC. We found significant downregulation of miR-384 in HCC tissues and cells compared with the corresponding adjacent non-tumor tissues and human hepatic cell lines THLE3. Additionally, functional experiments revealed that miR-384 suppressed cell proliferation in vitro through targeting IRS1 in HCC.
Recently, the function of miRNAs on cancer development is attracting increasing attention. Lots of reports have demonstrated that dysregulation of miRNAs have been observed in various kinds of malignancies and are closely related to tumorigenesis, invasion, migration, and prognosis. MiR-26a was reported to be downregulated and suppressed angiogenesis in human hepatocellular carcinoma [15]. MiR-708 was found to be downregulated in hepatocellular carcinoma and suppressed tumor invasion and migration [16]. Furthermore, miR-335 acted as a potential tumor suppressor and inhibited cell growth in hepatocellular carcinoma [17]. Finding by Xu C et al. demonstrated that miR-193b regulated proliferation, migration, and invasion in human hepatocellular carcinoma [18]. But the biological function of miR-384 and its mechanism in HCC development has not been elucidated. Our study demonstrated that miR-384 acted as tumor suppressor and suppressed HCC cell proliferation.
Increasing numbers of reports indicated that miRNAs silence the gene expression by targeting the 3′untranslated region (3′-UTR) of its targeted mRNAs. MiR-21 had been observed to promote cell proliferation in human hepatocellular carcinoma partly by targeting HEPN1 [19]. MiR-331-3p targeted PH domain and leucine-rich repeat protein phosphatase and then promoted proliferation and metastasis of hepatocellular carcinoma [4]. Finding of Imam JS et al. indicated that miR-185 targeted Six1 and suppressed tumor growth and progression in human cancers [20]. IRS1, the first identified member of IRS family, is interact with β-catenin activation induces and involved in tumor development [14]. Cao M et al. revealed that miR-23a-mediated migration/invasion was rescued by targeting IRS-1 in non-small cell lung cancer [21]. Finding of Su W et al. indicated that miR-200c targeted IRS1 and suppressed prostate cancer cell growth [22]. In this study, our data demonstrated that miR-384 could decrease IRS1 protein expression through binding to its 3′-UTR region. Moreover, IRS1 downregulation by using IRS1-siRNAs counteracted the promotion of cell proliferation by miR-384-in. Our findings revealed that IRS1 was a direct target of miR-384.
In summary, our study indicated that miR-384 suppressed cell proliferation by modulating IRS1 expression and provided more insight into the mechanisms of miR-384 involved in HCC, suggesting miR-384 could be a potential therapeutic target in HCC.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108.
Callegari E, Gramantieri L, Domenicali M, D’Abundo L, Sabbioni S, Negrini M. Micrornas in liver cancer: a model for investigating pathogenesis and novel therapeutic approaches. Cell Death Differ. 2015;22:46–57.
Zhang Y, Huang F, Wang J, Peng L, Luo H. Mir-15b mediates liver cancer cells proliferation through targeting bcl-2. Int J Clin Exp Pathol. 2015;8:15677–83.
Chang RM, Yang H, Fang F, Xu JF, Yang LY. Microrna-331-3p promotes proliferation and metastasis of hepatocellular carcinoma by targeting ph domain and leucine-rich repeat protein phosphatase. Hepatology. 2014;60:1251–63.
Fu X, Meng Z, Liang W, Tian Y, Wang X, Han W, et al. Mir-26a enhances mirna biogenesis by targeting lin28b and zcchc11 to suppress tumor growth and metastasis. Oncogene. 2014;33:4296–306.
Sun Z, Han Q, Zhou N, Wang S, Lu S, Bai C, et al. Microrna-9 enhances migration and invasion through klf17 in hepatocellular carcinoma. Mol Oncol. 2013;7:884–94.
Bai R, Weng C, Dong H, Li S, Chen G, Xu Z. Microrna-409-3p suppresses colorectal cancer invasion and metastasis partly by targeting gab1 expression. Int J Cancer. 2015;137:2310–22.
Xing F, Sharma S, Liu Y, Mo YY, Wu K, Zhang YY, et al. Mir-509 suppresses brain metastasis of breast cancer cells by modulating rhoc and tnf-alpha. Oncogene. 2015;34:4890–900.
Zhang H, Li S, Yang X, Qiao B, Zhang Z, Xu Y. Mir-539 inhibits prostate cancer progression by directly targeting spag5. J Exp Clin Cancer Res. 2016;35:60.
Tan G, Wu L, Tan J, Zhang B, Tai WC, Xiong S, et al. Mir-1180 promotes apoptotic resistance to human hepatocellular carcinoma via activation of nf-kappab signaling pathway. Sci Rep. 2016;6:22328.
Sun JJ, Chen GY, Xie ZT. Microrna-361-5p inhibits cancer cell growth by targeting cxcr6 in hepatocellular carcinoma. Cell Physiol Biochem. 2016;38:777–85.
Tu K, Liu Z, Yao B, Han S, Yang W. Microrna-519a promotes tumor growth by targeting pten/pi3k/akt signaling in hepatocellular carcinoma. Int J Oncol. 2016;48:965–74.
Song X, Wang Z, Jin Y, Wang Y, Duan W. Loss of mir-532-5p in vitro promotes cell proliferation and metastasis by influencing cxcl2 expression in hcc. Am J Transl Res. 2015;7:2254–61.
Zheng H, Zhang F, Lin X, Huang C, Zhang Y, Li Y, et al. Microrna-1225-5p inhibits proliferation and metastasis of gastric carcinoma through repressing insulin receptor substrate-1 and activation of beta-catenin signaling. Oncotarget. 2016;7:4647–63.
Yang X, Zhang XF, Lu X, Jia HL, Liang L, Dong QZ, et al. Microrna-26a suppresses angiogenesis in human hepatocellular carcinoma by targeting hepatocyte growth factor-cmet pathway. Hepatology. 2014;59:1874–85.
Li G, Yang F, Xu H, Yue Z, Fang X, Liu J. Microrna-708 is downregulated in hepatocellular carcinoma and suppresses tumor invasion and migration. Biomed Pharmacother. 2015;73:154–9.
Liu H, Li W, Chen C, Pei Y, Long X. Mir-335 acts as a potential tumor suppressor mirna via downregulating rock1 expression in hepatocellular carcinoma. Tumor Biol. 2015;36:6313–9.
Xu C, Liu S, Fu H, Li S, Tie Y, Zhu J, et al. Microrna-193b regulates proliferation, migration and invasion in human hepatocellular carcinoma cells. Eur J Cancer. 2010;46:2828–36.
Hu S, Tao R, Wang S, Wang C, Zhao X, Zhao H, et al. Microrna-21 promotes cell proliferation in human hepatocellular carcinoma partly by targeting hepn1. Tumor Biol. 2015;36:5467–72.
Imam JS, Buddavarapu K, Lee-Chang JS, Ganapathy S, Camosy C, Chen Y, et al. Microrna-185 suppresses tumor growth and progression by targeting the six1 oncogene in human cancers. Oncogene. 2010;29:4971–9.
Cao M, Li Y, Lu H, Meng Q, Wang L, Cai L, et al. Mir-23a-mediated migration/invasion is rescued by its target, irs-1, in non-small cell lung cancer cells. J Cancer Res Clin Oncol. 2014;140:1661–70.
Su W, Xu M, Chen X, Nie L, Chen N, Gong J, et al. Mir200c targets irs1 and suppresses prostate cancer cell growth. Prostate. 2015;75:855–62.
Acknowledgments
This work was supported by Guangzhou medicine and health care technology projects (20141A011011, 20151A011007 and 20161A011008), Guangzhou science and technology plan projects (201510010009) and Guangdong provincial science and technology plan projects (2014A020212033). All authors designed the study together, performed the experiment together, analyzed the data and wrote the paper; all authors approved the final manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflicts of interest
None.
Rights and permissions
About this article

Cite this article
Lai, Yy., Shen, F., Cai, WS. et al. MiR-384 regulated IRS1 expression and suppressed cell proliferation of human hepatocellular carcinoma. Tumor Biol. 37, 14165–14171 (2016). https://doi.org/10.1007/s13277-016-5233-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-016-5233-5