
Abstract
MicroRNAs (miRNAs) are a class of small non-coding RNAs and have critical roles in tumorigenesis and metastasis. A growing body of evidence showed that microRNA-133a (miR-133a) was downregulated and played tumor suppressor roles in gastric, colorectal, bladder, and lung cancer. However, the role and underlying molecular mechanism of miR-133a in hepatocellular carcinoma (HCC) remain unclear. In this study, we analyzed the expression of miR-133a in HCC tissues and HCC cell lines. We find that miR-133a was downregulated in HCC tissues and cell lines and that miR-133a expression negatively correlated with tumor differentiation (P < 0.01), TNM stage (P < 0.01), and lymph node metastasis (P < 0.01). Then, functional studies demonstrate that restoration of miR-133a in HepG2 cells significantly suppressed proliferation, colony formation, migration, and invasion, induced cell cycle arrest at G0/G1 stage and cell apoptosis in vitro, and decreased tumor size and weight in a nude mouse HepG2 xenograft model. Using bioinformatics method and dual luciferase assays identified insulin-like growth factor 1 receptor (IGF-1R) as a direct target of miR-133a in HCC cells. Furthermore, overexpression of miR-133a inhibited activation AKT and ERK signal pathway, which contributed to suppression of HCC cell growth. These findings suggest that miR-133a may act as a tumor suppressor and inhibited survival of HCC cells by targeting IGF-1R.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hepatocellular carcinoma (HCC) is the sixth most commonly malignant tumor and third leading cause of cancer-related death worldwide [1, 2], with very high morbidity and mortality [3]. Although improvements have been made in surgery and other treatments for HCC, the prognosis for HCC patients remains unsatisfactory, and that of the patients with frequent intra- or extra-hepatic metastasis is less than 5 % [4] since most HCC cases are diagnosed in an advanced stage with limited therapeutic options, leading to the poor prognosis [5]. There is an urgent need to understand the underlying molecular mechanisms of HCC to develop optimized strategies for the early diagnosis and therapy of HCC.
MicroRNAs (miRNAs), small non-coding endogenous RNA gene products consisting of 18 to 25 nucleotides in length, are responsible for posttranscriptional regulation by binding to the 3′-untranslated regions (3′-UTRs) of target messenger RNAs and causing either degradation or inhibition of translation, thus effectively silencing their messenger RNA (mRNA) target [6]. miRNA has been reported to be participating in various biological processes such as differentiation, morphogenesis, and tumorigenesis [7, 8]. Accumulating pieces of evidence have suggested that miRNAs could play oncogene or tumor suppressor roles in the etiology and pathogenesis of cancer by targeting tumor suppressors or oncogenes [9, 10]. Based on miRNA multifunctionality, some specific miRNAs could be utilized to distinguish benign from malignant lesions [11, 12], and others may be powerful predictors of clinical outcomes and therapeutic molecular targets in various cancers [13, 14].
MicroRNA-133a (miR-133a), together with miR-133b, belongs to the miR-133 family [15]. It was first identified as a muscle-specific miRNA and played a crucial role in myoblast proliferation and differentiation during embryonic muscle development [16]. Recently, growing pieces of evidence have shown that miR-133a acts as a tumor suppressor in prostate cancer [17], bladder cancer [18], head and neck cancer [19], esophageal cancer [20], and colorectal cancer [21], suggesting that miR-133a may serve as a potential therapeutic gene for the treatment of these cancers. Although a recent study has demonstrated that miR-133a could regulate multidrug resistance mediated by targeting multidrug-resistance-associated protein 1 (ABCC1) in HCC cell line HepG2 [22], the clinical importance of miR-133a in HCC progression, the detailed function, and underlying molecular mechanism of miR-133a in HCC have not yet been thoroughly validated.
Therefore, in this study, the miR-133a expression and its clinical diagnosis significance in patients with HCC were investigated. In addition, the role and underlying molecular mechanism of miR-133a on the carcinogenesis of HCC were also assessed.
Materials and methods
Tissue samples
Forty pairs of HCC tissues and adjacent non-tumor tissues were obtained from patients who underwent routine curative surgery at the Department of Thoracic Surgery, The First Hospital, Jilin University (Changchun, China) between July 2010 and September 2014. None of these patients received chemotherapy and radiotherapy before the surgery. Relevant corresponding clinical data of HCC patients were listed in Table 1. Informed consent was obtained from each patient, and all of the experiments were approved by the ethics committee of Jilin University.
Cell lines and cell cultures
HCC cell lines (SMMC-7721, Hep3B, HepG2, and Huh-7) and normal hepatic cell line, HL-7702, were purchased from the Institute of Cell Biology of the Chinese Academy of Science (Shanghai, China) and were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, USA) supplemented with 10 % fetal bovine serum (FBS, HyClone, USA), 100 U/ml penicillin, 100 μg/ml streptomycin, and 1 % non-essential amino acids (Invitrogen, Carlsbad, CA, USA) and cultured at 37 °C in an atmosphere of 5 % CO2 with a relative humidity of 95 %.
RNA preparation and quantitative real-time polymerase chain reaction
Total RNA was isolated from tissue or cells using TRIzol® reagent (Invitrogen) according to the manufacturer’s instructions and the concentration of RNA was determined using a NanoDrop 2000 (Thermo Fisher Scientific, Waltham, MA, USA). The complementary DNA (cDNA) was synthesized using One Step Prime script miRNA cDNA Synthesis Kit (Qiagen, Valencia, CA, USA) following the manufacturer’s instructions. Then, miR-133a was quantified as described previously [23]. U6 snRNA was used as an endogenous control.
For insulin-like growth factor 1 receptor (IGF-1R), first-strand cDNA was synthesized using the PrimeScript RT Reagent Kit (TaKaRa, Dalian, Japan). Quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) was performed with SYBR Green premix Ex Taq (TaKaRa) on an ABI 7900 Fast system (Applied Biosystems, CA, USA). The primer sequences of IGF-1R and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were as follows: IGFR (sense, 5′ CCGCTGCCAGAAAATGTGCCCA-3′ and anti-sense: 5′-TGTCGTTGTCAGGCGCGCTG-3′); GAPDH (sense: 5′-GAAGGTGAAGGTCGGAGTC-3′ and anti-sense: 5′-GAAGATGGTGATGGGATTTC-3′). GAPDH was used as an internal control. All reactions were run in triplicate and the fold changes of genes were calculated by the 2−ΔΔCt method.
Cell transfection
miR-133a mimics and corresponding miRNA negative control (miR-NC), the siRNAs targeting human IGF-1R (GenBank accession no. NM_000875.3, si-IGF-1R), and corresponding negative control (si-NC) were designed and synthesized by GenePharma Co., Ltd. (Shanghai, China), and were transfected into HepG2 cells using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s instructions at final concentration of 100 nM. Transfection efficiency values were evaluated in every experiment by qRT-PCR 48 h posttransfection.
Cell proliferation and colony formation assay
For analysis of cell proliferation, transfected HepG2 cells (5 × 103 cells/well) were seeded into 24-well plates, then were incubated in 10 % Cell Counting Kit-8 (CCK-8, Dojindo, Kumamoto, Japan), and diluted in normal culture medium at 37 °C until visual color conversion occurred. The proliferation rate was determined at 24, 48, and 72 h after transfection. The absorbance in each well was measured with a microplate reader set at 450 nm.
For colony formation, transfected HepG2 cells were resuspended and seeded onto six-well plates at a density of 1000 cells/well and cultured for 2 weeks and then were stained with 0.5 % crystal violet for 30 min. Excess dye was rinsed off twice with PBS. The percentage colony formation was calculated by adjusting control (untreated cells) to 100 %.
Cell cycle analysis and cell apoptosis analysis
Cells cycle analysis was performed on HepG2 cells 48 h posttransfection. In brief, cells were harvested and fixed in ice-cold 70 % ethanol, incubated with RNase A at 37 °C for 30 min, and then stained with propidium iodide (PI, Sigma, USA) at 4 °C for 30 min in the dark, and then, cells were analyzed by fluorescence-activated cell sorting (FACS).
For analysis of apoptosis, transfected HepG2 cells were determined by PE annexin-V Apoptosis Detection Kit I (BD Pharmingen, CA, USA) according to the manufacturer’s protocol. The cell cycle distribution and apoptotic rate were analyzed using CellQuest software (BD Biosciences San Jose, CA, USA).
Wound-healing assay
Transfected HepG2 cells were grown to confluence. Subsequently, an artificial homogenous wound was scratched into the monolayer using a sterile plastic micropipette tip, and then, cells were cultured under standard conditions for 24 h. Following several washes, plates were photographed and the cell migration was evaluated by counting cells that had migrated from the wound edge at five random fields.
Cell invasion assays
Invasion assays were performed using Transwell invasion chambers coated with Matrigel (50 μl per filter; BD Biosciences) following the manufacturer’s instructions. In brief, transfected cells (1 × 105 cells per Transwell) were placed into the top of Matrigel-coated invasion chambers in serum-free DMEM. DMEM containing 20 % FBS was added to the lower chambers. After cells had been cultured at 37 °C for 48 h, cells remaining on the top of the filter were removed and those that migrated to the lower surface were removed with 70 % ethanol for 30 min and stained with 0.2 % crystal violet for 10 min. The number of migrated cells on the lower surface of the membrane was counted under a microscope in five selected random fields with a magnification of ×200. The invasion assays were carried out in triplicate.
miRNA target prediction
Prediction of miR-133a target genes was performed by three publicly available algorithms: TargetScan (http://www.targetscan.org/), PicTar (http://pictar.mdc-berlin.de/), and miRanda (http://www.microrna.org/).
Vector construction and luciferase reporter assay
The 3′-UTR of IGF-1R and the mutant 3′-UTR of IGF-1R were amplified and cloned into the downstream of pGL3/luciferase vector (Ambion, Austin, TX, USA) at the NheI and XhoI sites. For the luciferase reporter assay, the HepG2 cells were co-transfected with miR-133a mimics or miR-NC control and pGL3/luciferase-IGF-1R 3′-UTR or the mutant 3’UTR, together with the controls. At 48 h after transfection, luciferase activity was detected using the dual-luciferase assay system (Promega, Madison, USA). Renilla luciferase was used for normalization.
Western blotting
Cells were harvested and were lysed in protein extraction buffer (50 mM HEPES, 5 mM EDTA, 50 mM NaCl, 1 % Triton X-100, 50 mM NaF, 10 mM Na2P2O7, 1 mM Na3VO4, 5 μg/ml aprotinin, 5 μg/ml leupeptin, 1 mM PMSF, and protease inhibitor cocktail). Concentrations of total cellular protein were determined using a BCA assay kit (Pierce, Rockford, IL, USA) based on the manufacturer’s instructions. Equal amounts of proteins (20 μg) were separated by sodium dodecyl sulfate–polyacrylamide gels (SDS-PAGE) and transferred onto polyvinylidene difluoride membrane (Bio-Rad, Hercules, CA, USA). The blots were blocked with a 5 % skim milk solution and incubated with the following antibodies overnight at 4 °C: anti-IGF-1R (1:500, Santa Cruz, USA), anti-phospho-AKT (p-AKT) (1:1000, Cell Signaling), anti-AKT (1:2000, Cell Signaling), anti-phospho-extracellular signal-regulated kinase (ERK) (p-ERK) (1:1000, Cell Signaling), anti-ERK (1:1500, Cell Signaling), anti-matrix metalloproteinase (MMP)-2 (1:1000, Santa Cruz), anti-MMP-9 (1:2000, Santa Cruz), and anti-GAPDH (1:5000, Santa Cruz, USA). The membrane was further probed with horseradish peroxidase (HRP)-conjugated corresponding second antibody for 2 h at room temperature. Proteins were visualized with an ECL chemiluminescent kit (ECL Plus, Thermo Scientific). GAPDH was used as an internal control. Quantification of protein bands was performed using the ImageJ software (National Institutes of Health, Bethesda, MD, USA).
In vivo nude mice tumorigenesis assay
Six-week-old nude mice (BALB/C-nu (nu/nu)) were obtained from the Experiments Animal Center of Changchun Biological Institute (Changchun, China) and maintained in specific pathogen-free conditions. All animals were undertaken accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. This study was approved by the Animal Ethics Committee of Jilin University (Changchun, China).
HepG2-miR-NC and HepG2-miR-133a cells (2 × 106 cells) were suspended in 100 μl of serum-free DMEM medium and injected subcutaneously into each side of the posterior flank of the nude mouse. Tumor volume was measured every 5 days using digital vernier calipers and was calculated following the formula: 1/2 × length × width2. The xenograft tumors were removed from the mice 30 days after the implantation, and weight was measured. Parts of the tumor tissues were snap frozen in liquid nitrogen and stored at −80 °C for analysis of miR-133a expression by qRT-PCR and determination of IGF-IR, p-AKT, and p-ERK expression by Western blot.
Statistical analysis
All data are showed as mean ± standard deviation (SD), and all experiments were repeated at least three times independently. Comparisons between the groups were analyzed with a two-tailed Student’s t test. The relationship between miR-133a expression level and clinical and pathological variables was analyzed using a Pearson’s χ 2 test. The GraphPad Prism version 5.01 (San Diego, CA, USA) was used for statistical analyses. P < 0.05 was considered significant.
Results
miR-133a is downregulated in HCC cell lines and tissues
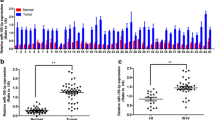
To investigate the level of miR-133a in hepatic carcinogenesis and progression, we detected the expression of miR-133a in HCC tissues and adjacent non-cancerous tissues from 40 patients with HCC using real-time RT quantitative PCR (qPCR) assay. Our results revealed that the mean expression level of miR-133a in HCC tissues (0.51 ± 0.11) was significantly lower than that in non-cancerous tissues (1.05 ± 0.24) (P < 0.05, Fig. 1a). RT-qPCR assays were further developed to quantify miR-133a levels in HCC cell lines, including SMMC-7721, Hep3B, HepG2, and Huh-7 cells, and the normal hepatic cell line HL-7702. A significantly lower expression of miR-133a was found in HCC cell lines compared to HL-7702 (all P < 0.05, Fig. 1b), and HepG2 cells exhibited the lowest expression of miR-133a and were selected for further studies.

miR-133a is downregulated in HCC cell lines and tissues. a Relative expression level of miR-133a in 40 pairs of HCC tissue samples (tumor) and matched adjacent non-tumor tissue samples (normal) was analyzed using real-time quantitative reverse transcriptase polymerase chain reaction (RT-qPCR). U6 was used as an endogenous control. b Relative expression of miR-133a in four HCC cell lines (SMMC-7721, Hep3B, HepG2, and Huh-7) and the normal hepatic cell line HL-7702. *P < 0.05, **P < 0.01
Based on the median (0.51) miR-133a level, all 40 HCC patients were divided into two subgroups according mean value: a low-miR-133a group (<0.51, 18 cases) and a high-miR-133a group (>0.51, 22 cases). The association between miR-133a expression and the clinicopathological parameters of the patients, including age; gender; metastasis (TNM) stage; differentiation; and lymph node metastasis, was investigated. As shown Table 1, miR-133a was significantly associated with TNM stage (P < 0.01), tumor differentiation (P < 0.01), and lymph node metastasis (P < 0.01) but not with other clinicopathological parameters including age and gender (both P > 0.05). These data indicated that miR-133a might be involved in HCC initiation and procession.
Ectopic expression of miR-133a inhibits cell proliferation and colony formation and induces cell cycle and apoptosis of HCC cells
To explore the relevance effect of miR-133a on HCC cell growth, miR-133a or miR-NC was transfected into HepG2 cells. Our results showed that transfection of miR-133a mimics in HepG2 cells significantly restored intracellular miR-133a expression (Fig. 2a). Then, cell proliferation rate was measured using CCK-8 assays. It was found that restored expression of miR-133a resulted in a significant decrease of proliferation in HepG2 cells (Fig. 2b). In addition, colony forming was performed to assess the role of miR-133a in HCC cell colony formation. As shown in Fig. 2c, restored expression of miR-133a resulted in a significant decrease of colony formation in HepG2 cells compared with the miR-NC group. As proliferation directly was linked to cell cycle distribution, we evaluated the effect of miR-133a on cell cycle progression. As expected, the percentage of S phase cells decreased, while the percentage of G1 phase cells increased in HepG2 cells in the miR-133a group compared to the miR-NC group (P < 0.05, Fig. 2d). Furthermore, to reveal the biological role of miR-133a on HCC cell apoptosis, cell apoptosis assays were performed by FACS. Our results showed that restored expression of miR-133a increased the proportion of Annexin-V-positive cells compared to the miR-NC group (P < 0.05, Fig. 2e), suggesting that miR-133a can efficiently induce apoptosis of HepG2 cells. Thus, these results indicated that miR-133a can efficiently inhibit cell proliferation and colony formation and induces cell cycle and apoptosis of HepG2 cells.

Ectopic expression of miR-133a inhibits cell proliferation and colony formation and induces cell cycle and apoptosis of HCC cells. a The relative expression levels of miR-133a in HepG2 cells were determined by RT-qPCR after transfection with miR-133a or miR-NC. b The proliferation was determined by CCK8 assay after transfection with miR-133a or miR-NC. c The capacity of colony formation was determined by colony formation assay after transfection with miR-133a or miR-NC. d The cell cycle was detected by PI staining after being transfected with miR-133a or miR-NC. The cell apoptosis was determined by PE annexin-V staining after being transfected with miR-133a or miR-NC. *P < 0.05, **P < 0.01 versus miR-NC
Ectopic expression of miR-133a inhibits the migration and invasion of HCC cells
To test the miR-133a effect on HCC cell migration and invasion, the migration and invasion of HepG2 cells were measured after transfection of miR-133a mimic or miR-NC by wound-healing assay and Transwell assay, respectively. Our results demonstrated that ectopic expression of miR-133a could remarkably suppress migration (Fig. 3a) and invasion (Fig. 3b) in HepG2 cells.

Ectopic expression of miR-133a inhibits migration and invasion of HCC cells. a The migration capacities were determined by wound scratch after being transfected with miR-133a or miR-NC (bar 50 μm). b The invasion capacities were determined by Transwell assay after being transfected with miR-133a or miR-NC(bar 50 μm). c The expression levels of MMP-2 and MMP-9 were determined by Western blot after being transfected with miR-133a or miR-NC. GAPDH was used as internal control. *P < 0.05, **P < 0.01 versus the miR-NC group
To investigate whether the inhibitory effect of miR-133a on migration and invasion of HCC was mediated by MMPs, we examined the expression of MMP-2 and MMP-9 of HepG2 cells by Western blot after being transfected with miR-133a or miR-NC. The results of Western blot showed that ectopic expression of miR-133a decreased the expression level of MMP-2 and MMP-9 in HepG2 cells (Fig. 4c). These findings suggest that miR-133a could impede migration and invasion, at least in part, by regulating MMP-2 and MMP-9 expression.

miR-133a targets the IGF-1R gene in HCC cells. a Bioinformatics analysis of the predicted interactions of miR-133a and its binding sites within the 3′-UTR of IGF-1R. b miR-133a mimic reduced luciferase activities controlled by wild type of R 3′-UTR IGF-1R but did not affect luciferase activity controlled by mutant 3′-UTR of IGF-1R. WT wild type, Mut mutant type. c Introduction of miR-133a reduced the levels of endogenous IGF-1R mRNA in HepG2 cells. d Introduction of miR-133a or si-IGF-1R reduced the levels of endogenous IGF-1R proteins in HepG2 cells. *P < 0.05, **P < 0.01 versus the miR-NC group
IGF-1R is a direct target of miR-133a
To understand the molecular mechanism of miR-133a action in HCC, we searched for miR-133a targets using the algorithms PicTar, miRBase, and TargetScan. IGF-1R was selected from several putative miR-133a target genes, since IGF-1R has been shown to be involved in tumorigenesis and metastasis in HCC [24, 25]. As shown in Fig. 4a, miR-133a owns one predicted binding site in the 3′-UTR of IGF-1R mRNA. Using the luciferase reporter system, we found that overexpression of miR-133a in HepG2 cells suppressed the activity of reporter gene (P < 0.05; Fig. 4b), whereas mutants plasmids showed no changes in the reporter gene activity in HepG2 cells, indicating that IGF-1R may be a target gene of miR-133a.
To determine whether overexpression of miR-133a downregulated endogenous IGF-1R expression, miR-133a mimics or miR-NC was transfected into HepG2 cells; then, IGF-1R mRNA and protein expression were determined by RT-qPCR and Western blot, respectively. Consistent with the dual-luciferase reporter assays, we observed that the mRNA and protein expression of IGF-1R were both decreased in HepG2 cells transfected with miR-133a mimic (Fig. 4c, d). Furthermore, knockdown of IGF-1R significantly inhibited the IGF-1R level in HepG2 (Fig. 4d), which phenocopied the effect of miR-133a. These results indicate that miR-133a directly binds to the 3′-UTR of IGF-1R and inhibits it expression.
Ectopic expression of miR-133a suppresses AKT and ERK signaling pathways
It has been shown that IGF-1R played key roles in survival, proliferation, differentiation, and motility of tumors because IGF-1R activates multiple downstream signaling cascades, including phosphoinositide 3-kinase (PI3K)/AKT and mitogen-activated protein kinase (MAPK)/ERK signaling pathways [26, 27]. Here, we investigated whether miR-133a affects activation of AKT and EKR pathways. The AKT, p-AKT, ERK, and p-ERK expression was determined in HepG2 cells transfected with miR-133a or miR-NC by Western blot. The results of Western blot analysis showed that the levels of p-AKT and p-ERK were decreased in the miR-133a group compared with the miR-NC group, while the levels of total AKT and ERK were not significantly changed in both groups (Fig. 5). These results suggested that upregulation of miR-133a could suppress HCC growth by regulating the AKT and ERK pathways.

Ectopic expression of miR-133a suppresses AKT and ERK signaling pathways. p-AKT, AKT, p-ERK1/2, and ERK1/2 protein expression was detected in HepG2 cells by Western blot analysis after being transfected with miR-133a or miR-NC. GAPDH was used as internal control
miR-133a suppresses tumor growth in a mouse xenograft model
Next, an in vivo model was applied to evaluate the effect of miR-133a restoration on tumorigenicity. HepG2-miR-NC and HepG2-miR-133a cells were subcutaneously inoculated in nude mice (n = 10 for each group). It was found that the miR-133a-expressing HepG2 tumors were significantly smaller than that of the HepG2-miR-NC group (Fig. 6a). Additionally, the average weight and volume of the miR-133a-expressing tumors were also markedly reduced compared with the control group and miR-NC group (both P < 0.01, Fig. 6b, c). Furthermore, we determined miR-133a expression of tumor tissue by qRT-PCR. Our result demonstrated that the miR-133a expression significantly increased in the HepG2-miR-133a group compared to the HepG2-miR-NC group (P < 0.01, Fig. 6d). We also investigated the IGF-1R, p-AKT, and p-ERK expression in tumor tissue. The results of Western blot showed that the IGF-1R, p-AKT, and p-ERK expression significantly increased in the HepG2-miR-133a group compared to the HepG2-miR-NC group. These data indicated that miR-133a could suppress tumor growth of HCC in vivo, at least in part, by targeting IGR-IR via regulating AKT and ERK signaling pathways.

miR-133a suppressed tumor growth in a xenograft model. a Representative graphs of tumor masses harvested from nude mice 30 days after inoculation are shown. b Growth curves for tumor volumes in xenografts of nude mice were established based on the tumor volume measured every 5 days until 30 days. c Tumor weights were measured when mice were killed. d miR-133a expression level of tumor tissue was determined by RT-qPCR. e IGF-1R, p-AKT, and p-ERK protein expression was determined by Western blot. GAPDH was used as internal control. *P < 0.05, **P < 0.01 versus the miR-NC group
Discussion
HCC is currently the sixth most commonly malignant and is the third leading cause of cancer death worldwide, with very high morbidity and mortality [1–3]. It is an urgent need to understand the precise molecular mechanisms underlying HCC tumorigenesis to diagnosis and therapeutic strategy for HCC and to further improve overall survival [28]. In recent years, it has become one of the most promising fields to identification and investigation of the deregulated miRNAs in HCC development, which contribute to understand molecular mechanisms of HCC carcinogenesis and progression [29]. Aberrant miRNA expression is closely associated with various types of cancer, including HCC, and numerous miRNAs play crucial roles in cancer cell proliferation, apoptosis, migration, invasion, and neoplastic transformation [30], suggesting that miRNA acts as a new direction in HCC diagnosis and treatment. Several deregulated miRNAs, such as upregulated miR-21, miR-222 and miR-221 and downregulated miR-122, miR-145, and miR-34, have been reported in HCC development [28, 31]. However, it is still needed to find new important deregulated miRNAs and their detailed roles in HCC carcinogenesis and progression. Here, we first showed the miR-133a expression was decreased in most HCC tumor tissues and its expression level correlated with tumor differentiation, TNM stage, and lymph node metastasis. Our result also confirmed that miR-133a functions as a tumor suppressor and suppresses tumor growth of HCC in vitro and in vivo. These data provided new insights into HCC research and therapeutic strategies for HCC.
Recently, growing pieces of evidence have suggested that miR-133a acts as a tumor suppressor in several cancers [17–21]. For example, Qiu et al. [32] found that miR-133a was downregulated in gastric cancer tissues and cell lines and that overexpression of miR-133a inhibited proliferation, migration, invasion, and cell cycle progression in vitro. Mataki et al. [33] reported that miR-133a was significantly downregulated in clinical lung cancer specimens and functions as tumor suppressors in lung cancer through regulation of oncogenic CORO1C. Ji et al. [34] found that restoration of miR-133a expression in osteosarcoma cells significantly reduces cell proliferation, promotes cell apoptosis, and suppresses tumorigenicity via inhibiting Bcl-xl and Mcl-1. Chiyomaru et al. [18] reported that miR-133a functions as a tumor suppressor and directly regulates fscn1 expression in bladder cancer. However, the molecular mechanism of miR-133a-involved HCC progression has not been clarified until now. Here, we found that miR-133a expression level is significantly downregulated in HCC tissue and cell lines, and its expression level correlated with key pathological characteristics including tumor differentiation, TNM stage, and lymph node metastasis. Further, our in vitro studies revealed that overexpression of miR-133a inhibited cell proliferation, colony formation, migration, and invasion and induced cell cycle at G1 stage and apoptosis in HepG2 cells. In addition, our in vivo studies found that overexpression of miR-133a suppressed tumor growth in a nude mice model. These results suggested that miR-133a functions as a tumor suppressor in HCC and plays a crucial role in HCC progression.
IGF-1R, a member of the insulin receptor family of receptor tyrosine kinases, played the pivotal role in growth and development of the cells [35]. The IGF-1R protein itself was frequently found to be overexpressed in cancer, including HCC [36]. Recently, several lines of evidence have supported the role of IGF-1R in promoting carcinogenesis and act as an oncogene [35, 37]. IGF-1R has been reported to be a target of miR-133a in lung cancer [38] and gastric cancer [39]; however, the interaction between miR-133a and IGF-1R has not been experimentally validated in HCC. Here, using luciferase reporter assays, we verified that the IGF-1R gene is a direct target of miR-133a. Overexpression of miR-133a directly suppresses the expression of IGF-1R at the mRNA level and protein level. In addition, it has been reported that abnormal expression of IGF-1R could affect multiple downstream signal pathways, PI3K/AKT and MAPK/ERK signaling pathways, which were involved in tumorigenesis and development [27, 40]. Therefore, we investigated whether miR-133a affects activation of AKT and EKR pathways. We found that overexpression of miR-133a could inhibit p-AKT and p-ERK protein expression in HCC cells (Fig. 5) and tumor tissue (Fig. 6e). These results suggested that upregulation of miR-133a could suppress HCC growth through regulating IGF-1R and its downstream AKT and ERK signaling pathways.
Taken together, the results presented here first demonstrate that miR-133a expression level was downregulated in HCC tissue and cell lines, and its expression level was significantly associated with tumor differentiation, TNM stage, and lymph node metastasis, and that overexpression of miR-133a inhibited cell proliferation, colony formation, migration, and invasion and induced cell apoptosis and cell cycle at G0/G1stage, as well as suppressed tumor growth in vivo. Moreover, we identified IGF-1R as a crucial target gene of miR-133a and found that miR-133a could regulate AKT and ERK signaling pathways. These findings indicated that miR-133a functions as a tumor suppressor in HCC and blocks tumor growth at least partially through regulating IGF-1R and its downstream AKT and ERK signaling pathways, suggesting that miR-133a could be a potential target for the treatment of HCC in the future.
Change history
21 December 2021
This article has been retracted. Please see the retraction notice for more detail: https://doi.org/10.3233/TUB-219010"
References
Waly Raphael S, Yangde Z, Yuxiang C. Hepatocellular carcinoma: focus on different aspects of management. ISRN Oncol. 2012;2012:421673.
Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379:1245–55.
Bosch FX, Ribes J, Diaz M, Cleries R. Primary liver cancer: worldwide incidence and trends. Gastroenterology. 2004;127:S5–16.
Zhou YM, Zhang XF, Yu F, Liu XB, Wu LP, Li B, et al. Efficacy of surgical resection for pulmonary metastases from hepatocellular carcinoma. Med Sci Monit Int Med J Exp Clin Res. 2014;20:1544–9.
Raza A, Sood GK. Hepatocellular carcinoma review: current treatment, and evidence-based medicine. World J Gastroenterol WJG. 2014;20:4115–27.
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97.
Brennecke J, Hipfner DR, Stark A, Russell RB, Cohen SM. Bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell. 2003;113:25–36.
Sun K, Lai EC. Adult-specific functions of animal microRNAs. Nat Rev Genet. 2013;14:535–48.
Shenouda SK, Alahari SK. MicroRNA function in cancer: oncogene or a tumor suppressor? Cancer Metastasis Rev. 2009;28:369–78.
Zhang X, Han C, He J. Research progress of oncogene and tumor suppressor gene in bladder cancer. Panminerva Med. 2015.
Tian T, Wang J, Zhou X. A review: microRNA detection methods. Org Biomol Chem. 2015;13:2226–38.
Saumet A, Mathelier A, Lecellier CH. The potential of microRNAs in personalized medicine against cancers. BioMed Res Int. 2014;2014:642916.
Cheng G. Circulating miRNAs: roles in cancer diagnosis, prognosis and therapy. Adv Drug Deliv Rev. 2015;81:75–93.
Hayes J, Peruzzi PP, Lawler S. MicroRNAs in cancer: biomarkers, functions and therapy. Trends Mol Med. 2014;20:460–9.
Yu H, Lu Y, Li Z, Wang Q. MicroRNA-133: expression, function and therapeutic potential in muscle diseases and cancer. Curr Drug Targets. 2014;15:817–28.
Feng Y, Niu LL, Wei W, Zhang WY, Li XY, Cao JH, et al. A feedback circuit between miR-133 and the erk1/2 pathway involving an exquisite mechanism for regulating myoblast proliferation and differentiation. Cell Death Dis. 2013;4:e934.
Kojima S, Chiyomaru T, Kawakami K, Yoshino H, Enokida H, Nohata N, et al. Tumour suppressors miR-1 and miR-133a target the oncogenic function of purine nucleoside phosphorylase (PNP) in prostate cancer. Br J Cancer. 2012;106:405–13.
Chiyomaru T, Enokida H, Tatarano S, Kawahara K, Uchida Y, Nishiyama K, et al. miR-145 and miR-133a function as tumour suppressors and directly regulate fscn1 expression in bladder cancer. Br J Cancer. 2010;102:883–91.
Nohata N, Hanazawa T, Kikkawa N, Mutallip M, Fujimura L, Yoshino H, et al. Caveolin-1 mediates tumor cell migration and invasion and its regulation by miR-133a in head and neck squamous cell carcinoma. Int J Oncol. 2011;38:209–17.
Kano M, Seki N, Kikkawa N, Fujimura L, Hoshino I, Akutsu Y, et al. miR-145, miR-133a and miR-133b: tumor-suppressive miRNAs target fscn1 in esophageal squamous cell carcinoma. Int J Cancer. 2010;127:2804–14.
Dong Y, Zhao J, Wu CW, Zhang L, Liu X, Kang W, et al. Tumor suppressor functions of miR-133a in colorectal cancer. Mol Cancer Res MCR. 2013;11:1051–60.
Ma J, Wang T, Guo R, Yang X, Yin J, Yu J, et al. Involvement of miR-133a and miR-326 in ADM resistance of HepG2 through modulating expression of abcc1. J Drug Target. 2015;25:1–6.
Wan TM, Lam CS, Ng L, Chow AK, Wong SK, Li HS, et al. The clinicopathological significance of miR-133a in colorectal cancer. Dis Markers. 2014;2014:919283.
Chun YS, Huang M, Rink L, Von Mehren M. Expression levels of insulin-like growth factors and receptors in hepatocellular carcinoma: a retrospective study. World J Surg Oncol. 2014;12:231.
Wu J, Zhu AX. Targeting insulin-like growth factor axis in hepatocellular carcinoma. J Hematol Oncol. 2011;4:30.
Osaki LH, Gama P. MAPKs and signal transduction in the control of gastrointestinal epithelial cell proliferation and differentiation. Int J Mol Sci. 2013;14:10143–61.
Cao Z, Liu LZ, Dixon DA, Zheng JZ, Chandran B, Jiang BH. Insulin-like growth factor-I induces cyclooxygenase-2 expression via PI3K, MAPK and PKC signaling pathways in human ovarian cancer cells. Cell Signal. 2007;19:1542–53.
Oishi N, Yamashita T, Kaneko S. Molecular biology of liver cancer stem cells. Liver Cancer. 2014;3:71–84.
Khare S, Zhang Q, Ibdah JA. Epigenetics of hepatocellular carcinoma: role of microRNA. World J Gastroenterol WJG. 2013;19:5439–45.
Gramantieri L, Fornari F, Callegari E, Sabbioni S, Lanza G, Croce CM, et al. MicroRNA involvement in hepatocellular carcinoma. J Cell Mol Med. 2008;12:2189–204.
Negrini M, Gramantieri L, Sabbioni S, Croce CM. MicroRNA involvement in hepatocellular carcinoma. Anti Cancer Agents Med Chem. 2011;11:500–21.
Qiu T, Zhou X, Wang J, Du Y, Xu J, Huang Z, et al. miR-145, miR-133a and miR-133b inhibit proliferation, migration, invasion and cell cycle progression via targeting transcription factor Sp1 in gastric cancer. FEBS Lett. 2014;588:1168–77.
Mataki H, Enokida H, Chiyomaru T, Mizuno K, Matsushita R, Goto Y, et al. Downregulation of the microRNA-1/133a cluster enhances cancer cell migration and invasion in lung-squamous cell carcinoma via regulation of coronin1c. J Hum Genet. 2015;60:53–61.
Ji F, Zhang H, Wang Y, Li M, Xu W, Kang Y, et al. MicroRNA-133a, downregulated in osteosarcoma, suppresses proliferation and promotes apoptosis by targeting Bcl-xl and Mcl-1. Bone. 2013;56:220–6.
King H, Aleksic T, Haluska P, Macaulay VM. Can we unlock the potential of IGF-1R inhibition in cancer therapy? Cancer Treat Rev. 2014;40:1096–105.
Scharf JG, Braulke T. The role of the igf axis in hepatocarcinogenesis. Horm Metab Res Horm Stoffwechselforschung Horm Metab. 2003;35:685–93.
Singh P, Alex JM, Bast F. Insulin receptor (IR) and insulin-like growth factor receptor 1 (IGF-1R) signaling systems: novel treatment strategies for cancer. Med Oncol. 2014;31:805.
Wang LK, Hsiao TH, Hong TM, Chen HY, Kao SH, Wang WL, et al. MicroRNA-133a suppresses multiple oncogenic membrane receptors and cell invasion in non-small cell lung carcinoma. PLoS One. 2014;9:e96765.
Gong Y, Ren J, Liu K, Tang LM. Tumor suppressor role of miR-133a in gastric cancer by repressing IGF1R. World J Gastroenterol WJG. 2015;21:2949–58.
Guo T, Feng Y, Liu Q, Yang X, Jiang T, Chen Y, et al. MicroRNA-320a suppresses in GBM patients and modulates glioma cell functions by targeting IGF-1R. Tumour Biol J Int Soc Oncodevelopmental Biol Med. 2014;35:11269–75.
Conflicts of interest
None
Author information
Authors and Affiliations
Corresponding author
Additional information
This article has been retracted. Please see the retraction notice for more detail: https://doi.org/10.3233/TUB-219010"
About this article

Cite this article
Zhang, W., Liu, K., Liu, S. et al. RETRACTED ARTICLE: MicroRNA-133a functions as a tumor suppressor by targeting IGF-1R in hepatocellular carcinoma. Tumor Biol. 36, 9779–9788 (2015). https://doi.org/10.1007/s13277-015-3749-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-015-3749-8