
Abstract
In the last several decades, the number of people dying from cancer-related deaths has not reduced significantly despite phenomenal advances in the technologies related to diagnosis and therapeutic modalities. The principal cause behind limitations in the curability of this disease is the reducing sensitivity of the cancer cells towards conventional anticancer therapeutic modalities, particularly in advance stages of the disease. Amongst several reasons, certain secretory factors released by the tumour cells into the microenvironment have been found to confer resistance towards chemo- and radiotherapy, besides promoting growth. Interleukin-6 (IL-6), one of the major cytokines in the tumour microenvironment, is an important factor which is found at high concentrations and known to be deregulated in cancer. Its overexpression has been reported in almost all types of tumours. The strong association between inflammation and cancer is reflected by the high IL-6 levels in the tumour microenvironment, where it promotes tumorigenesis by regulating all hallmarks of cancer and multiple signalling pathways, including apoptosis, survival, proliferation, angiogenesis, invasiveness and metastasis, and, most importantly, the metabolism. Moreover, IL-6 protects the cancer cells from therapy-induced DNA damage, oxidative stress and apoptosis by facilitating the repair and induction of countersignalling (antioxidant and anti-apoptotic/pro-survival) pathways. Therefore, blocking IL-6 or inhibiting its associated signalling independently or in combination with conventional anticancer therapies could be a potential therapeutic strategy for the treatment of cancers with IL-6-dominated signalling.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Inflammation has a very strong link with various types of cancer. Malignant cells are highly proliferative in nature, which is facilitated by the inflammatory molecules that are continuously being secreted by other cells and/or tumour cells themselves in a microenvironment [1, 2]. Interleukin-6 (IL-6) is one such inflammatory molecule, which is produced and secreted by various types of cells including the tumour cells. It is involved in the proliferation and differentiation of malignant cells and found to be high in serum and tumour tissues of a majority of cancers, viz. colorectal cancer [3], breast cancer [4], prostate cancer [5], ovarian carcinoma [6], pancreatic cancer [7], lung cancer [8], renal cell carcinoma [9], cervical cancer [10] and multiple myeloma [11]. Elevated levels of IL-6 are associated with aggressive tumour growth and response to therapies in many types of cancer [12, 13]. Patients with high levels of circulating IL-6 are generally associated with poor prognosis and shorter survival, whilst a lower level of IL-6 is associated with better response to therapy [14, 15].
Anticancer drugs and ionizing radiation used during cancer therapy induce inflammatory signalling, mainly in the form of the nuclear factor-kappa B (NF-kB) pathway [16, 17]. NF-kB regulates the expression of different pro-inflammatory cytokines, chemokines and anti-apoptotic genes and acts as a key molecular link between inflammation and initiation as well as progression of oncogenesis [18]. Chemotherapeutic drugs and radiation also induce IL-6 expression in tumour and stromal cells [12, 13] through the activation of NF-kB signalling, leading to therapeutic resistance [19, 20]. These evidences suggest that blocking IL-6 or inhibiting the IL-6 downstream signalling pathways may provide therapeutic gain in those cancers which are associated with a higher level of IL-6. This review provides an insight into the current understanding of the role of IL-6 in the regulation of various hallmarks and associated signalling in cancer as well as its contribution to therapeutic resistance. It also provides an insight into how the anti-IL-6 antibody or the inhibitors of pathways downstream to IL-6 signalling can improve the effectiveness of cancer radio- and/or chemotherapy.
Interleukin-6
IL-6 is a glycosylated polypeptide chain having a molecular weight of nearly 25 kDa, depending on the glycosylation and the species. It has a characteristic structure made up of four long α-helices arranged in an up–up–down–down topology [21]. It was first discovered as a B cell differentiation factor (BSF-2) which induces the maturation of B cells into antibody-producing cells [22]. Besides its role in immune regulation, it plays an important role in the maintenance of hepatocytes, haematopoietic progenitor cells, the skeleton, the placenta, the cardiovascular system and the endocrine as well as nervous systems. In the murine haematopoietic system, IL-6 induces the expansion of progenitor cells by stimulating cells from the resting stage to enter the G1 phase [23]. IL-6 also supports various physiological functions by acting as a hepatocyte stimulatory factor and by inducing the acute-phase protein synthesis. It is also known to stimulate osteoclast formation, induce bone resorption and is responsible for neural differentiation [24]. IL-6 supports the survival of cholinergic neurons, induces adrenocorticotropic hormone synthesis, and, in placenta, causes the secretion of chorionic gonadotropin from trophoblasts [23]. IL-6 also plays a very important role in metabolism. For example, in the absence of IL-6, mice develop glucose intolerance and insulin resistance, whilst IL-6−/− mice exhibit signs of liver inflammation [25]. The secretion and availability of IL-6 is ubiquitous, and it can bind to various types of cells in different tissues. However, its binding on different cell types may differ, resulting in two different types of IL-6-dependent cell signalling (Fig. 1).

Classical and trans-signalling of IL-6: In classical signalling, which occurs mainly in leukocytes and liver cells, IL-6 binds to the membrane-bound receptor mbIL-6Rα, which then forms a complex with the ubiquitously present cell receptor gp130 (IL-6Rβ). Trans-signalling can occur in any cell expressing gp130. In trans-signalling, IL-6 forms a complex with sIL-6R, which is a small part of mbIL-6Rα produced by either metalloproteinase or by alternative splicing. Furthermore, the IL-6–sIL-6R complex binds with gp130 on cells which do not express mbIL-6R. The inflammatory reactions induce the production of sIL-6R, which elicits response to IL-6 in cells that do not express IL-6 receptor (mbIL-6Rα) and/or remain inert to IL-6 signalling in normal physiological conditions. Classical signalling activates the anti-inflammatory pathways and promotes the regeneration of tissues, whereas trans-signalling activates pro-inflammatory pathways and is known to play a significant role in many diseases such as sepsis and cancer
IL-6 binds to the IL-6 receptor (IL-6R) on the plasma membrane, and the resultant IL-6/IL-6R complex associates with gp130 and causes gp130 homodimerization to form an activated IL-6 receptor complex, which is a hexameric structure consisting of two molecules each of IL-6, IL-6R and gp130 [26, 27]. The binding of IL-6 to IL-6R occurs at three distinct receptor-binding sites of IL-6R and gp130. However, the Ig-like domain of the human IL-6R is not involved in the direct binding of IL-6 [28]. Upon binding to the receptor and gp130, IL-6 induces various functions by activating cell signalling events [24]. IL-6 triggers signal transduction via two forms of IL-6R: one a transmembrane 80-kDa receptor with a short cytoplasmic domain (mbIL-6R, also known as IL-6Rα, gp80 or CD126) and the other a small, extracellular, secretory soluble receptor (sIL-6R) [29]. Classical IL-6 signalling, which is the predominant form of IL-6 signalling, requires membrane-bound IL-6R (mbIL-6R) and is restricted to hepatocytes, some epithelial cells and certain leukocytes (Fig. 1) [26]. IL-6R contains a very short cytosolic domain that lacks the major potential motifs for transduction of intracellular cell signalling. However, gp130 (also known as IL-6Rβ or CD130) in the same hexameric complex is rich in all these potential motifs required for intracellular signalling, such as SHP-2 domain and YXXQ motif for JAK/STAT signalling. Upon binding with IL-6/IL-6R, the dimerization of gp130 leads to the activation of associated cytoplasmic tyrosine kinases, resulting in the phosphorylation of various transcription factors [24]. gp130 is expressed in almost all organs, including the brain, heart, lung, liver, kidney, spleen and placenta, where it plays an indispensable role in their development, cell survival, growth and tissue homeostasis [30]. gp130 is a common signal transducing receptor and is also used by other members of the IL-6 family cytokines, such as IL-11, IL-12, IL-27, leukaemia inhibitory factor, oncostatin M, etc. [31]. Although the expression of transmembrane IL-6R is limited to the hepatocytes and subsets of leukocytes, gp130 is expressed ubiquitously. Therefore, the IL-6/sIL-6R complex can transduce the IL-6 signal in various cells, which do not express transmembrane IL-6R but express gp130, through a trans-signalling mechanism. sIL-6R is generated by alternative splicing of IL-6R mRNA or by limited proteolysis of mbIL-6R by Zn-dependent metalloproteinase (ADAM10 and ADAM17, a disintegrin and metalloproteinases 10 and 17; Fig. 1) [29–32]. sIL-6R is devoid of the cytoplasmic and transmembrane domains and binds to IL-6 with comparable affinity as the membrane-bound form, thereby mediating gp130 activation in an autocrine or paracrine manner [21]. Consequently, by binding to sIL-6R, IL-6 increases its reach to a wide variety of cells. There is enough evidence to suggest that neural cell, neural stem cells, haematopoietic stem cells, liver progenitor cells and embryonic stem cells depend on sIL-6R in their response to IL-6 [33–36]. The level of sIL-6R present in the human sera increases during inflammation [32, 37]. Knockdown of the IL-6R gene in hepatocytes reduces the levels of sIL-6R by 32 % in the serum, whilst ablation of the IL-6R gene in haematopoietic cells reduces the sIL-6R serum levels by 60 % [38]. These observations suggest that hepatocytes and haematopoietic cells are the main sources of sIL-6R found in the circulation [38]. Like sIL-6R, a soluble form of the signal transducer protein gp130 (sgp130) is also present in the circulation at relatively high concentrations during inflammation and cancer [37, 39]. sgp130 is mainly produced by alternative splicing rather than limited proteolysis, as in the case of sIL-6R generation. Since sgp130 binds to the IL-6/sIL-6R complex in the circulation, it acts as a specific inhibitor of IL-6-mediated trans-signalling [40]. Classic signalling via the mbIL-6R is not affected by sgp130. Its inhibitory action depends on the IL-6/sIL-6R ratio, with trans-signalling inhibition at low concentrations [37].
IL6 expression and secretion
The common characteristic of many of the stimuli that activate IL-6 is that they are associated with tissue damage or stress (e.g. ionizing radiation, UV, reactive oxygen species, viruses, microbial products and other pro-inflammatory cytokines) [12, 41, 42]. IL-6 production is predominantly regulated by changes in the gene expression of various transcription factors such as NF-kB, CCAAT/enhancer-binding protein a and activator protein 1, the major transcriptional regulator, although posttranscriptional modifications have also been identified [41, 43]. Though the activation of these transcription factors leads to the overexpression of this cytokine during inflammation, its expression is also known to be regulated epigenetically in breast cancer, hepatocellular carcinoma, colon cancer, prostate cancer and lung cancer through miRNAs (Lin28 and Let-7) [44].
The normal blood circulating level of IL-6 is nearly 1 pg/ml [45, 46], but an increase in its level is found under several conditions such as acute hyperglycemia [47], high-fat meal [48], normal menstrual cycle [49], physical activity [50] and during/after surgery [51]. Inconsistent levels of IL-6 have also been observed during pregnancy, with median values around 128 pg/ml registered at delivery, which drop by more than two fold (∼58 pg/ml) immediately afterward [52]. Furthermore, serum IL-6 levels have been found to increase drastically during sepsis [53].
Many physiological factors such as diet, exercise and stress are known to regulate the secretion of IL-6 [47–53]. Exercise is an important stimulus for increased gene expression and production of IL-6 in skeletal muscle, and the majority of circulating IL-6 during exercise originates from contracting muscle, resulting in a 100-fold increase over the normal physiological level [50, 54]. IL-6 produced in the working muscle during physical activity acts as an energy sensor that activates AMP-activated kinase and enhances glucose uptake, metabolism, lipolysis and fat oxidation [50]. IL-6 is also known to sensitize myotubes to insulin and enhances glucose uptake in muscles for high glycogen synthesis. Moreover, the reduced level of muscle glycogen also augments IL-6 production and secretion from muscle cells [55]. In addition to exercise, the expression of IL-6 increases in skeletal muscles under other conditions as well, such as denervation of muscles and muscular dystrophy, also resulting in the upregulated expression of muscle IL-6 [54]. The adipose tissue produces nearly 30 % of circulating systemic IL-6, where it is closely associated with obesity, impaired glucose tolerance and insulin resistance [56]. Plasma IL-6 concentrations are a predictor of the development of type 2 diabetes, and peripheral administration of IL-6 results in insulin resistance in rodents and humans by causing hyperlipidaemia and hyperglycaemia [56]. Besides muscle cells, e.g. macrophages, mast cells, dendritic cells, B cells and CD4 effector T helper cells in the immune system are amongst the major sources of IL-6 production [22, 57–59]. In addition, IL-6 is also secreted by a variety of non-haematopoietic cells such as fibroblasts, endothelial cells, epithelial cells, astrocytes and malignant cells [2, 33, 42, 60]. Enhanced levels of IL-6 have been found in many cancers, with an inverse relationship between IL-6 levels and response to chemotherapy and hormone therapy [61]. Furthermore, IL-6 expression has also been found higher in recurrent tumours as compared to primary tumours, as well as in recurrent metastatic lesion as compared to primary metastasis [15].
The primary sources of IL-6 in the tumour microenvironment are tumour cells as well as tumour-associated macrophages (TAMs), CD4+ T cells, myeloid-derived suppressor cells (MDSCs) and fibroblasts [59–62]. In the tumour microenvironment, IL-6 supports tumorigenesis by directly affecting cancer cells through the modulation of both the intrinsic and extrinsic activities of tumour cells as well as by influencing stromal cells that indirectly support tumorigenesis [63]. For example, in skin and prostate cancer, the autocrine and paracrine secretion of IL-6 induces a complex of cytokine, growth factors and protease network consisting of granulocyte macrophage colony-stimulating factor (GM-CSF), IL-8, MCP-1, vascular endothelial growth factor (VEGF) and MMP-1 and stimulates malignant progression [64]. Basically, tumour cells produce IL-6 for promoting their survival and progression and do not depend on paracrine release of IL-6 by stromal cells [63]. However, both autocrine and paracrine mechanisms of IL-6 are known to influence tumour progression and metastasis through IL-6 trans-signalling [2, 64].
Pleiotropic role of IL-6
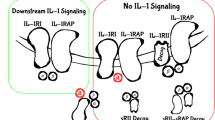
Cancer is an inflammatory disease, and the key feature of cancer-related inflammation is the expression of cytokines. Different cytokines play different roles in the onset and resolution of inflammation. However, a ubiquitous and functionally diverse cytokine, IL-6 is a pleiotropic cytokine with pro- and anti-inflammatory properties (Fig. 1). It is an important cytokine regulating the acute-phase response of inflammation [21]. During inflammatory response, tumour necrosis factor alpha (TNFα) induces the expression of IL-6 together with other inflammatory alarm cytokines, such as IL-1β, which are involved in the elicitation of acute-phase inflammatory reactions/responses (Fig. 1) [65]. Furthermore, IL-6 controls the level of acute inflammatory responses by downregulating the expression of pro-inflammatory cytokines and upregulating anti-inflammatory molecules, including IL-1 receptor antagonist protein, TNF-soluble receptor and extrahepatic protease inhibitors (Fig. 1) [66]. IL-6 has also been found to counter inflammation by inhibiting TNFα release in experimental endotoxemia [67]. This pleiotropic nature of IL-6 maintains the host–tumour homeostasis.
During switch between pro- and anti-inflammatory roles, TNFα and IL-1β negatively regulate IL-6 signalling at different levels by enhancing the IL-6-induced expression of the suppressor of cytokine signalling (SOCS3, feedback inhibitor) and/or targeting IL-6-induced gene expression via its action on target gene promoters [59, 68–70]. IL-1β also counteracts IL-6-mediated STAT-3 activation independent of SOCS3 expression [71]. IL-1β is the major regulator of the pro- and anti-inflammatory nature of IL-6; on the one hand, it reduces the pro-inflammatory activity of IL-6 that results in the inhibition of overshooting immunological reactivity, such as in inflammatory bowel disease or autoimmune arthritis, whilst on the other hand, it delays the anti-inflammatory effects of IL-6 to reinforce the pro-inflammatory processes in the initial phase of inflammation [70]. Similarly, the high concentration of IL-1β in the tumour microenvironment must maintain the chronic inflammatory environment by suppressing the anti-inflammatory processes of IL-6.
Emerging evidences suggest that IL-6 plays key roles in the acute as well as the transition (resolution) phase of inflammation [68]. Furthermore, IL-6 trans-signalling recruits T cells at the site of inflammation by triggering the expression of T cell-attracting chemokines (CCL4, CCL5, CCL17 and CXCL10) [72]. Moreover, IL-6 also rescues T cells from entering apoptosis by STAT-3-dependent upregulation of anti-apoptotic regulators (Bcl-2 and Bcl-xL) and modulation of Fas surface expression [73, 74]. IL-6 also regulates the differentiation of recruited T cells towards TH2 by inducing the expression of IL-4. Thus, IL-6 regulates some of the key steps in controlling inflammation and sets the anti-inflammatory environment by promoting TH2 response [57]. Collectively, these evidences suggest that endogenous IL-6 plays a vital anti-inflammatory role in both local and systemic acute inflammatory responses by controlling the level of pro-inflammatory cytokines, mainly. The trans-signalling of IL-6 regulates mainly the pro-inflammatory response; however, IL-6 classical signalling imparts its anti-inflammatory or regenerative activity (Fig. 1) such as regeneration [75], inhibition of epithelial apoptosis and the activation of the acute-phase response [76]. Understanding of the pleiotropic role of IL-6 in cancer is not very clear; however, the information available from other inflammatory diseases suggest that IL-6 might play both pro- and anti-inflammatory roles in the tumour microenvironment, which is crucial for host–tumour interaction.
Role of IL-6 and associated signalling in cancer
The notion that inflammation drives cancer is now well established. One of the major drivers of this link is NF-kB, which plays a central role in the secretion and activation of numerous pro-inflammatory cytokines from multiple cell types in the tumour microenvironment, including macrophages, T cells and epithelial cells [43, 57, 58]. Several pro-inflammatory cytokines released by innate and adaptive immune cells regulate cancer cell growth and thereby contribute to tumour promotion and progression. Amongst these, IL-6 is important in the development of human cancer and activates oncogenic pathways, and it is known to be deregulated in cancer [77]. Overexpression of IL-6 in many types of tumours, such as colorectal cancer [3], prostate cancer [5], breast cancer [4], ovarian carcinoma [6], pancreatic cancer [7], lung cancer [8], renal cell carcinoma [9], cervical cancer [10], multiple myeloma [11] and lymphomas [78], suggests a strong link between this cytokine and cancer. The high susceptibility and incidence of liver cancer in males is also found to be associated with high levels of IL-6 [79]. However, in females, oestrogen steroid hormones inhibit IL-6 production and so protect female mice from cancer [79, 80]. Activation of the IL-6/STAT-3 signalling axis is an important event in cancer which promotes tumorigenesis by regulating multiple survival signalling pathways in cancer cells [24]. IL-6 regulates nearly all hallmarks of cancer, such as inhibition of apoptosis [81, 82], promotion of survival [8, 75], proliferation [35, 83], angiogenesis [10], invasiveness and metastasis [62, 84], and is also known to regulate cancer cell metabolism (Fig. 2) [85, 86]. Therefore, there exists a strong link between IL-6 and cancer, similar to the link between cancer and inflammation. Majority of the phenotypes or hallmarks of cancer which are influenced by IL-6 comprise many biological capabilities that are acquired during tumour development. The role of IL-6 in the regulation of hallmarks of cancer will be discussed in detail later.

IL-6 and cancer hallmarks: IL-6 plays an important role in cancer growth and progression by influencing nearly all hallmarks of cancer. The picture illustrates the contribution of IL-6 in some major characteristics of cancer, which are known as cancer hallmarks
Evasion of growth suppressors
Cancer cells evade some powerful programmes that negatively regulate cell proliferation. Many of these programmes depend on the action of tumour suppressor genes such as p53 and RB (retinoblastoma gene), which undergo either loss or gain of function, respectively, to limit cell growth and proliferation. Rb (retinoblastoma-associated) and TP53 proteins are the essential regulatory nodes within two key complementary cellular regulatory circuits that govern the decisions of cells to proliferate or activate senescence and apoptotic programmes [87]. The hypermethylation of CpG islands in the promoter regions of tumour suppressor genes has been found in many tumours, which allows cancer cells to bypass crucial checkpoints in cell cycle progression and evade apoptotic signals (Fig. 3) [88, 89]. IL-6 is known to increase both the expression of DNA methyltransferase (DNMT-1) [90] and its translocation to the nucleus by DNMT-1 nuclear localizing signal’s phosphorylation via PI-3K/AKT signalling [91], thereby increasing the activity of DNMT-1, resulting in CpG island methylation of the promoter region of the p53 gene (Fig. 3) [89]. Contrary to the hypermethylation of tumour suppressor genes, IL-6 is also involved in causing global hypomethylation of retrotransposon long interspersed nuclear element-1 in oral squamous cell carcinoma cell lines, which promotes tumorigenesis in the oral cavity [92]. These epigenetic alterations in tumour cells contribute to the epigenetic silencing of tumour suppressor genes and lead to enhanced tumorigenesis.

Role of IL-6 in evading growth suppressors: Normally, E2F remains bound with Rb and localized to the cytosol. IL-6 signalling either induced by mbIL-6R or sIL-6R activates JAK/STAT-3 phosphorylation, which then phosphorylates Rb, resulting in the dissociation of E2F from Rb. The free E2F translocates to the nucleus, where it induces the expression of genes (cyclin E and E2F itself) responsible for the proliferation of cells. Similarly, IL-6 via PI3K/AKT signalling causes the activation of DNMT-1 by its phosphorylation. After phosphorylation, DNMT-1 translocates to the nucleus and hypermethylates the p53 promoter, resulting in silencing of tumour suppressor, pro-apoptotic and other p53 target genes
Mutations in the RB gene contribute to cellular transformation in various types of malignancies [93]. Normal retinoblastoma protein suppresses the transition from the G1 to the S phase of the cell cycle, which is regulated by the phosphorylation of Rb protein. The active, hypo- or dephosphorylated form of Rb binds with E2F and induces G1 growth arrest. On the contrary, the phosphorylated Rb, which is inactive, cannot bind E2F and activates CDK, thereby facilitating entry of cells into the S phase [94]. In multiple myeloma (MM) cells, IL-6 facilitates the phosphorylation of Rb and, thus, promotes cell growth (Fig. 3). Furthermore, Rb phosphorylation also upregulates IL-6 secretion by MM cells and IL-6-mediated autocrine tumour cell growth [95].
Resistance against cell death
Cancer cells evolve a variety of cytoprotective approaches to limit or circumvent cell death programmes, mainly apoptosis. Besides evading growth suppression signalling, by loss of TP53 function, tumours are also associated with an increase in the expression of anti-apoptotic regulators (Bcl-2, Bcl-xL and Mcl-1) and survival signals (Igf1/2) or downregulated pro-apoptotic factors (Bax, Bim and Puma) [96, 97]. IL-6 regulates the process of apoptosis by activating STAT-3 and NF-kB signalling (Fig. 4), which transactivates the expression of many anti-apoptotic proteins such as Bcl-2, Bcl-xL, Mcl-1, etc., in cholangiocarcinoma cells [98], cervical cancer [99], gastric cancer cells [81], myeloma cells [82], basal cell carcinoma cells [100] and esophageal carcinoma [101]. In addition, these pro-survival proteins, mainly Bcl-2, promote cell proliferation [102]. As the balance between pro-apoptotic and anti-apoptotic proteins is important for apoptotic decision, the ratio of pro-apoptotic to anti-apoptotic factors is increased with oxidative stress, but the increased levels of IL-6 may alter this ratio in favour of anti-apoptotic signalling, leading to cell survival, both in IL-6-treated cells and IL-6-expressing transgenic mice [103]. Besides this, IL-6-induced Bcl-2 regulates Bak interactions with mitofusins via inhibition of Bak dissociation from Mfn2 and also inhibits the interaction of Bak with Mfn1. These two mitochondrial events are the major determinants of cell death pathways as they prevent mitochondrial fragmentation during apoptosis [103, 104]. Therefore, Bcl-2 appears to be an essential mediator of IL-6-induced cytoprotection (Fig. 4).

IL-6 in resisting cell death: IL-6-induced JAK/STAT-3 and NF-kB signalling facilitates the translocation of STAT-3 and NF-kB in the nucleus. Activation of these signalling pathways results in the expression of anti-apoptotic genes (Bcl-2, Bcl-xL, Mcl-1, survivin, etc.) and IL-6 for the constitutive activation of IL-6-dependent signalling in cancer cells. IL-6-induced Bcl-2 expression inhibits stress (endogenous and therapeutic)-induced mitochondrial fragmentation and protects the cells from apoptosis
Besides Bcl-2 and Bcl-xL, IL-6 also supports tumour cell survival by inducing the expression of survivin through direct binding of STAT-3 to the survivin promoter [105]. Furthermore, downregulation of survivin at the gene expression level by inhibiting STAT-3 induces apoptosis in tumour cells [105]. IL-6 triggers PI3K/Akt, NF-kB and MAPK/ERK signalling in prostate cancer cells and results in the upregulation of cyclin A1 expression that promotes tumour cell proliferation in hepatoma, prostate cancer, bladder cancer and in multiple myeloma. IL-6-induced activation of PI3K/Akt signalling further activates IkB kinase (IKK), which initiates NF-kB signalling, leading to transactivation of pro-survival and proliferation-inducing proteins (Fig. 4) [83, 106–109]. IL-6/STAT-3 signalling is also required for the survival of intestinal epithelial cells in colitis-associated cancer, where IL-6 produced from lamina propria myeloid cells protects normal and pre-malignant epithelial cells from cell death [75].
Collectively, it appears that IL-6 facilitates tumour growth primarily by inhibiting apoptosis and enhancing cell proliferation. Besides deriving the growth potential, cancer cells exploit IL-6 for inducing resistance towards anticancer therapy-induced death pathways. For example, IL-6 confers protection from dexamethasone-induced apoptosis by activating PI3K/AKT signalling and inactivating casapase-9, thereby inhibiting apoptosis in multiple myeloma cells [109]. It is also known to induce resistance in cisplatin-mediated cytotoxicity in prostate cancer cell lines and esophageal squamous cell carcinoma [110, 111]. Furthermore, IL-6-induced Bcl-2 confers protection against hyperoxic damage and oxidant (H2O2) injury [103]. Thus, enhanced IL-6 levels appear to confer resistance against chemotherapy in cancer by downregulating cell death pathways.
Induction of proliferation/replicative immortality
The potential growth stimulatory effect of IL-6 in tumour cells is due to the activation of several signalling pathways. IL-6 stimulates tumour cell proliferation and survival by activating the Ras/Raf/MEK/MAPK, PI3K/AKT and JAK/STAT pathways via gp130 tyrosine phosphorylation [83, 101, 108]. In colitis-associated cancer, IL-6 produced by myeloid cells stimulates the proliferation of malignant epithelial cells via NF-kB/IL-6/STAT-3 cascade [75]. These signalling pathways help tumours in the acquisition of unlimited replication potential, which is essentially required to generate large tumours.
Majority of the genes that regulate cell survival and proliferation, such as Bcl-2, Bcl-xL, Mcl-1, Fas, cyclin D1, cyclin E1 and p21, are direct targets of STAT-3. In addition, other transcription factors which promote proliferation, including c-Myc, c-Jun and c-Fos, are also targets of STAT-3 [112]. In tumour cells, STAT-3 activation is mediated through autocrine production and paracrine secretion of IL-6 from stroma and infiltrating inflammatory cells [58–62]. IL-6/STAT-3 signalling also functions as a transcriptional repressor of p53 expression, whilst blocking STAT-3 upregulates the expression of p53, leading to p53-mediated apoptosis [113].
IL-6 has also been found to mediate its multi-lineage haematopoietic effects by shifting stem cells from the G0 to the G1 stage of the cell cycle, thereby inducing the proliferation and making stem cells more responsive to additional haematopoietic growth factors such as IL-3, IL-4, G-CSF, M-CSF or GM-CSF [114]. The autocrine production of IL-6 by non-stem cells activates the JAK1/STAT-3 signal transduction pathway which plays an important role in the conversion of non-stem cells into stem-like cells through the upregulation of Oct-4 (a stem cell marker) [115]. Therefore, IL-6 not only induces the proliferation of cancer cells but also maintains the population of cancer stem cells that induce the reoccurrence of tumours. Since only the cancer stem cells have tumorigenic potential amongst the heterogeneous mass of tumours [116], IL-6 seems to play an important role in the maintenance of equilibrium between non-cancer and cancer stem cells, as observed in breast and prostate cancers [44]. Thus, IL-6 has been suggested as a potential regulator of normal and tumour stem cell self-renewal.
Cancer-related inflammation
Accumulating evidences suggest that chronic inflammation predisposes cells and tissues to different forms of cancer [117]. Thus, cancer and inflammation have a strong connection, which prompted the use of anti-inflammatory drugs for cancer prevention. Cancer-related inflammation involves the infiltration of TAMs, white blood cells and inflammatory messengers like cytokines, such as TNF, IL-1 and IL-6, and chemokines (CCL2 and CXCL8), which facilitate tissue remodelling and angiogenesis [118]. IL-6 is one of the most highly expressed mediators of inflammation in the tumour microenvironment, and STAT-3-dependent tumorigenesis has been associated with the local secretion of IL-6 and its related trans-signalling within the tumour microenvironment in inflammation-induced colorectal cancer [75]. In addition, production of IL-6 by M2-type macrophages in ulcerative colitis supports the development of colon tumours [119]. These studies have identified a link between IL-6 and tumour-associated inflammation. The primary players in inflammation include transcription factors such as NF-kB, STAT-3 and primary inflammatory cytokines (IL-1b, IL-6 and TNFα) [1, 120]. NF-kB is the major regulator of inflammation which is deregulated in many cancers [120]. As a major effector molecule of NF-kB activation through the STAT-3 pathway, IL-6 appears to be an important component of the NF-kB/IL-6/STAT-3 cascade involved in tumorigenesis [75]. STAT-3 is required for the maintenance of NF-kB activation in tumours [121], whilst IL-6 promotes carcinogenesis through inflammation and cell proliferation [35, 44, 106]. Since inflammation enhances the growth and progression of gastrointestinal tumours via the activation of IL-6-mediated STAT-3 signalling, it appears that there is a strong link between IL-6, inflammation and tumour promotion [1, 117, 118].
Metabolic remodelling
Enhanced aerobic glycolysis is one of the prominent phenotypes of a majority of cancer cells which facilitate proliferation and confer protection against death, besides energy production [122, 123]. This induced glycolysis is one of the major factors that contribute to IL-6-induced therapeutic resistance in cancer. IL-6-mediated stimulation of glucose metabolism is dependent on the signal transduction involving the PI-3 kinase and JAK/STAT pathways through the enhanced expression of major glycolytic enzymes hexokinase 2 and PFKFB-3 [124]. It also enhances glucose transport by inducing the expression of glucose transporters GLUT-1 and GLUT-4, which further translocate to the plasma membrane, resulting in a higher glucose influx in cells (Fig. 5) [124]. In addition, it also increases fatty acid oxidation (Fig. 5) [85]. Furthermore, IL-6-induced p53 regulates glycolytic metabolism through NF-kB-mediated mechanism, which also increases GLUT-2 and GLUT-4 receptors on the cells to enhance glycolysis [125].

IL-6 in metabolic remodelling: IL-6-induced JAK/STAT-3 signalling (both classical and trans) induces the expression of major glycolytic enzymes (HK2 and PFKB3) and glucose transporters (GLUT-1 and GLUT-4). The expression of these glycolytic genes ensures aerobic glycolysis in tumour cells. IL-6 also induces fatty acid oxidation in the mitochondria by activating the AMPK pathway
IL-6 also causes 5′ AMP-activated protein kinase (AMPK) phosphorylation, which is important for IL-6-mediated glucose uptake and lipid oxidation (Fig. 5) [126] and is known to be involved in obesity-associated cancers [127]. In sickle cell disease, elevated plasma IL-6 levels are correlated with increased rates of glycolysis in red blood cells, as evidenced by the increase in lactic acid and higher 2,3-bisphosphoglycerate levels [128]. More recently, treatment of IL-6−/− mice with diethylnitrosamine (a carcinogenic agent) after a high-fat diet has been shown not to support faster development of hepatocellular carcinoma (HCC) as compared to a low-fat diet, suggesting that IL-6 preferentially may render obese individuals susceptible to HCC [106]. In cachectic tumour-bearing mice, a high level of IL-6 has been found to suppress mTORC1 activity by AMPK activation, thereby rendering the mice irresponsive to glucose administration [86]. p62 is a scaffold protein that binds to the nutrient-sensing component of mTOR, crucial for metabolic reprogramming during cell transformation. Loss of p62 has been found to reduce mTOR activity, resulting in impaired metabolism and higher IL-6 release, causing tumorigenesis [129].
Maintenance of redox potential
Oxidative stress induced either by therapeutic agents or metabolic alterations induces damage to macromolecules, viz. proteins, lipids, membranes and DNA, that play a key role in the development of cancer. NF-kB is a critical transcription factor that senses redox imbalance and facilitates cytokine gene induction during cellular stress [130]. Endogenous or induced oxidative stress activates transcription factor NF-kB, which regulates the expression of IL-6 by binding to the promoter region of the IL-6 gene [131, 132]. IL-6, which is the major effector molecule of NF-kB, itself causes NF-kB activation in cancer cells, which results in more IL-6 production. Furthermore, this enhanced IL-6 concentration in the tumour microenvironment constitutively activates NF-kB signalling in the same or neighbouring cells (autocrine/paracrine) [131, 133]. Aberrant NF-kB regulation has been observed in many cancers, whose sustained activation requires STAT-3. Since NF-kB and STAT-3 are regulated by IL-6, it appears that the NF-kB/STAT-3/IL-6 signalling cascade plays an important role in oncogenesis [134]. IL-6 is known to protect cardiac myocytes from oxidative stress-induced apoptosis through STAT-3 signalling and gastric cancer cells by upregulating Mcl-1 expression [81]. In multiple myeloma cells, IL-6 causes radio-resistance by activating NF-kB in an autocrine manner, which results in the activation of antioxidant defence system enzymes such as glutathione peroxidase (GPx), superoxide dismutase II (MnSOD) and catalase (Fig. 6) [135].

IL-6 maintains redox balance: p38 senses oxidative stress through ROS-induced phosphorylation of MKK3/6. p38 activation (phosphorylation) induces NF-kB signalling, leading to the enhanced expression and secretion of IL-6 from affected cells. IL-6 (in classical/trans-signalling fashion) further activates JAK/STAT-3 signalling in both paracrine and autocrine manner, resulting in the overexpression of the antioxidant enzymes MnSOD, glutathione peroxidase (GPx) and catalase. Altered favourable redox balance also enhances the mitochondrial membrane potential
Apoptosis plays a very crucial role in maintaining genomic integrity by selectively removing the population of heavily damaged cells. Reactive oxygen species (ROS) and pro-inflammatory cytokines are generally elevated following exposure to ionizing radiation and during human carcinogenic processes [136]. Hydrogen peroxide (H2O2) is a potent reactive oxygen species that causes mitochondrial dysfunction and cell death. Preconditioning cells with IL-6 decreases H2O2-induced cell death by increasing the expression of prohibitin, which is involved in mitochondrial biogenesis and metabolism, apoptosis and replicative senescence [137]. In addition to signalling through STAT-3, the stress-induced activation (phosphorylation) of p38 also induces IL-6 release from cells via the activation of NF-kB (Fig. 6) [138]. All these studies showed the antioxidant potential of IL-6.
Invasion, metastasis and angiogenesis
During tumour metastasis, cancer cells invade surrounding local tissues to migrate to distant organs by acquiring a mesenchymal phenotype that allows the metastatic cancer cells to migrate from the site of the primary tumour. Upon lodging into the new organ, tumour cells lodge themselves by switching back to an epithelial phenotype and proliferate to form metastatic tumours. The processes by which cells switch between epithelial and mesenchymal phenotypes are widely known as the epithelial-to-mesenchymal transition (EMT) and its counterpart, the mesenchymal-to-epithelial transition [139]. It is well established that inflammation promotes EMT [139, 140]. Elevated levels of IL-6 in the serum have been associated with EMT and invasion along with increased size of tumours, metastasis and decreased survival of colorectal cancer patients [60]. Available evidences suggest that IL-6 facilitates the metastasis of many tumours (e.g. breast, lung, prostate, renal carcinomas, neuroblastoma, melanoma and multiple myeloma) to the bone [141] by increasing CXCR4 expression via STAT-3 and c-Jun [142]. Furthermore, IL-6-activated STAT-3 plays a role in EMT, invasiveness and angiogenesis in bladder cancer by increasing VEGF, MMP9 and DNMT1 expressions [107]. Autocrine production of IL-6 has also been shown to enhance the capacity to invade the extracellular matrix in breast cancer [143].
Supply of nutrients and oxygen as well as the clearance of metabolic by-products required for tumour sustenance are provided by tumour-associated neovasculature. The formation and maintenance of this neovasculature remains always activated as an ‘angiogenic switch’ and helps tumour development [144]. STAT-3 activation by IL-6 facilitates angiogenesis in many cancers by inducing the expressions of VEGF, bFGF and MMP9 in tumour-associated endothelial cells, TAMs and MDSCs [60, 84, 143, 145]. In addition, the Notch ligand, JAG-2, has been reported to be overexpressed in malignant plasma cells from MM patients, which induces the secretion of IL-6 and VEGF [146], whilst in breast cancer, Notch-3-dependent ERK activation via IL-6 appears to activate JAG-1 (Notch ligand) and CA-IX (a hypoxia survival gene). Furthermore, this CA-IX upregulated by IL-6 has also been found to maintain the invasive potential of breast cancer cells and mammospheres [143].
DNA damage and repair
DNA is the primary target of majority of the established anticancer therapies which induce cell death processes in tumour cells. DNA damage has been shown to induce the expression and secretion of IL-6 from cancer cells, resulting in the activation of the JAK1/STAT-3 signalling pathway. STAT-3 activation not only protects tumour cells against DNA damage but also facilitates the growth of damaged cells by inhibiting induced senescence [147, 148]. Furthermore, inhibition of the IL-6/STAT-3 signalling pathway by the STAT-3 inhibitors, knockdown of gp130, or the neutralization of IL-6 impairs the growth of tumour cells exposed to DNA damage [147]. IL-6 secreted by tumour-associated endothelial cells in the tumour microenvironment has been suggested to protect lymphoma cells from genotoxic chemotherapy [149], whilst persistent DNA damage response has been shown to induce IL-6 secretion in stromal cells such as fibroblasts [148]. The ATM/NEMO/ELKS complex formed by DNA damage-induced phosphorylation of ATM promotes IKK-mediated IkBα degradation, leading to NF-kB activation, which further induces the expression of IL-6 (Fig. 7) [150]. In the partial hepatectomy (PH) model of DNA repair, wild-type mice were found to express some of the key DNA repair enzymes such as OGG-1, 8-oxo-GTP, Neil-1 and PARP, whereas these enzymes were absent in IL-6 knockout mice, suggesting a direct role of IL-6 in the repair of DNA damage [151]. Thus, in PH, it appears that IL-6 facilitates the restoration of hepatic mass by activating DNA repair enzymes, followed by accurate replication in proliferating hepatic cells [151]. Loss of Rrm2b function (a key enzyme in de novo deoxyribonucleotide synthesis) is known to cause severe numerical and structural chromosomal abnormalities, which leads to the activation of NF-kB through ATM phosphorylation and IKK activation, leading to enhanced IL-6 expression and constitutive activation of STAT-3 [152]. Since IL-6 signalling functions by the activation of transcription factors such as STAT-3 and NF-kB, inhibition of these transactivators or neutralizing the IL-6 may enhance the efficacy of DNA damage-causing chemotherapeutic drugs [153].

Role of IL-6 in DNA repair: IL-6 facilitates DNA repair by inducing the expression of DNA repair enzymes in cancer cells. The induction of DNA damage phosphorylates the ATM, leading to the phosphorylation of IKK through the phosphorylation of various downstream kinases. IKK phosphorylation activates NF-kB signalling, resulting in sustained IL-6 expression and constitutive activation of STAT-3 signalling, which induces the expression of DNA repair enzymes
IL-6 induces therapeutic resistance in cancer
Accumulating evidences suggest that inflammatory signals from tumour cells and surrounding microenvironment facilitate tumour growth [63, 117]. Furthermore, most anticancer therapies induce inflammation by killing tumour cells and normal tissues [154]. Several inflammatory cytokines are believed to play key roles in therapeutic resistance and lead to tumour regrowth, invasion and angiogenesis. Anticancer therapies induce upregulation of the levels of a variety of inflammatory cytokines, including IL-6, IL-8 and TNFα [8, 12, 13]. Amongst these, IL-6 is known to contribute to poor therapeutic gain, tumour relapse and aggressive tumour growth [2, 8, 12]. IL-6 is also recognised as a key regulator of immunosuppression in patients with advanced cancer [1, 154]. Elevated IL-6 serum levels have been correlated with metastasis and morbidity in prostate cancer and therapeutic resistance in ovarian cancer, whilst patients with reduced levels of IL-6 respond better to therapy [14, 15]. It appears that tumour cells produce IL-6 as a protective mechanism against drug-induced death, as in the case of prostate cancer where inhibition of IL-6 secretion increases the sensitivity of prostate cancer cells to anticancer drugs [12, 110]. Clinical studies on combinations of docetaxel and zolendronic acid in prostate cancer patients with bone metastasis have shown interesting data in relation to IL-6. Patients who responded to therapy had a 35 % decrease in overall serum IL-6 levels, whilst patients who did not respond had a 76 % increase in serum IL-6 levels [155], lending support to the notion that IL-6 confers therapeutic resistance in prostate cancer [5, 12]. The autocrine secretion of IL-6 by breast cancer cells is also shown to confer therapeutic resistance; however, it does not affect their growth. Further studies showed that drug-sensitive breast cancer cells do not express IL-6, whereas multidrug-resistant breast cancer cells produced high levels of IL-6 [156]. IL-6-mediated STAT3 activation has been reported to cause therapeutic resistance in tumours by inducing several pro-survival pathways [12, 105, 110]. However, IL-6-induced drug resistance is associated with increased expression of the multidrug resistance gene, mdr1, and upregulation of C/EBPβ and C/EBPδ (CCAAT enhancer-binding protein family of transcription factors) [156]. Moreover, in colorectal cancer, IL-6 secreted from stromal cells induces CYP2E1 and CYP1B1 expression (CYP450 enzymes have a significant role in xenobiotic activation) through the JAK/STAT and PI3K/AKT pathways, which causes tumour initiation and promotion via the activation of chemical carcinogens [157].
IL-6-induced modifications of the stromal cell function appear to be important for tumour growth and angiogenesis as an anti-IL-6 receptor antibody has been found to inhibit tumour angiogenesis and growth by blocking tumour–stroma interaction [60]. Cancer-associated adipocytes can also promote radio-resistance by secreting IL-6 [158], whilst IL-6 and IL-8 secreted by mesenchymal stem cells activate macrophages in the microenvironment of human colorectal and ovarian cancers, causing chemoresistance [159, 160]. Besides causing resistance to chemo- and radiotherapy, autocrine secretion of IL-6 (induced by therapy) also confers resistance to some targeted therapies; for example, aflibercept (anti-VEGF, inhibitor)-resistant epidermoid carcinoma cells and herceptin (trastuzumab, anti-HER2)-resistant breast cancer cells secrete a large amount of IL-6 and show hyperactivation of STAT-3 signalling, causing resistance to therapy [161, 162].
Besides therapeutic resistance, IL-6 also minimizes clinical outcome by promoting the maintenance of a highly therapeutic-resistant cancer stem cell population which is mainly responsible for tumour reoccurrence [116]. IL-6 secreted from either cancer cells or tumour microenvironment (immune cells and tumour stromal cells) not only facilitates tumour growth but also acts as a major obstacle in obtaining therapeutic gain and tumour-free survival [163]. Therefore, targeting IL-6 and its associated signalling may hold greater promise in minimizing therapeutic resistance in cancer. IL-6 inhibition could sensitize tumour cells to anticancer drugs and radiation by increasing DNA damage and cell death, and mitigate tumour regrowth by inhibiting subsequent angiogenesis and reducing cancer stem cell population.
Targeting IL-6 for therapeutic gain
IL-6 plays an important role in tumour progression and therapeutic resistance through inhibition of cancer cell apoptosis and stimulation of tumour-promoting factors such as proliferation, angiogenesis, etc. These effects are mediated by several signalling pathways; however, STAT-3 plays a central and major role [1, 112]. Targeting the IL-6/JAK/STAT-3 pathway has shown promising results in many types of cancer and various inflammation-related diseases. The clinical correlation of increased serum IL-6 concentrations and advanced tumour stages in various malignancies, as discussed earlier in the review, makes a strong case of blocking IL-6 signalling for therapeutic gain. Therefore, inhibiting IL-6 signalling or minimizing the level of IL-6 can be a potential therapeutic strategy for those cancers which are characterized by overproduction of IL-6. IL-6 signalling can be targeted in multiple ways, such as the use of specific monoclonal antibodies against IL-6 or IL-6R [15, 19, 31], by using synthetic/semi-synthetic compounds as specific inhibitors of IL-6 downstream signalling molecules or by kinase inhibitors (like JAK inhibitor) [164]. All these approaches evaluated in experimental models and reached clinical trials are summarized in Table 1.
Antibodies targeting IL-6 or IL-6 signalling have been extensively investigated in experimental tumour models and clinical trials for a variety of cancers. Elsilimomab (BE-8), a murine monoclonal antibody, and siltuximab (CNTO 328), a chimeric antibody with strong affinity to IL-6 and licenced for the treatment of Castleman’s disease, are used for the treatment of prostate cancer, ovarian cancer, renal cell carcinoma and colorectal cancer in combination with other chemotherapeutics [3, 6, 166]. In prostate cancer, siltuximab has been shown to downregulate the genes downstream of IL-6 signalling along with decreased STAT-3 expression [165]. Tocilizumab, a FDA-approved drug for rheumatoid arthritis and Crohn’s disease, is the humanized antibody specific for IL-6R that recognises both soluble and membrane-bound receptors and blocks its signalling [195]. BE-8, a murine anti-IL-6 monoclonal antibody, has also been used in the treatment of lymphoma and multiple myeloma [196]. Sequestering the enhanced IL-6 showed a significant increase in the therapeutic efficacy of paclitaxel in mouse models of epithelial ovarian cancer by reducing tumour angiogenesis [197]. Tocilizumab has also been used in the treatment of oral cell carcinoma, prostate cancer, renal cancer, multiple myeloma and breast cancer [15]. It inhibits the growth of prostate and breast cancer cells by reducing STAT-3-induced VEGF expression. In addition, it reduces bone metastasis of breast cancer cells [142]. Tocilizumab has been shown to improve cachexia developed by IL-6 overexpression in lung cancer patients [175]. The combination of anti-IL-6R with herceptin and aflibercept enhances therapeutic gain, also in targeted therapy-resistant breast cancer and epidermoid carcinoma, respectively [161, 162]. However, blocking IL-6 signalling can be harmful and may result in some adverse effects, such as gastrointestinal, nasopharyngeal and upper respiratory tract infections, gastrointestinal haemorrhage, thrombocytopenia and neutropenia [198]. Therefore, selective blocking of trans-signalling (the major pathway involved in inflammation and related diseases like cancer) using sgp130Fc (a recombinant fusion protein of soluble gp130 and human IgG1 Fc) may be useful in achieving therapeutic gain in cancer [76].
Besides antibodies, a number of other compounds are currently available that inhibit IL-6 signalling. Zoledronic acid (ZA) is the most potent nitrogen-containing bisphosphonate compound which has been used in adjuvant therapies to inhibit bone metastasis caused by multiple cancers [172]. It inhibits growth, besides inducing apoptosis by reducing IL-6 secretion in prostate cancer cell lines, which is suggestive of its use in the treatment of prostate cancer either alone or in combination with chemotherapy [171]. Radiation activates IL-6/STAT-3 signalling, which stimulates tumour invasion and EMT changes and promotes the survival of tumour cells after therapy, thereby conferring resistance to therapy [12, 84, 156]. However, use of siRNA against IL-6 inhibits tumour regrowth after radiotherapy in prostate cancer and sensitizes tumour cells to radiation by increasing cell death and DNA damage [12]. Its inhibition also mitigates tumour regrowth by eliminating radiotherapy-triggered MDSC and subsequent angiogenesis after radiation exposure [199]. Therefore, inhibition of IL-6 could be a potential therapeutic strategy for increasing the radiation response of tumours. Similarly, medroxyprogesterone acetate (MPA) is a synthetic compound used as an endocrine therapeutic agent for patients with breast cancer which is known to reduce serum IL-6 levels [173]. Several studies have shown that the use of an inhibitor for downstream targets of IL-6 signalling can also be a good approach for enhancing therapeutic gain. For example, the use of JAK inhibitor (TG101209) enhances the efficacy of radiotherapy in lung cancer [180]. Similarly, WP1066 and CEP3379, other JAK2 inhibitors, suppress the growth of gastric cancer and colorectal tumours by inhibiting IL-6/JAK2/STAT-3 signalling [184, 200]. Sorafenib, which is a multiple kinase inhibitor, causes dephosphorylation of STAT-3 and prevents AKT signalling in glioblastoma cells and prostate cancer, respectively [186]. AG490, a potent JAK inhibitor, targets STAT-3 signalling, reduces the invasion of human pancreatic cancer cells in vitro and induces apoptosis in gastric cancer cells [201, 202]. Moreover, AG490 is also effective in murine ovarian cancer, where it induces the expression of anti-tumour cytokines [182]. Another JAK inhibitor, ruxolitinib, is currently in clinical trials for leukaemia [183].
Inhibition of STAT-3 phosphorylation by the use of STAT-3 inhibitors can also be a good approach to block IL-6 signalling. JSI-124 (STAT-3 inhibitor) inhibits STAT-3 phosphorylation at serine 727 and sensitizes B-leukaemia cells to apoptosis [203]. JSI-124 suppresses breast cancer cell growth by downregulating STAT-3 activation in tumour-associated B cells. Stattic and Eriocalyxin B (a diterpenoid) also inhibit STAT-3 phosphorylation and induce apoptosis in MDA-MB-231 and HepG2 cells [204, 205]. Besides inhibiting STAT-3 phosphorylation and transcriptional activation, Decoy oligodeoxynucleotides target the DNA-binding domain of STAT-3 by competing against endogenous DNA cis element, which results in reduced cell growth and increased apoptosis [206]. G-quartet oligodeoxynucleotides can also be used as a STAT-3 inhibitor which suppresses the growth of prostate, breast, and head and neck cancers in nude mice [206, 207]. Instead of inhibiting STAT-3 phosphorylation, these oligodeoxynucleotides inhibit the binding of STAT-3 dimer on DNA. Moreover, small-molecule inhibitors can also be used to inhibit STAT-3. A small-molecule non-peptide STAT-3 inhibitor named S3I-201 (a.k.a. NSC74859) selectively inhibits STAT-3 DNA-binding activity in vitro and blocks the formation of STAT-3:STAT-3 dimer, which leads to the inhibition of STAT-3-dependent gene transcription and the blockade of the proliferation and survival of human breast carcinoma cells [208]. STA-21 also selectively inhibits STAT-3 DNA-binding activity in vitro, disrupts STAT-3:STAT-3 dimerization and suppresses STAT-3-mediated gene transcription along with cell growth inhibition and apoptosis through the caspase pathway in a human breast carcinoma and rhabdomyosarcoma model [209, 210]. Catechol-containing compounds, IS3 295, galiellalactone and peptide aptamers have been found to inhibit the DNA-binding ability of STAT-3 and further signalling [208, 211].
Conclusion
IL-6, which is produced by tumours and many other cells in the tumour microenvironment, facilitates tumour growth and sustenance by influencing and regulating nearly all hallmarks of cancer, besides contributing to therapeutic resistance. Recent studies have enhanced our knowledge regarding the potential role of IL-6 in the cross talk between tumour and its microenvironment. IL-6/JAK/STAT-3 appears to be the primary pathway through which IL-6 regulates majority of the tumour-promoting functions. Therefore, neutralizing IL-6 or the IL-6 receptor to prevent the initiation of signalling or inhibiting the activity of two other members, JAK and STAT-3, to prevent the execution in the end has established therapeutic efficacy in cellular and systemic models of cancer. Blocking IL-6 using monoclonal antibodies against either IL-6 or IL-6R has shown promising results in preclinical studies and clinical trials. Phase I and II clinical trials have established the efficacy of monoclonal antibodies either as a single agent or in combination with other chemotherapeutic drugs, radiation and targeted therapies in various types of cancer. Subsequently, certain synthetic molecules, such as ZA, MPA, polyphenols, etc., have also shown promising results in various cancers by regulating IL-6 levels. The use of small-molecule inhibitors of JAK and STAT-3 alone or in combination with radiation or anticancer agents has also resulted in promising anti-tumour effects in various cancers. However, undesirable effects (such as infections, gastrointestinal haemorrhage, thrombocytopenia and neutropenia) of anti-IL-6 therapy have also been reported in patients with various inflammatory diseases. Therefore, selective inhibition of IL-6 trans-signalling using molecules such as sgp130Fc in the future may result in better therapeutic gain without the side effects associated with anti-IL-6 therapy currently under evaluation.
References
Bromberg J, Wang TC. Inflammation and cancer: IL-6 and STAT3 complete the link. Cancer Cell. 2009;15:79–80.
Grivennikov S, Karin M. Autocrine IL-6 signaling: a key event in tumorigenesis? Cancer Cell. 2008;13:7–9.
Waldner MJ, Foersch S, Neurath MF. Interleukin-6—a key regulator of colorectal cancer development. Int J Biol Sci. 2012;8(9):1248–53.
Dethlefsen C, Højfeldt G, Hojman P. The role of intratumoral and systemic IL-6 in breast cancer. Breast Cancer Res Treat. 2013;138(3):657–64.
Culig Z, Puhr M. Interleukin-6: a multifunctional targetable cytokine in human prostate cancer. Mol Cell Endocrinol. 2012;360(1–2):52–8.
Macciò A, Madeddu C. The role of interleukin-6 in the evolution of ovarian cancer: clinical and prognostic implications—a review. J Mol Med. 2013;91:1355–68.
Miura T, Mitsunaga S, Ikeda M, Shimizu S, Ohno I, Takahashi H, et al. Characterization of patients with advanced pancreatic cancer and high serum interleukin-6 levels. Pancreas. 2015;44(5):756–63.
Chang CH, Hsiao CF, Yeh YM, Chang GC, Tsai YH, Chen YM, et al. Circulating interleukin-6 level is a prognostic marker for survival in advanced nonsmall cell lung cancer patients treated with chemotherapy. Int J Cancer. 2013;132(9):1977–85.
Altundag O, Altundag K, Gunduz E. Interleukin-6 and C-reactive protein in metastatic renal cell carcinoma. J Clin Oncol. 2005;23:1044.
Wei LH, Kuo ML, Chen CA, Chou CH, Lai KB, Lee CN, et al. Interleukin-6 promotes cervical tumor growth by VEGF-dependent angiogenesis via a STAT3 pathway. Oncogene. 2003;22:1517–27.
Singh U, Shevra CR, Singh S, Singh N, Kumar S, Rai M. Interleukin-6 and interleukin-4 levels in multiple myeloma and correlation of interleukin-6 with β2 microglobulin and serum creatinine. Clin Cancer Investig J. 2015;4:211–5.
Wu CT, Chen MF, Chen WC, Hsieh CC. The role of IL-6 in the radiation response of prostate cancer. Radiat Oncol. 2013;8:159.
Chen MF, Chen PT, Lu MS, Lin PY, Chen WC, Lee KD. IL-6 expression predicts treatment response and outcome in squamous cell carcinoma of the esophagus. Mol Cancer. 2013;12:26.
Shibayama O, Yoshiuchi K, Inagaki M, Matsuoka Y, Yoshikawa E, Sugawara Y, et al. Association between adjuvant regional radiotherapy and cognitive function in breast cancer patients treated with conservation therapy. Cancer Med. 2014;3(3):702–9.
Guo Y, Xu F, Lu T, Duan Z, Zhang Z. Interleukin-6 signaling pathway in targeted therapy for cancer. Cancer Treat Rev. 2012;38(7):904–10.
Veuger SJ, Hunter JE, Durkacz BW. Ionizing radiation-induced NF-kappaB activation requires PARP-1 function to confer radioresistance. Oncogene. 2009;28:832–42.
Kozakai N, Kikuchi E, Hasegawa M, Suzuki E, Ide H, Miyajima A, et al. Enhancement of radiosensitivity by a unique novel NF-kB inhibitor, DHMEQ, in prostate cancer. Br J Cancer. 2012;107:652–7.
Naugler WE, Karin M. NF-kappaB and cancer-identifying targets and mechanisms. Curr Opin Genet Dev. 2008;18(1):19–26.
Wolf J, Rose-John S, Garbers C. Interleukin-6 and its receptors: a highly regulated and dynamic system. Cytokine. 2014;70(1):11–20.
Yoon S, Woo SU, Kang JH, Kim K, Shin HJ, Gwak HS, et al. NF-kB and STAT3 cooperatively induce IL6 in starved cancer cells. Oncogene. 2012;31(29):3467–81.
Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta. 2011;1813(5):878–88.
Hirano T, Taga T, Nakano N, Yasukawa K, Kashiwamura S, Shimizu K, et al. Purification to homogeneity and characterization of human B-cell differentiation factor (BCDF or BSFp-2). Proc Natl Acad Sci U S A. 1985;82(16):5490–4.
Kishimoto T, Akira S, Narazaki M, Taga T. Interleukin-6 family of cytokines and gp130. Blood. 1995;86:1243–54.
Mihara M, Hashizume M, Yoshida H, Suzuki M, Shiina M. IL-6/IL-6 receptor system and its role in physiological and pathological conditions. Clin Sci (Lond). 2012;122(4):143–59.
Wunderlich FT, Ströhle P, Könner AC, Gruber S, Tovar S, Brönneke HS, et al. Interleukin-6 signaling in liver-parenchymal cells suppresses hepatic inflammation and improves systemic insulin action. Cell Metab. 2010;12(3):237–49.
Taga T, Kishimoto T. Gp130 and the interleukin-6 family of cytokines. Annu Rev Immunol. 1997;15:797–819.
Hibi M, Murakami M, Saito M, Hirano T, Taga T, Kishimoto T. Molecular cloning and expression of an IL-6 signal transducer, gp130. Cell. 1990;63:1149–57.
Yawata H, Yasukawa K, Natsuka S, Murakami M, Yamasaki K, Hibi M, et al. Structure–function analysis of human IL-6 receptor: dissociation of amino acid residues required for IL-6-binding and for IL-6 signal transduction through gp130. EMBO J. 1993;12(4):1705–12.
Jones SA, Horiuchi S, Topley N, Yamamoto N, Fuller GM. The soluble interleukin 6 receptor: mechanisms of production and implications in disease. FASEB J. 2001;15(1):43–58.
Yoshida K, Taga T, Saito M, Suematsu S, Kumanogoh A, Tanaka T, et al. Targeted disruption of gp130, a common signal transducer for the interleukin 6 family of cytokines, leads to myocardial and hematological disorders. Proc Natl Acad Sci U S A. 1996;93(1):407–11.
Jones SA, Scheller J, Rose-John S. Therapeutic strategies for the clinical blockade of IL-6/gp130 signaling. J Clin Invest. 2011;121:3375–83.
Chalaris A, Garbers C, Rabe B, Rose-John S, Scheller J. The soluble interleukin 6 receptor: generation and role in inflammation and cancer. Eur J Cell Biol. 2011;90(6–7):484–94.
Islam O, Gong X, Rose-John S, Heese K. Interleukin-6 and neural stem cells: more than gliogenesis. Mol Biol Cell. 2009;20(1):188–99.
Peters M, Solem F, Goldschmidt J, Schirmacher P, Rose-John S. Interleukin-6 and the soluble interleukin-6 receptor induce stem cell factor and Flt-3L expression in vivo and in vitro. Exp Hematol. 2001;29(2):146–55.
Yeoh GC, Ernst M, Rose-John S, Akhurst B, Payne C, Long S, et al. Opposing roles of gp130-mediated STAT-3 and ERK-1/2 signaling in liver progenitor cell migration and proliferation. Hepatology. 2007;45(2):486–94.
Humphrey RK, Beattie GM, Lopez AD, Bucay N, King CC, Firpo MT, et al. Maintenance of pluripotency in human embryonic stem cells is STAT3 independent. Stem Cells. 2004;22(4):522–30.
Rose-John S. IL-6 trans-signaling via the soluble IL-6 receptor: importance for the pro-inflammatory activities of IL-6. Int J Biol Sci. 2012;8(9):1237–47.
McFarland-Mancini MM, Funk HM, Paluch AM, Zhou M, Giridhar PV, Mercer CA, et al. Differences in wound healing in mice with deficiency of IL-6 versus IL-6 receptor. J Immunol. 2010;184(12):7219–28.
Kovacs E. Investigation of interleukin-6 (IL-6), soluble IL-6 receptor (sIL-6R) and soluble gp130 (sgp130) in sera of cancer patients. Biomed Pharmacother. 2001;55(7):391–6.
Jostock T, Müllberg J, Ozbek S, Atreya R, Blinn G, Voltz N, et al. Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor transsignaling responses. Eur J Biochem. 2001;268(1):160–7.
Akira S, Kishimoto T. IL-6 and NF-IL6 in acute-phase response and viral infection. Immunol Rev. 1992;127:25–50.
Yang R, Lin Q, Gao HB, Zhang P. Stress-related hormone norepinephrine induces interleukin-6 expression in GES-1 cells. Braz J Med Biol Res. 2014;47(2):101–9.
Zhao W, Liu M, Kirkwood KL. p38alpha stabilizes interleukin-6 mRNA via multiple AU-rich elements. J Biol Chem. 2008;283(4):1778–85.
Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 microRNA, and IL6 links inflammation to cell transformation. Cell. 2009;139(4):693–706.
Narbutt J, Lukamowicz J, Bogaczewicz J, Sysa-Jedrzejowska A, Torzecka JD, Lesiak A. Serum concentration of interleukin-6 is increased both in active and remission stages of pemphigus vulgaris. Mediat Inflamm. 2008;2008:875394.
D’Auria L, Bonifati C, Mussi A, D’Agosto G, De Simone C, Giacalone B, et al. Cytokines in the sera of patients with pemphigus vulgaris: interleukin-6 and tumour necrosis factor-alpha levels are significantly increased as compared with healthy subjects and correlate with disease activity. Eur Cytokine Netw. 1997;8(4):383–7.
Devaraj S, Venugopal SK, Singh U, Jialal I. Hyperglycemia induces monocytic release of interleukin-6 via induction of protein kinase c-{alpha} and -{beta}. Diabetes. 2005;54(1):85–91.
Blackburn P, Després JP, Lamarche B, Tremblay A, Bergeron J, Lemieux I, et al. Postprandial variations of plasma inflammatory markers in abdominally obese men. Obesity (Silver Spring). 2006;14(10):1747–54.
Angstwurm MW, Gartner R, Ziegler-Heitbrock HW. Cyclic plasma IL-6 levels during normal menstrual cycle. Cytokine. 1997;9:370–4.
Reihmane D, Dela F. Interleukin-6: possible biological roles during exercise. Eur J Sport Sci. 2014;14(3):242–50.
Sakamoto K, Arakawa H, Mita S, Ishiko T, Ikei S, Egami H, et al. Elevation of circulating interleukin 6 after surgery: factors influencing the serum level. Cytokine. 1994;6:181–6.
Keski-Nisula L, Hirvonen MR, Roponen M, Heinonen S, Pekkanen J. Spontaneous and stimulated interleukin-6 and tumor necrosis factor-alpha production at delivery and three months after birth. Eur Cytokine Netw. 2004;15:67–72.
Naffaa M, Makhoul BF, Tobia A, Kaplan M, Aronson D, Saliba W, et al. Interleukin-6 at discharge predicts all-cause mortality in patients with sepsis. Am J Emerg Med. 2013;31(9):1361–4.
Muñoz‐Cánoves P, Scheele C, Pedersen BK, Serrano AL. Interleukin‐6 myokine signaling in skeletal muscle: a double‐edged sword? FEBS. 2013;280(17):4131–48.
Steensberg A, Febbraio MA, Osada T, Schjerling P, van Hall G, Saltin B, et al. Interleukin-6 production in contracting human skeletal muscle is influenced by pre-exercise muscle glycogen content. J Physiol. 2001;537:633–9.
Bastard JP, Maachi M, Van Nhieu JT, Jardel C, Bruckert E, Grimaldi A, et al. Adipose tissue IL-6 content correlates with resistance to insulin activation of glucose uptake both in vivo and in vitro. J Clin Endocrinol Metab. 2002;87(5):2084–9.
Krishnamoorthy N, Oriss T, Pagila M, Ray A, Ray P. A critical role for IL-6 secretion by dendritic cells promoting Th2 and limiting Th1 response. J Immunol. 2007;178:S181.
Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013;73(3):1128–4.
Bode JG, Nimmesgern A, Schmitz J, Schaper F, Schmitt M, Frisch W, et al. LPS and TNFalpha induce SOCS3 mRNA and inhibit IL-6-induced activation of STAT3 in macrophages. FEBS Lett. 1999;463:365–70.
Nagasaki T, Hara M, Nakanishi H, Takahashi H, Sato M, Takeyama H. Interleukin-6 released by colon cancer-associated fibroblasts is critical for tumour angiogenesis: anti-interleukin-6 receptor antibody suppressed angiogenesis and inhibited tumour–stroma interaction. Br J Cancer. 2014;110(2):469–78.
Nolen BM, Marks JR, Ta’san S, Rand A, Luong TM, Wang Y, et al. Serum biomarker profiles and response to neoadjuvant chemotherapy for locally advanced breast cancer. Breast Cancer Res. 2008;10(3):R45.
Oh K, Lee OY, Shon SY, Nam O, Ryu PM, Seo MW, et al. A mutual activation loop between breast cancer cells and myeloid-derived suppressor cells facilitates spontaneous metastasis through IL-6 trans-signaling in a murine model. Breast Cancer Res. 2013;15(5):R79.
Fisher DT, Appenheimer MM, Evans SS. The two faces of IL-6 in the tumor microenvironment. Semin Immunol. 2014;26(1):38–47.
Lederle W, Depner S, Schnur S, Obermueller E, Catone N, Just A, et al. IL-6 promotes malignant growth of skin SCCs by regulating a network of autocrine and paracrine cytokines. Int J Cancer. 2011;128(12):2803–14.
Gruys E, Toussaint MJM, Niewold TA, Koopmans SJ. Acute phase reaction and acute phase proteins. J Zhejiang Univ Sci B. 2005;6(11):1045–56.
Tilg H, Trehu E, Atkins MB, Dinarello CA, Mier JW. Interleukin-6 (IL-6) as an anti-inflammatory cytokine: induction of circulating IL-1 receptor antagonist and soluble tumor necrosis factor receptor p55. Blood. 1994;83(1):113–8.
Aderka D, Le JM, Vilcek J. IL-6 inhibits lipopolysaccharide induced tumor necrosis factor production in cultured human monocytes, U937 cells and in mice. J Immunol. 1989;143:3517–23.
Xing Z, Gauldie J, Cox G, Baumann H, Jordana M, Lei XF, et al. IL-6 is an antiinflammatory cytokine required for controlling local or systemic acute inflammatory responses. J Clin Invest. 1998;101(2):311–20.
Croker BA, Krebs DL, Zhang JG, Wormald S, Willson TA, Stanley EG, et al. SOCS3 negatively regulates IL-6 signaling in vivo. Nat Immunol. 2003;4:540–5.
Radtke S, Wüller S, Yang XP, Lippok BE, Mütze B, Mais C, et al. Cross-regulation of cytokine signalling: pro-inflammatory cytokines restrict IL-6 signalling through receptor internalisation and degradation. J Cell Sci. 2010;123(6):947–59.
Ahmed ST, Mayer A, Ji JD, Ivashkiv LB. Inhibition of IL-6 signaling by a p38-dependent pathway occurs in the absence of new protein synthesis. J Leukoc Biol. 2002;72:154–62.
McLoughlin RM, Jenkins BJ, Grail D, Williams AS, Fielding CA, Parker CR, et al. IL-6 trans-signaling via STAT3 directs T cell infiltration in acute inflammation. Proc Natl Acad Sci U S A. 2005;102(27):9589–94.
Curnow SJ, Scheel-Toellner D, Jenkinson W, Raza K, Durrani OM, Faint JM, et al. Inhibition of T cell apoptosis in the aqueous humor of patients with uveitis by IL-6/soluble IL-6 receptor trans-signaling. J Immunol. 2004;173(8):5290–7.
Atreya R, Mudter J, Finotto S, Müllberg J, Jostock T, Wirtz S, et al. Blockade of IL-6 transsignaling abrogates established experimental colitis in mice by suppression of the antiapoptotic resistance of lamina propria T cells. Nat Med. 2000;6(5):583–8.
Grivennikov S, Karin E, Terzic J, Mucida D, Yu G-Y, Vallabhapurapu S, et al. IL-6 and STAT3 signaling is required for survival of intestinal epithelial cells and colitis associated cancer. Cancer Cell. 2009;16:103–13.
Barkhausen T, Tschernig T, Rosenstiel P, van Griensven M, Vonberg RP, Dorsch M, et al. Selective blockade of interleukin-6 trans-signaling improves survival in a murine polymicrobial sepsis model. Crit Care Med. 2011;39(6):1407–13.
Grivennikov SI, Karin M. Inflammatory cytokines in cancer: tumour necrosis factor and interleukin 6 take the stage. Ann Rheum Dis. 2011;70 Suppl 1:i104–8.
Cozen W, Gill PS, Ingles SA, Masood R, Martinez-Maza O, Cockburn MG, et al. IL-6 levels and genotype are associated with risk of young adult Hodgkin lymphoma. Blood. 2004;103:3216–21.
Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317(5834):121–4.
Wei Q, Guo P, Mu K, Zhang Y, Zhao W, Huai W, et al. Estrogen suppresses hepatocellular carcinoma cells through ERβ-mediated upregulation of the NLRP3 inflammasome. Lab Investig. 2015;95(7):804–16.
Lin MT, Juan CY, Chang KJ, Chen WJ, Kuo ML. IL-6 inhibits apoptosis and retains oxidative DNA lesions in human gastric cancer AGS cells through up-regulation of anti-apoptotic gene mcl-1. Carcinogenesis. 2001;22(12):1947–53.
Jourdan M, Veyrune JL, Vos JD, Redal N, Couderc G, Klein B. A major role for Mcl-1 antiapoptotic protein in the IL-6-induced survival of human myeloma cells. Oncogene. 2003;22(19):2950–9.
Wegiel B, Bjartell A, Culig Z, Persson JL. Interleukin-6 activates PI3K/Akt pathway and regulates cyclin A1 to promote prostate cancer cell survival. Int J Cancer. 2008;122(7):1521–9.
Zou M, Zhang X, Xu C. IL6-induced metastasis modulators p-STAT3, MMP-2 and MMP-9 are targets of 3,3′-diindolylmethane in ovarian cancer cells. Cell Oncol (Dordr). 2015;39:47–57.
Mauer J, Denson JL, Brüning JC. Versatile functions for IL-6 in metabolism and cancer. Trends Immunol. 2015;36(2):92–101.
White JP, Puppa MJ, Gao S, Sato S, Welle SL, Carson JA. Muscle mTORC1 suppression by IL-6 during cancer cachexia: a role for AMPK. Am J Physiol Endocrinol Metab. 2013;304(10):E1042–52.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Herman JG. Hypermethylation of tumor suppressor genes in cancer. Semin Cancer Biol. 1999;9:359–67.
Hodge DR, Peng B, Cherry JC, Hurt EM, Fox SD, Kelley JA, et al. Interleukin 6 supports the maintenance of p53 tumor suppressor gene promoter methylation. Cancer Res. 2005;65(11):4673–82.
Liu CC, Lin JH, Hsu TW, Su K, Li AF, Hsu HS, et al. IL-6 enriched lung cancer stem-like cell population by inhibition of cell cycle regulators via DNMT1 upregulation. Int J Cancer. 2015;136(3):547–59.
Hodge DR, Cho E, Copeland TD, Guszczynski T, Yang E, Seth AK, et al. IL-6 enhances the nuclear translocation of DNA cytosine-5-methyltransferase 1 (DNMT1) via phosphorylation of the nuclear localization sequence by the AKT kinase. Cancer Genomics Proteomics. 2007;4(6):387–98.
Gasche JA, Hoffmann J, Boland CR, Goel A. Interleukin-6 promotes tumorigenesis by altering DNA methylation in oral cancer cells. Int J Cancer. 2011;129(5):1053–63.
Nevins JR. The Rb/E2F pathway and cancer. Hum Mol Genet. 2001;10(7):699–703.
Giacinti C, Giordano A. RB and cell cycle progression. Oncogene. 2006;25(38):5220–7.
Urashima M, Ogata A, Chauhan D, Vidriales MB, Teoh G, Hoshi Y, et al. Interleukin-6 promotes multiple myeloma cell growth via phosphorylation of retinoblastoma protein. Blood. 1996;88(6):2219–27.
Zhuang L, Lee CS, Scolyer RA, McCarthy SW, Zhang XD, Thompson JF, et al. Mcl-1, Bcl-XL and Stat3 expression are associated with progression of melanoma whereas Bcl-2, AP-2 and MITF levels decrease during progression of melanoma. Mod Pathol. 2007;20(4):416–26.
Borhani N, Manoochehri M, Saleh-Gargari S, Ghaffari Novin M, Mansouri A, Omrani MD. Decreased expression of proapoptotic genes caspase-8- and BCL2-associated agonist of cell death (BAD) in ovarian cancer. Clin Ovarian Other Gynecol Cancer. 2014;7:8–23.
Isomoto H, Kobayashi S, Werneburg NW, Bronk SF, Guicciardi ME, Frank DA, et al. Interleukin 6 upregulates myeloid cell leukemia-1 expression through a STAT3 pathway in cholangiocarcinoma cells. Hepatology. 2005;42:1329–38.
Wei LH, Kuo ML, Chen CA, Chou CH, Cheng WF, Chang MC, et al. The anti-apoptotic role of interleukin-6 in human cervical cancer is mediated by up-regulation of Mcl-1 through a PI 3-K/Akt pathway. Oncogene. 2001;20(41):5799–809.
Jee SH, Chiu HC, Tsai TF, Tsai WL, Liao YH, Chu CY, et al. The phosphotidyl inositol 3-kinase/Akt signal pathway is involved in interleukin-6-mediated Mcl-1 upregulation and anti-apoptosis activity in basal cell carcinoma cells. J Investig Dermatol. 2002;119(5):1121–7.
Leu CM, Wong FH, Chang C, Huang SF, Hu CP. Interleukin-6 acts as an antiapoptotic factor in human esophageal carcinoma cells through the activation of both STAT3 and mitogen-activated protein kinase pathways. Oncogene. 2003;22(49):7809–18.
Garcia-Tuñón I, Ricote M, Ruiz A, Fraile B, Paniagua R, Royuela M. IL-6, its receptors and its relationship with bcl-2 and bax proteins in infiltrating and in situ human breast carcinoma. Histopathology. 2005;47(1):82–9.
Waxman AB, Kolliputi N. IL-6 protects against hyperoxia-induced mitochondrial damage via Bcl-2-induced Bak interactions with mitofusions. Am J Respir Cell Mol Biol. 2009;41(4):385–96.
Brooks C, Wei Q, Feng L, Dong G, Tao Y, Mei L, et al. Bak regulates mitochondrial morphology and pathology during apoptosis by interacting with mitofusins. Proc Natl Acad Sci U S A. 2007;104:11649–54.
Gritsko T, Williams A, Turkson J, Kaneko S, Bowman T, Huang M, et al. Persistent activation of stat3 signaling induces survivin gene expression and confers resistance to apoptosis in human breast cancer cells. Clin Cancer Res. 2006;12(1):11–9.
Johnson C, Han YY, Nathan H, McCarra J, Alpini G, Meng F. Interleukin-6 and its receptor, key players in hepatobiliary inflammation and cancer. Transl Gastrointest Cancer. 2012;1:58–70.
Chen M-F, Lin P-Y, Wu C-F, Chen W-C, Wu C-T. IL-6 expression regulates tumorigenicity and correlates with prognosis in bladder cancer. PLoS ONE. 2013;8(4):e61901.
Hsu JH, Shi Y, Frost P, Yan H, Hoang B, Sharma S, et al. Interleukin-6 activates phosphoinositol-3′ kinase in multiple myeloma tumor cells by signaling through RAS-dependent, and separately, through p85-dependent pathways. Oncogene. 2004;23(19):3368–75.
Hideshima T, Nakamura N, Chauhan D, Anderson KC. Biologic sequelae of interleukin-6 induced PI3-K/Akt signaling in multiple myeloma. Oncogene. 2001;20(42):5991–6000.
Borsellino N, Belldegrun A, Bonavida B. Endogenous interleukin 6 is a resistance factor for cis-diamminedichloroplatinum and etoposide-mediated cytotoxicity of human prostate carcinoma cell lines. Cancer Res. 1995;55:4633–9.
Suchi K, Fujiwara H, Okamura S, Okamura H, Umehara S, Todo M, et al. Overexpression of interleukin-6 suppresses cisplatin-induced cytotoxicity in esophageal squamous cell carcinoma cells. Anticancer Res. 2011;31(1):67–75.
Hirano T, Ishihara K, Hibi M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene. 2000;19:2548–56.
Niu G, Wright KL, Ma Y, Wright GM, Huang M, Irby R, et al. Role of Stat3 in regulating p53 expression and function. Mol Cell Biol. 2005;25(17):7432–40.
Patchen ML, MacVittie TJ, Williams JL, Schwartz GN, Souza LM. Administration of interleukin-6 stimulates multilineage hematopoiesis and accelerates recovery from radiation-induced hematopoietic depression. Blood. 1991;77(3):472–80.
Kim SY, Kang JW, Song X, Kim BK, Yoo YD, Kwon YT, et al. Role of the IL-6–JAK1–STAT3–Oct-4 pathway in the conversion of non-stem cancer cells into cancer stem-like cells. Cell Signal. 2013;25(4):961–9.
Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, Kuperwasser C, et al. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. 2011;146(4):633–44.
Landskron G, De la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA. Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res. 2014;2014:149185.
Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 2009;30(7):1073–81.
Schiechl G, Bauer B, Fuss I, Lang SA, Moser C, Ruemmele P, et al. Tumor development in murine ulcerative colitis depends on MyD88 signaling of colonic F4/80+CD11bhighGr1low macrophages. J Clin Invest. 2011;121(5):1692–708.
Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441(7092):431–6.
Kesanakurti D, Chetty C, Maddirela DR, Gujrati M, Rao JS. Essential role of cooperative NF-kB and Stat3 recruitment to ICAM-1 intronic consensus elements in the regulation of radiation-induced invasion and migration in glioma. Oncogene. 2013;32(43):10.
Warburg O. On the origin of cancer cells. Science. 1956;123:309–14.
Bhatt AN, Chauhan A, Khanna S, Rai Y, Singh S, Soni R, et al. Transient elevation of glycolysis confers radio-resistance by facilitating DNA repair in cells. BMC Cancer. 2015;15:335.
Ando M, Uehara I, Kogure K, Asano Y, Nakajima W, Abe Y, et al. Interleukin 6 enhances glycolysis through expression of the glycolytic enzymes hexokinase 2 and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3. J Nippon Med Sch. 2010;77(2):97–105.
Kawauchi K, Araki K, Tobiume K, Tanaka N. p53 regulates glucose metabolism through an IKK–NF-kappaB pathway and inhibits cell transformation. Nat Cell Biol. 2008;10:611–8.
Carey AL, Steinberg GR, Macaulay SL, Thomas WG, Holmes AG, Ramm G, et al. IL-6 increases insulin-stimulated glucose disposal in humans and glucose uptake and fatty acid oxidation in vitro via AMP activated protein kinase. Diabetes. 2006;55:2688–97.
Ghosh S, Ashcraft K. An IL-6 link between obesity and cancer. Front Biosci. 2013;5:461–78.
Sarkar PD, Gopinath A, Skaria LK. Association of serum interleukin-6 and glycolysis in sickle cell disease patients. Med J DPU. 2014;7(3):317–20.
Valencia T, Kim JY, Abu-Baker S, Moscat-Pardos J, Ahn CS, Reina-Campos M, et al. Metabolic reprogramming of stromal fibroblasts through p62-mTORC1 signaling promotes inflammation and tumorigenesis. Cancer Cell. 2014;26(1):121–35.
Mercurio F, Manning AM. NF-kappaB as a primary regulator of the stress response. Oncogene. 1999;18(45):6163–71.
Kratsovnik E, Bromberg Y, Sperling O, Zoref-Shani E. Oxidative stress activates transcription factor NF-kB-mediated protective signaling in primary rat neuronal cultures. J Mol Neurosci. 2005;26(1):27–32.
Libermann TA, Baltimore D. Activation of interleukin-6 gene expression through the NF-kappa B transcription factor. Mol Cell Biol. 1990;10(5):2327–34.
Wang L, Walia B, Evans J, Gewirtz AT, Merlin D, Sitaraman SV. IL-6 induces NF-kappa B activation in the intestinal epithelia. J Immunol. 2003;171(6):3194–201.
Grivennikov S, Karin M. Dangerous liaisons: STAT3 and NF-kB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010;21(1):11–9.
Brown CO, Salem K, Wagner BA, Bera S, Singh N, Tiwari A, et al. Interleukin-6 counteracts therapy-induced cellular oxidative stress in multiple myeloma by up-regulating manganese superoxide dismutase. Biochem J. 2012;444(3):515–27.
Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med. 2010;49(11):1603–16.
Jia Y, Zhou F, Deng P, Fan Q, Li C, Liu Y, et al. Interleukin 6 protects H(2)O(2)-induced cardiomyocytes injury through upregulation of prohibitin via STAT3 phosphorylation. Cell Biochem Funct. 2012;30(5):426–31.
Craig R, Larkin A, Mingo AM, Thuerauf DJ, Andrews C, McDonough PM, et al. p38 MAPK and NF-kappa B collaborate to induce interleukin-6 gene expression and release. Evidence for a cytoprotective autocrine signaling pathway in a cardiac myocyte model system. J Biol Chem. 2000;275(31):23814–24.
Kalluri R, Weinberg RA. The basics of epithelial–mesenchymal transition. J Clin Invest. 2009;119(6):1420–8.
Ricciardi M, Zanotto M, Malpeli G, Bassi G, Perbellini O, Chilosi M, et al. Epithelial-to-mesenchymal transition (EMT) induced by inflammatory priming elicits mesenchymal stromal cell-like immune-modulatory properties in cancer cells. Br J Cancer. 2015;112(6):1067–75.
Tawara K, Oxford JT, Jorcyk CL. Clinical significance of interleukin (IL)-6 in cancer metastasis to bone: potential of anti-IL-6 therapies. Cancer Manag Res. 2011;3:177–89.
Helbig G, Christopherson 2nd KW, Bhat-Nakshatri P, Kumar S, Kishimoto H, Miller KD, et al. NF-kappaB promotes breast cancer cell migration and metastasis by inducing the expression of the chemokine receptor CXCR4. J Biol Chem. 2003;278(24):21631–8.
Sansone P, Storci G, Tavolari S, Guarnieri T, Giovannini C, Taffurelli M, et al. IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J Clin Invest. 2007;117(12):3988–4002.
Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86(3):353–64.
Kujawski M, Kortylewski M, Lee H, Herrmann A, Kay H, Yu H. Stat3 mediates myeloid cell dependent tumor angiogenesis in mice. J Clin Invest. 2008;118(10):3367–77.
Houde C, Li Y, Song L, Barton K, Zhang Q, Godwin J, et al. Overexpression of the NOTCH ligand JAG2 in malignant plasma cells from multiple myeloma patients and cell lines. Blood. 2004;104(12):3697–704.
Yun UJ, Park SE, Jo YS, Kim J, Shin DY. DNA damage induces the IL-6/STAT3 signaling pathway, which has anti-senescence and growth-promoting functions in human tumors. Cancer Lett. 2012;323(2):155–60.
Rodier F, Coppé JP, Patil CK, Hoeijmakers WA, Muñoz DP, Raza SR, et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11:973–9.
Gilbert LA, Hemann MT. DNA damage-mediated induction of a chemoresistant niche. Cell. 2010;143:355–66.
Wu ZH, Wong ET, Shi Y, Niu J, Chen Z, Miyamoto S, et al. ATM- and NEMO-dependent ELKS ubiquitination coordinates TAK1-mediated IKK activation in response to genotoxic stress. Mol Cell. 2010;40:75–86.
Tachibana S, Zhang X, Ito K, Ota Y, Cameron AM, Williams GM, et al. Interleukin-6 is required for cell cycle arrest and activation of DNA repair enzymes after partial hepatectomy in mice. Cell Biosci. 2014;4(1):6.
Chang L, Guo R, Huang Q, Yen Y. Chromosomal instability triggered by Rrm2b loss leads to IL-6 secretion and plasmacytic neoplasms. Cell Rep. 2013;3(5):1389–97.
Deorukhkar A, Krishnan S. Targeting inflammatory pathways for tumor radiosensitization. Biochem Pharmacol. 2010;80(12):1904–14.
Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–99.
Woods Ignatoski KM, Friedman J, Escara-Wilke J, Zhang X, Daignault S, Dunn RL, et al. Change in markers of bone metabolism with chemotherapy for advanced prostate cancer: interleukin-6 response is a potential early indicator of response to therapy. J Interferon Cytokine Res. 2008;29(2):105–12.
Conze D, Weiss L, Regen PS, Bhushan A, Weaver D, Johnson P, et al. Autocrine production of interleukin 6 causes multidrug resistance in breast cancer cells. Cancer Res. 2001;61:8851–8.
Patel SAA, Bhambra U, Charalambous MP, David RM, Edwards RJ, Lightfoot T, et al. Interleukin-6 mediated upregulation of CYP1B1 and CYP2E1 in colorectal cancer involves DNA methylation, miR27b and STAT3. Br J Cancer. 2014;111(12):2287–96.
Bochet L, Meulle A, Imbert S, Salles B, Valet P, Muller C. Cancer-associated adipocytes promotes breast tumor radioresistance. Biochem Biophys Res Commun. 2011;1:102–6.
Xu H, Lai W, Zhang Y, Liu L, Luo X, Zeng Y, et al. Tumor-associated macrophage-derived IL-6 and IL-8 enhance invasive activity of LoVo cells induced by PRL-3 in a KCNN4 channel-dependent manner. BMC Cancer. 2014;14:330.
Touboul C, Lis R, Al Farsi H, Raynoud CM, Warfa M, Althawadi H, et al. Mesenchymal stem cells enhances ovarian cancer cell infiltration through IL-6 secretion in an amniochorionic membrane based 3D model. J Transl Med. 2013;11:28.
Eicthen A, Su J, Adler A, Zhang L, Loffe E, Parveen AA, et al. Resistance to anti-VEGF therapy mediated by autocrine IL-6/STAT3 signaling and overcome by IL-6 blockade. Cancer Res. 2016;76:2327–39.
Korkaya H, Kim GI, Davis A, Malik F, Henry NL, Ithimakin S, et al. Activation of IL-6 inflammatory loop mediates transtuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol Cell. 2012;47(4):570–84.
Chen F, Zhuang X, Lin L, Yu P, Wang Y, Shi Y, et al. New horizons in tumor microenvironment biology: challenges and opportunities. BMC Med. 2015;13:45.
Kontzias A, Kotlyar A, Laurence A, Changelian P, O’Shea JJ. Jakinibs: a new class of kinase inhibitors in cancer and autoimmune disease. Curr Opin Pharmacol. 2012;12(4):464–70.
Karkera J, Steiner H, Li W, Skradski V, Moser PL, Riethdorf S, et al. The anti-interleukin-6 antibody siltuximab down-regulates genes implicated in tumorigenesis in prostate cancer patients from a phase I study. Prostate. 2011;71(13):1455–65.
Chen R, Chen B. Siltuximab (CNTO 328): a promising option for human malignancies. Drug Des Devel Ther. 2015;9:3455–8.
Chou CH, Wei LH, Kuo ML, Huang YJ, Lai KP, Chen CA, et al. Up-regulation of interleukin-6 in human ovarian cancer cell via a Gi/PI3K-Akt/NF-kappaB pathway by lysophosphatidic acid, an ovarian cancer-activating factor. Carcinogenesis. 2005;26(1):45–52.
Song L, Smith MA, Doshi P, Sasser K, Fulp W, Altiok S, et al. Antitumor efficacy of the anti-interleukin-6 (IL-6) antibody siltuximab in mouse xenograft models of lung cancer. J Thorac Oncol. 2014;9(7):974–82.
Bagcchi S. Siltuximab in transplant-ineligible patients with myeloma. Lancet Oncol. 2014;15(8):e309.
Emilie D, Wijdenes J, Gisselbrecht C, Jarrousse B, Billaud E, Blay JY, et al. Administration of an anti-interleukin-6 monoclonal antibody to patients with acquired immunodeficiency syndrome and lymphoma: effect on lymphoma growth and on B clinical symptoms. Blood. 1994;84:2472–9.
Asbagh LA, Uzunoglu S, Cal C. Zoledronic acid effects interleukin-6 expression in hormone findependent prostate cancer cell lines. Int Braz J Urol. 2008;34(3):355–64.
Gnant M. Zoledronic acid in breast cancer: latest findings and interpretations. Ther Adv Med Oncol. 2011;3(6):293–301.
Kurebayashi J, Yamamoto S, Otsuki T, Sonoo H. Medroxyprogesterone acetate inhibits interleukin 6 secretion from KPL-4 human breast cancer cells both in vitro and in vivo: a possible mechanism of the anticachectic effect. Br J Cancer. 1999;79(3/4):631–6.
Shinriki S, Jono H, Ota K, Ueda M, Kudo M, Ota T, et al. Humanized anti-interleukin-6 receptor antibody suppresses tumor angiogenesis and in vivo growth of human oral squamous cell carcinoma. Clin Cancer Res. 2009;15(17):5426–34.
Ando K, Takahashi F, Kato M, Kaneko N, Doi T, Ohe Y, et al. Tocilizumab, a proposed therapy for the cachexia of interleukin6-expressing lung cancer. PLoS One. 2014;9(7):e102436.
Moreau P, Harousseau JL, Wijdenes J, Morineau N, Milpied N, Bataille R. A combination of anti-interleukin 6 murine monoclonal antibody with dexamethasone and high-dose melphalan induces high complete response rates in advanced multiple myeloma. Br J Haematol. 2000;109:661–4.
Tai YT, Anderson KC. Antibody-based therapies in multiple myeloma. Bone Marrow Res. 2011;2011:924058.
Isobe A, Sawada K, Kinose Y, Ohyagi-Hara C, Nakatsuka E, Makino H, et al. Interleukin 6 receptor is an independent prognostic factor and a potential therapeutic target of ovarian cancer. PLoS ONE. 2015;10(2):e0118080.
Jiang XP, Yang DC, Elliott RL, Head JF. Down-regulation of expression of interleukin-6 and its receptor results in growth inhibition of MCF-7 breast cancer cells. Anticancer Res. 2011;31:2899–906.
Sun Y, Moretti L, Giacalone NJ, Schleicher S, Speirs CK, Carbone DP, et al. Inhibition of JAK2 signaling by TG101209 enhances radiotherapy in lung cancer models. J Thorac Oncol. 2011;6(4):699–706.
Ramakrishnan V, Kimlinger T, Haug J, Timm M, Wellik L, Halling T, et al. TG101209, a novel JAK2 inhibitor, has significant in-vitro activity in multiple myeloma and displays preferential cytotoxicity for CD45+ myeloma cells. Am J Hematol. 2010;85(9):675–86.
Kobayashi A, Tanizaki Y, Kimura A, Ishida Y, Nosaka M, Toujima S, et al. AG490, a Jak2 inhibitor, suppressed the progression of murine ovarian cancer. Eur J Pharmacol. 2015;766:63–75.
Eghtedar A, Verstovsek S, Estrov Z, Burger J, Cortes J, Bivins C, et al. Phase II study of the JAK kinase inhibitor ruxolitinib in patients with refractory leukemias, including post myeloproliferative neoplasms (MPN) acute myeloid leukemia (AML). Blood. 2012;119(20):4614–8.
Seavey MM, Lu LD, Stump KL, Wallace NH, Hockeimer W, O’Kane TM, et al. Therapeutic efficacy of CEP-33779, a novel selective JAK2 inhibitor, in a mouse model of colitis-induced colorectal cancer. Mol Cancer Ther. 2012;11(4):984–93.
Hart S, Goh KC, Novotny-Diermayr V, Tan YC, Madan B, Amalini C, et al. Pacritinib (SB1518), a JAK2/FLT3 inhibitor for the treatment of acute myeloid leukemia. Blood Cancer J. 2011;1(11):e44.
Yang F, Brown C, Buettner R, Hedvat M, Starr R, Scuto A, et al. Sorafenib induces growth arrest and apoptosis of human glioblastoma cells through the dephosphorylation of signal transducers and activators of transcription 3. Mol Cancer Ther. 2010;9(4):953–62.
Duan Z, Bradner JE, Greenberg E, Levine R, Foster R, Mahoney J, et al. SD-1029 inhibits signal transducer and activator of transcription 3 nuclear translocation. Clin Cancer Res. 2006;12(22):6844–52.
Shi X, Franko B, Frantz C, Amin HM, Lai R, et al. JSI-124 (cucurbitacin I) inhibits Janus kinase-3/signal transducer and activator of transcription-3 signalling, downregulates nucleophosmin-anaplastic lymphoma kinase (ALK), and induces apoptosis in ALK-positive anaplastic large cell lymphoma cells. Br J Haematol. 2006;135(1):26–32.
Su Y, Li G, Zhang X, Gu J, Zhang C, Tian Z, et al. JSI-124 inhibits glioblastoma multiforme cell proliferation through G2/M cell cycle arrest and apoptosis augment. Cancer Biol Ther. 2008;7(8):1243–9.
Blaskovich MA, Sun J, Cantor A, Turkson J, Jove R, Sebti SM. Discovery of JSI-124 (cucurbitacin I), a selective Janus kinase/signal transducer and activator of transcription 3 signaling pathway inhibitor with potent antitumor activity against human and murine cancer cells in mice. Cancer Res. 2003;63(6):1270–9.
Gholam P. The role of sorafenib in hepatocellular carcinoma. Clin Adv Hematol Oncol. 2015;13(4):232–4.
Moreno-Aspitia A. Clinical overview of sorafenib in breast cancer. Future Oncol. 2010;6(5):655–63.
Jäger D, Ma JH, Mardiak J, Ye DW, Korbenfeld E, Zemanova M, et al. Sorafenib treatment of advanced renal cell carcinoma patients in daily practice: the large international PREDICT Study. Clin Genitourin Cancer. 2015;13(2):156–64.e1.
Moreira RB, Peixoto RD, de Sousa Cruz MR. Clinical response to sorafenib in a patient with metastatic colorectal cancer and FLT3 amplification. Case Rep Oncol. 2015;8(1):83–7.
Tanaka T, Narazaki M, Kishimoto T. Anti-interleukin-6 receptor antibody, tocilizumab, for the treatment of autoimmune diseases. FEBS Lett. 2011;585(23):3699–709.
Trikha M, Corringham R, Klein B, Rossi J-F. Targeted anti-interleukin-6 monoclonal antibody therapy for cancer: a review of the rationale and clinical evidence. Clin Cancer Res. 2003;9(13):4653–65.
Coward J, Kulbe H, Chakravarty P, Leader D, Vassileva V, Leinster DA, et al. Interleukin-6 as a therapeutic target in human ovarian cancer. Clin Cancer Res. 2011;17(18):6083–96.
Guan M, Zhou YP, Sun JL, Chen SC. Adverse events of monoclonal antibodies used for cancer therapy. Biomed Res Int. 2015;2015:428169.
Chen MF, Hsieh CC, Chen WC, Lai CH. Role of interleukin-6 in the radiation response of liver tumors. Int J Radiat Oncol Biol Phys. 2012;84(5):e621–30.
Judd LM, Menheniott TR, Ling H, Jackson CB, Howlett M, Kalantzis A, et al. Inhibition of the JAK2/STAT3 pathway reduces gastric cancer growth in vitro and in vivo. PLoS One. 2014;9(5):e95993.
Huang C, Yang G, Jiang T, Huang K, Cao J, Qiu Z. Effects of IL-6 and AG490 on regulation of Stat3 signaling pathway and invasion of human pancreatic cancer cells in vitro. J Exp Clin Cancer Res. 2010;29(1):51.
Xiong A, Yang Z, Shen Y, Zhou J, Shen Q. Transcription factor STAT3 as a novel molecular target for cancer prevention. Cancers. 2014;6(2):926–57.
Ishdorj G, Johnston JB, Gibson SB. Inhibition of constitutive activation of STAT3 by curcurbitacin-I (JSI-124) sensitized human B-leukemia cells to apoptosis. Mol Cancer Ther. 2010;9(12):3302–14.
Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol. 2006;13(11):1235–42.
Yu X, He L, Cao P, Yu Q. Eriocalyxin B inhibits STAT3 signaling by covalently targeting STAT3 and blocking phosphorylation and activation of STAT3. PLoS ONE. 2015;10(5):e0128406.
Jing N, Li Y, Xiong W, Sha W, Jing L, Tweardy DJ. G-quartet oligonucleotides: a new class of signal transducer and activator of transcription 3 inhibitors that suppresses growth of prostate and breast tumors through induction of apoptosis. Cancer Res. 2004;64(18):6603–9.
Jing N, Zhu Q, Yuan P, Li Y, Mao L, Tweardy DJ. Targeting signal transducer and activator of transcription 3 with G-quartet oligonucleotides: a potential novel therapy for head and neck cancer. Mol Cancer Ther. 2006;5(2):279–86.
Yue P, Turkson J. Targeting STAT3 in cancer: how successful are we? Expert Opin Investig Drugs. 2009;18(1):45–56.
Chen CL, Loy A, Cen L, Chan C, Hsieh FC, Cheng G, et al. Signal transducer and activator of transcription 3 is involved in cell growth and survival of human rhabdomyosarcoma and osteosarcoma cells. BMC Cancer. 2007;7:111.
Song H, Wang R, Wang S, Lin J. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc Natl Acad Sci U S A. 2005;102(13):4700–5.
Weidler M, Rether J, Anke T, Erkel G. Inhibition of interleukin-6 signaling by galiellalactone. FEBS Lett. 2000;484(1):1–6.
Acknowledgments
NK thanks the Defence Research and Development Organization, Government of India, for fellowship support. We acknowledge Director INMAS for constant support and encouragement.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kumari, N., Dwarakanath, B.S., Das, A. et al. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumor Biol. 37, 11553–11572 (2016). https://doi.org/10.1007/s13277-016-5098-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-016-5098-7