
Abstract
As a pleiotropic cytokine, interleukin (IL)-6 plays a key role in mediating the host’s immune defense. Multiple cell types are involved in IL-6 production, including macrophages, dendritic cells, fibroblasts, mast cells, vascular endothelial cells, monocytes, dendritic cells, T cells, B cells, etc. Furthermore, stromal cells, immune cells that infiltrate tumors, and cancer cells present in the tumor microenvironment are also in charge of producing IL-6. Although the blood levels of IL-6 are barely noticeable at physiological levels (1–5 pg/ml), they have been observed to increase by more than 100,000 times during an immediate inflammatory reaction. Elevated intensities of IL-6 have been identified in almost all human cancers. Overexpression of IL-6 in the tumor microenvironment stimulates carcinogenesis by controlling all the pathways that lead to cancer. There are two categories of interleukin-6 receptors: the first one comprises the cell membrane-bound receptor of IL-6 (IL-6R). Upon binding with IL-6R, IL-6 forms a complex with glycoprotein 130 (gp130) (a signal transducer), and, thus, the classical signaling pathway that exhibits anti-inflammatory activity gets activated. The second category comprises a soluble derivative of the interleukin-6 receptor, i.e., soluble IL-6R (sIL-6R). Upon IL-6 binding to sIL-6R, a membrane-associated signal transducer (gp130) complex is formed and trans-signaling gets activated. Signaling pathways, for instance, Ras/Raf/mitogen-activated protein kinase (MAPK), Janus kinase/signal transducer and activator of transcription (JAK/STAT), phosphatidylinositol-3 kinase (PI3K), or Src/yes-associated protein (Src/YAP), exhibit inflammatory activity, as a result of which they get activated. Almost all human malignancies are related to higher IL-6 levels in the blood. In this chapter we have summarized the function of interleukin-6 and its associated signaling pathways that promote cancer progression via various processes such as angiogenesis and increased expression of antiapoptotic genes known to promote cancer. Moreover, we have discussed the potential agents targeting IL-6R/IL-6/JAK/STAT3 that restores the anti-tumor activity by inhibiting IL-6/JAK/STAT3 axis.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Interleukin-6
- Glycoprotein 130
- mbIL-6Rα
- sIL-6R
- IL-6/JAK/STAT3 signaling
- ADAM17
- Tumorigenesis
- Tumor microenvironment
- Angiogenesis
- Metastasis
- Invasiveness
- Chronic inflammation
5.1 Introduction
Interleukins are a type of cytokines, which are cellular messengers with a low molecular weight of about 15–20 kDa, produced by both immune and nonimmune cells in response to pathogens and other antigens that mediate communication between cells as they bind to their receptors and trigger a cellular response in a target cell (Schafer and Brugge 2007; Gandhi et al. 2016; Takeuchi and Akira 2010). In 1979, the term “interleukin” was initially coined and used for several proteins secreted by leukocytes, which primarily act as a mediator between leukocytes, and were thus named “interleukins,” meaning “between leukocytes.” It is known that IL is also secreted by a variety of body cells (Brocker et al. 2010; Mir 2015a, b). The human genome encodes more than 50 different interleukins; they vary significantly in terms of structure and tend to overlap when it comes to their functions. Interleukins play a part in the differentiation, proliferation, and activation of immune cells (T and B lymphocytes) during inflammatory and immune responses. They exhibit properties of both anti- and pro-inflammatory cytokines (Scheller et al. 2011; Mir 2015a, b, c). Interleukin signaling mainly occurs through autocrine and paracrine mechanisms. Interleukins are not stockpiled within cells; rather, in response to a stimulus, they are rapidly secreted as they move toward their intended cell targets and elicit many cellular responses by binding to high-affinity receptor molecules on the cell surface. This interaction initiates a series of signal transduction pathways within the target cell that eventually change the cellular response (Vaillant and Qurie 2021).
A family of signaling molecules known as the cytokine family of interleukin-6 (IL-6)/glycoprotein 130 (gp130) came about as a consequence of infections and tissue injuries (Tanaka et al. 2014; Rose-John et al. 2015). IL-6 family members exhibit functional pleiotropy as well as redundancy. In their receptor complex, they have a core structure in common (Taga and Kishimoto 1997) and a signal transducer known as gp130 that performs a variety of functions in the body. Moreover, several molecules, such as tyrosine kinases of the JAK family and a STAT family member that communicates directly with the cytoplasmic domains of these receptors, have been identified. Amongst the members of this family, IL-6 deregulation is responsible for many diseases. Inhibiting IL-6 signaling is extremely effective against diseases like cytokine release syndrome, Castleman disease, and rheumatoid arthritis (RA). The axis of IL-6/IL-6R is known to contribute to the growth of a number of autoimmune disorders and cancer as well (Hirano et al. 1988; Hodge et al. 2005; Hirano et al. 1987; Schafer and Brugge 2007). Undoubtedly, inhibitors of IL-6, IL-6R, or JAK family proteins are useful in the treatment of various immunological conditions. Several cell types are linked to the secretion of the cytokine interleukin-6, including keratinocytes, mesangial cells, fibroblasts, mast cells, macrophages, vascular endothelial cells, monocytes, dendritic cells, B cells, and T cells (Mir 2015a, b, c). Furthermore, stromal cells, tumor-infiltrating immune cells, and the tumor cells located in the tumor niche are also responsible for IL-6 production (Fig. 5.1).

The major cell types linked to the secretion of IL-6
As a pleiotropic cytokine, interleukin-6 has many different names depending on the type of biological activity it carries out. It was discovered nearly 50 years ago in 1973 and was originally identified as a T-cell-derived soluble mediator essential for B cells to produce antibodies (Kishimoto et al. 1978). IL-6 was first identified as a B-cell differentiation factor or a B-cell stimulatory factor 2 (BSF-2) (Howard et al. 1982). It was formerly referred to as interferon-2 (IFN-2) due to its antiviral interferon-like action (Zilberstein et al. 1986). Because of its capacity to encourage the generation of acute-phase proteins like fibrinogen, serum amyloid A (SAA), and C-reactive protein (CRP), interleukin-6 became characterized as a hepatocyte-stimulating factor shortly after (Gauldie et al. 1987). Furthermore, it was reported to be identical to MGI-2A, also known as myeloid blood cell differentiation-inducing protein, macrophage and granulocyte inducer, type 2A, reflecting the cytokine's myeloid-differentiating capabilities (Shabo et al. 1988). Lastly, in 1988, this multifunctional cytokine was finally named “IL-6” (Hirano 2014).
IL-6 influences various metabolic and immune system functions, such as inflammation, generation of the acute-phase protein (CRP) in hepatocytes, antigen-specific immune responses, apoptosis, hematopoiesis, mitochondrial activity, insulin resistance (for instance, mice develop glucose intolerance and insulin resistance in the absence of IL-6, and, at the same time, liver inflammation is also seen) (Wunderlich et al. 2010), cell differentiation, vascular disease, and lipid metabolism (Ji et al. 2011), and it has also been reported to be associated with the neuroendocrine and neuropsychological systems (Rohleder et al. 2012; Rose-John et al. 2015). Moreover, as mentioned, it contributes to the progression of various disease types such as cancers and autoimmune diseases (Hirano et al. 1987, 1988; Hodge et al. 2005; Schafer and Brugge 2007).
In this chapter, we will discuss the function of interleukin-6 (IL-6) and its signaling, how it helps in regulating tumor progression, and learn exactly how agents targeting the IL-6/IL-6R/JAK/STAT3 or JAK/STAT signaling inhibitors can be therapeutic for various cancers.
5.2 IL-6 and Its Receptor Structure
IL-6 belongs to the hematopoietic cytokine family and is a glycosylated protein with a molecular weight of about 21–26 kDa (Fig. 5.2). In 1986, the IL-6 gene was cloned that encoded IL-6 precursor protein containing 212 amino acids that comprised of a mature segment of 184 amino acids and a signal sequence of 28 amino acids (Hirano et al. 1986). In 1986, the IL-6 gene was cloned that encoded IL-6 precursor protein containing 212 amino acids that comprised of a mature segment of 184 amino acids and a signal sequence of 28 amino acids (Simpson et al. 1997). The mature segment of IL-6 consists of long antiparallel α-helices A, B, C, and D connected through various loops. These helices are structured in such a way that they have an up-up-down-down geometry (Scheller et al. 2011), resulting in the formation of three binding sites for epitopes, namely, sites I, II, and III, at which binding of the receptor occurs (Brakenhoff et al. 1990, 1994; Ehlers et al. 1994).

Helix D and AB loop’s C-terminal residues form site I, which serves as a requisite site for a nonsignaling component of the interluekin-6 receptor (Savino et al. 1994a, b; Paonessa et al. 1995). Site II is produced by the dual association of IL-6 and IL-6R with the signal transducer glycoprotein (gp130), which is located on helices A and C and in the middle of domains 2 and 3 (Yawata et al. 1993; Savino et al. 1994a, b; Boulanger et al. 2003). Site III is composed of residues found at the C-terminal of helix D and at the N-terminal of the AB loop. This location is known as domain 1 (D1) because it interacts with immunoglobulin G (IgG) to produce a reliable signaling complex, much like the gp130 (glycoprotein 130) domain does (Yawata et al. 1993).
The IL-6 receptor is a member of the family of cytokine receptors that comprise of IL-6 binding chain domain and a signal transducing chain (gp180) both of these proteins have a Trp-Ser-X-Trp-Ser motif and are cytokine receptors. Two isoforms of the IL-6-binding chain have been found: one is a transmembrane receptor (mbIL-6R) with a molecular weight of 80 kDa and the other is soluble IL-6R (sIL-6R) of molecular weight 50–55 kDa. The interleukin-6 receptor consists of an 80-kDa IL-6-binding protein (α-chain) (CD126), with a cytoplasmic domain containing only 82 amino acids, and gp80, which is present only in limited cell types. IL-6 binds to low-affinity receptors by means of the residues present in the interior of domains 2 and 3, which subsequently recruit the gp130 signal transducer, a β-subunit of the interluekin-6 receptor, to create a high-affinity IL-6R complex that transmits signals and carries out signal transduction. It is worth mentioning that the cytoplasmic and the transmembrane domains of the interluekin-6 receptor are not competent for signaling, and, as such, if both these domains are removed, it will not affect IL-6 signaling (Yamasaki et al. 1988; Taga et al. 1989). Interleukin-6 is not able to transduce signals alone. The IL-6/IL-6R complex is associated with the signal transducer glycoprotein 130 (gp130), also known as CD130, and IL-6Rβ, a glycoprotein of molecular weight of about 130 kDa, has about 277 amino acids in its cytoplasmic domain containing several motifs and sites for phosphorylation. It is universally expressed as the IL-6-type cytokine family’s signal transducer, which comprises leukemia inhibitory factor (LIF), OSM (oncostatin M), IL-11 (interleukin-11), ciliary neurotrophic factor (CNTF) , and cardiotrophin-1 (CT-1) (Sprang and Bazan 1993). Since the gp130 subunit exclusively interacts with the complex of IL-6/IL-6R through the generated dual site II engages with gp130’s multiple CBDs ( cytokine-binding domains) D2 & D3 as well as via site III is present at D1 of gp130, neither IL-6 nor IL-6R can be activated by themselves (Hibi et al. 1990). Meanwhile, functional investigations indicate that two distinctive copies of IL-6, IL-6R, and gp130 combine to form the hexameric structure of the high-affinity IL-6/IL-6R complex. One molecule of IL-6, one molecule of IL-6R, and two molecules of gp130 are already part of a tetrameric model (Grötzinger et al. 1999; Takeuchi and Akira 2010).
5.3 IL-6 Signaling in Cancer
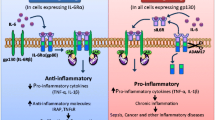
Malignant cells as well as lymphoid and nonlymphoid cells exhibit the type I cytokine receptor on their cell surfaces, and this is how IL-6 communicates. There are two categories of IL-6 receptors: the first is the cell membrane-bound IL-6 receptor (IL-6R), which has a low affinity and binds to gp130 to activate a classical signaling pathway, and the second is the soluble IL-6 receptor isoform that activates trans-signaling.
By attaching to a soluble variant of the IL-6 receptor (sIL-6R), IL-6 may be able to "trans-signal" in a way that is more convenient (sIL-6R). It first attaches to IL-6 and then to the membrane receptor b-chain (gp130), resulting in an intracellular signal. An IL-6/IL-6R or an IL-6/sIL-6R complex, formed with the membrane-associated signal transducer, triggers signal transduction. The stimulation of particular signaling, including JAK/STAT3 (signal transducer and activator of transcription 3)/IL-6, as well as other signaling pathways involved, leads to the amplification of pro-inflammatory cytokine IL-6. This condition is a hallmark of chronic inflammatory conditions, autoimmune diseases, vascular diseases, and, in a large percentage of patients, several neoplastic disorders, such as hematological malignancies, ovarian cancer, colon cancer, breast cancer, and prostate cancer (Fig. 5.3) (Mir and Mehraj 2019).

Modes of IL-6 signaling
5.4 Classical Signaling and Trans-Signaling
The two known isoforms of the interleukin-6 receptor, namely, mbIL-6R (a membrane-bound receptor) and sIL-6R (a soluble isoform), are the factors that will determine whether classical signaling or trans-signaling will occur. The proteolysis of the ectodomain of mbIL-6R by a disintegrin and metalloproteases 17 and 10 (ADAM17 and ADAM10, respectively) results in the shedding of the soluble receptors of IL-6, ADAM17, and ADAM10. These are zinc (Zn dependent metalloproteinases. sIL-6R is produced via alternative splicing of the messenger RNA (mRNA) of IL-6 and also that of IL-6R, but the latter occurs less commonly, i.e., about 1–20% (Mülberg et al. 1993; Lust et al. 1992; Riethmueller et al. 2017). Both these isoforms of IL-6R have a similar affinity toward IL-6 binding (Fig. 5.4) (Narazaki et al. 1993).

Classical signaling and trans-signaling via ADAM17-mediated cleavage of IL-6R or other events; soluble isoforms of IL-6R are produced
It is notable that an IL-6/sIL-6R complex can induce IL-6 signaling through autocrine and paracrine mechanisms in any cell type with a gp130 signal transducer, which results in activation of the trans-signaling of sIL-6R. On the other hand, only few epithelial cells, hepatocytes, and few leukocytes express cell membrane-bound IL-6R (mbIL-6R), and, as such, classical signaling of IL-6 gets activated. Classical signaling is anti-inflammatory, whereas trans-signaling stimulates pro-inflammatory processes. Both times, signaling is brought on by gp130 homodimerization.
The term "IL-6 trans-presentation," which has recently been established, was used to describe the third way of IL-6 signaling. Once dendritic cells (DCs) specifically interact with an antigen, IL-6 binds intracellularly to its receptor and moves to the cell membrane, where the membrane-anchored receptor of IL-6 interacts with the gp130 subunit of T cells when combined with transforming growth factor-beta 2 (TGF-β2) to form prime T-helper (Th)17 cells that lead to chronic inflammation and regulatory T (Treg) cell suppression by IL-27 (Qayoom et al. 2023; Narazaki et al. 1993; Heink et al. 2017). It is still not known whether this novel IL-6 signaling mechanism works in humans because this study was conducted experimentally using a murine model of multiple sclerosis of humans. gp130 dimerization is necessary for IL-6 signaling through the traditional or trans-signaling pathways because it activates the JAK protein that is linked to gp130 (Heink et al. 2017).
Trans-signaling plays an important role in the tumor microenvironment by regulating leukocyte recruitment and inducing the inflammatory activation of stromal cells that are linked to the tumor (Mehraj et al. 2021a, b, c). It is notable that alternative splicing of gp130 mRNA generates four different soluble isoforms of gp130 (sgp130). This adds to the complexity, given that when secreted sgp130 proteins interact with the IL-6/sIL-6R complex, they specifically prevent the trans-signaling facilitated by IL-6 (Baran et al. 2018). As a result of its binding, an inactive ternary complex is formed. The inhibitory activity of trans-signaling occurs at low doses and also depends on the IL-6/sIL-6R ratio because the classical signaling mediated by IL-6/mIL-6R is not affected by it (Nakajima et al. 1993; Jostock et al. 2001). Numerous downstream signaling processes occur after the formation of the IL-6/IL-6R/gp130 complex, enabling IL-6 to exhibit cell differentiation, proliferation, survival, and death, among a wide range of other biological processes. The signaling of IL-6 involves the Ras/Raf/mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase (PI3K), JAK/STAT, and Src/yes-associated protein (Src/YAP) pathways (Fig. 5.5) (Nakajima et al. 1993; Taniguchi et al. 2015, 2017).

Inhibition of trans-signaling
5.5 Signaling Pathway for IL-6/JAK/STAT3
The JAK/STAT3 signaling pathway is essential for mediating IL-6's diverse effects on tumor promotion, i.e., cancer cell survival, proliferation, invasion, metastasis, and immunity-suppressing properties (Kumari et al. 2016; Qayoom et al. 2021). The JAK/STAT (Janus kinase/signal transducer and activator of transcription) signaling pathway is highly conserved. IL-6 signaling can happen via traditional or trans-signaling routes, and the heterohexameric complex formed between IL-6R, IL-6, and gp130 is what initiates intracellular signaling. This complex activates JAKs that are linked to gp130. There are four mammalian JAKs: tyrosine kinase 2 (TYK2), JAK1, JAK2, and JAK3. There are seven members of the STAT family: STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6 (Seif et al. 2017).
The JAK Family: This consists of protein tyrosine kinases that are not receptors. JAK tyrosine kinases TYK2, JAK1, JAK2, and JAK3 are stimulated when cytokines connect to their receptors. The other members of JAK3 are uniformly present in all tissues, whereas JAK3 is restricted only to the lymphatic system, bone marrow, vascular smooth muscle cells, and endothelial cells (Rane and Reddy 1994; Takahashi and Shirasaw 1994; Lai et al. 1995; Verbsky et al. 1996). Members of the JAK family consist of seven different JAK homology domains (JH). There is the first JH, JH1, which originates from the carboxyl terminus and has roughly 250 amino acid residues. JH1 is also known as the kinase domain because it creates the kinase structural domain and phosphorylates a substrate by encoding a kinase protein. Although JH2 shares structural similarities with the kinase domain, it lacks kinase activity. It is known as the Pk domain or pseudo kinase domain and takes part in the interaction between JAK and STAT. Its major aim is to regulate the kinase domain's activity, and, by binding to the kinase domain, it can also inhibit Tyr kinase activity. JH3 and one-half of JH4 make up the Src homology 2 (SH2) domain, whereas JH4 and one of each of JH5, JH6, and JH7 make up the FERM (F for four point one protein, E for ezrin, R for radixin and M for moesin) domain. When the IL-6/IL-6R complex homodimerizes gp130, JAK1, JAK2, and/or TYK2 are selectively activated through the SH2 and FERM domains, mostly through interactions of these enzymes with gp130's Box domains 1 and 2, which are membrane-proximal areas. Due to the disruption of JH2-mediated inhibition of JH1 kinase activity caused by these contacts, reciprocal transphosphorylation that results in complete activation of JAKs takes place. After the JAK enzyme is activated in the cytoplasm of gp130, numerous tyrosine residues undergo phosphorylation. These residues later act as docking sites for proteins that start the IL-6/JAK/STAT3 signaling pathway and additional distinct signaling pathways (Frank et al. 1995; Velazquez et al. 1995; Ferrao and Lupardus 2017).
The STAT family members include 750–900 amino acid residues. The STAT structure consists of a coiled-coil domain (CCD), a linker domain (LD), an amino terminal domain, an Src homology 2 domain, a DNA-binding domain (DBD), and a transactivation domain. STAT3 is closely linked to the stimulation of tumor progression and immune suppression among the STAT protein family members (STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6). STAT3 normally resides in the cytoplasm of cells in the dormant state as a monomer. When JAK enzymes get activated, they subsequently phosphorylate multiple tyrosine residues of growth factor receptors like gp130. This places STAT3 in close proximity to active JAK enzymes, which then phosphorylate STAT3 at Tyr705. The phosphorylation of Tyr705 residues results in dimerization of the STAT3 protein in a head-to-tail manner, which is mediated by the Src homology 2 domain. The STAT3 dimer translocates from the cytoplasm to the nucleus through an importin-α–importin-β1-dependent mechanism (Chang et al. 2013a, b).
The STAT3 dimer attaches to the target genes' promoter consensus response elements inside the nucleus. As a result, transcription of genes in charge of controlling cell proliferation (like cyclin D1 and Myc) and cell survival (like survivin and Bcl-xL) as well as transcription of angiogenesis-promoting genes (like vascular endothelial growth factor (VEGF)), immunosuppressive growth factors, and cytokines are stimulated to be expressed (such as IL-6). The capability to phosphorylate or activate STAT3 is not restricted to only JAKs, as the tyrosine kinases Src and BCR–ABL1 can also directly activate STAT3. In addition, it has been reported that posttranslational changes, including phosphorylation of the Ser727 residues, and methylation, acetylation, SUMOylation, and ubiquitylation of other residues, may influence the stability or function of STAT3. Compared to Tyr705 phosphorylation, Ser727 phosphorylation has received less attention in research. It can be regulated by a number of kinases such as various MAPKs, JNKs, and protein kinase C, which typically increases STAT3 action. When nonmalignant cells are stimulated with IL-6 or other cytokines, STAT3 is momentarily phosphorylated and/or activated (Chang et al. 2013a, b).
To regulate the termination of cytokine signaling transduction few protein families are involved that have been documented quite recently and are known to downregulate the signal transduction that include: Several cellular phosphatases, including dual-specificity phosphatase 22 (DUSP22), Src homology 2 protein phosphatase 1 (SHP1), protein tyrosine phosphatase receptor type D (PTPRD), Src homology 2 protein phosphatase 2 (SHP2), protein tyrosine phosphatase nonreceptor types 1 and 2 (PTPN1 and PTPN2, respectively), and protein tyrosine phosphatase receptor type T (PTPRT), as well as a minimum of three distinct types of negative regulators comprising suppressors of cytokine signaling (SOCS1, SOCS2, and SOCS3) proteins, protein inhibitors of activated STAT (PIAS) proteins, and cytokine-inducible SH2 (Src homology 2) domain-containing proteins (CISH), can control STAT. All of them function as feedforward regulators, which eventually reduce signaling of IL-6 (Tartaglia et al. 2003; Nozawa et al. 2006; Zhang et al. 2007; Buchert et al. 2016; Peyser et al. 2016). According to reports, mice with mutant gp130 omit their ability to bind to SOCS3, which results in continuous stimulation of STAT3 and STAT1, leading to the onset of cancer and chronic inflammatory illness (Nozawa et al. 2006).
5.6 How IL-6 Regulates Tumor Progression
Sources of IL-6 that are most prevalent in the tumor microenvironment include tumor stromal cells, tumor-infiltrating immune cells, and tumor cells along with CD4+ T cells, TAMs (tumor-associated macrophages), fibroblasts, and MDSCs (myeloid-derived suppressor cells) (Nolen et al. 2008; Oh et al. 2013; Nagasaki et al. 2014; Mehraj et al. 2021a, b, c). In the tumor microenvironment, IL-6 directly affects cancer cells by modulating both their intrinsic and extrinsic actions, thereby directly supporting tumorigenesis and influencing stromal cells, and by indirectly supporting tumorigenesis (Fisher et al. 2014). We can say that IL-6 creates a complex of cytokines, proteases, and growth factors consisting of granulocyte macrophage colony-stimulating factor (GM-CSF), VEGF, and matrix metalloproteinase-1 (MMP-1) in prostate and skin cancer, which promotes the advancement of the malignancy.
As IL-6 is made by tumor cells to aid in their survival and growth, stromal cells' paracrine release of IL-6 is not necessary for the autocrine action of interluekin-6 on tumor cells. However, it is found that interluekin-6 trans-signaling affects tumor growth and metastasis through both paracrine and autocrine processes of IL-6 (Lederle et al. 2011; Fisher et al. 2014). It is generally acknowledged that cancer is driven by inflammation. One of the main factors driving this association is nuclear factor-kappa B (NF-κB) signaling, which plays a major role in the overexpression, and many pro-inflammatory cytokines are stimulated by a range of cell types in the tumor microenvironment. Both adaptive and inborn immunity are regulated by interleukin-6. Certainly, IL-6 knockout mice have altered inborn and acquired immune responses to bacteria, parasites, and viruses. IL-6 regulates the natural immune response in many ways. Upregulation of receptors that recognize patterns, including toll-like receptors (TLRs), causes macrophages, monocytes, or neutrophils to produce IL-6. Furthermore, when pattern recognition receptors are activated, stromal cells such as endothelial cells, fibroblasts, epithelial cells, mesothelial cells, smooth muscle cells, and myofibroblasts produce interleukin-6. IL-6 controls the action of cells of innate immunity as it recruits neutrophils, cells of myeloid lineage, and tissue-resident monocytes and also promotes their differentiation, adhesion, activation, and survival. IL-6's ability to regulate acquired immunity depends on its capacity to regulate T-helper cell differentiation. There are a few potent cytokines that promote inflammation caused by immune cells of adaptive or innate immunity, which control the progression of cancer cells and tumors (Krishnamoorthy et al. 2007; Zhao et al. 2008; Mitchem et al. 2013; Mehraj et al. 2021a, b, c). IL-6, one of these cytokines, is vital for the development of human cancer and is found to be dysregulated in cancer. It activates oncogenic pathways. In many types of cancers, elevated levels of IL-6 are found, for example, in colorectal cancer(CRC), ovarian carcinoma, prostate cancer, breast cancer, pancreatic cancer, pulmonary cancer, cervical cancer, renal cell carcinoma, multiple myeloma (MM), and lymphomas (Altundag et al. 2004; Culig and Puhr 2012; Waldner et al. 2012; Chang et al. 2013a, b; Dethlefsen et al. 2013; Miura et al. 2015). This shows a strong relationship between the IL-6 cytokine and cancer. It is found that males with higher levels of IL-6 show high susceptibility to the occurrence of a liver malignancy. As opposed to this, IL-6 production is inhibited by estrogen and steroid hormones to guard female mice from developing cancer (Naugler et al. 2007; Wei et al. 2015). The hyperactivation of the JAK/STAT3/IL-6 signaling axis regulates various signaling pathways, which is responsible for survival of cancer cells, and thus promotes tumorigenesis (Landskron et al. 2014).
IL-6 controls almost all hallmarks of cancer, such as cell survival, cell proliferation, angiogenesis, inhibition of apoptosis, and metastasis. It is also known for regulating tumor cell metabolism. The bulk of the characteristics of a tumor are regulated by interluekin-6, including a range of biological abilities developed during tumor growth. IL-6's involvement in controlling cancer cell growth stems from the fact that some regulatory mechanisms that downregulate or negatively influence cell growth are avoided by cancer cells. These mechanisms primarily rely on the activity of tumor suppressor genes like the retinoblastoma gene (Rb) and the p53 gene, which go through either gain of function or loss of function to control cell proliferation and cell growth, respectively (Mir et al. 2020). The Rb protein, which is controlled by phosphorylation, interferes with the cell cycle passage from the G1 phase to the S phase (Fig. 5.6). Normally, the retinoblastoma protein is in its active form in the dephosphorylated state, which is bound by E2F, and leads to cell arrest at the G1 phase. On the other hand, Rb is in its inactive form after phosphorylation. In this state, Rb is unable to bind to E2F that triggers cyclin-dependent kinase (CDK) activation in such a manner that it facilitates cell transition into the S phase (Qayoom et al. 2022; Sofi et al. 2022). IL-6 facilitates the phosphorylation of Rb in multiple myeloma (MM) cells, thereby promoting cell growth. Moreover, in MM cells, phosphorylation of Rb also enhances the generation of interluekin-6 (Urashima et al. 1996; Nevins 2001; Giacinti and Giordano 2006).

Downregulation of p53 by overexpression of IL-6
In cervical cancer, esophageal carcinoma, gastric cancer, myeloma cells, basal cell carcinoma cells, and myeloma cells, IL-6 controls the process of apoptosis by stimulating NF-κB and STAT3 signaling, which trans-activates many antiapoptotic proteins like B-cell lymphoma-2 (Bcl-2), Bcl-xL, and Mcl-1, among others. Additionally, these pro-survival or antiapoptotic proteins, particularly Bcl-2, encourage cell division. It has been suggested that increased amounts of IL-6, both in IL-6-treated cells and in transgenic mice that express IL-6, may alter the proportion of pro- and antiapoptotic proteins, which is crucial for the decision to undergo apoptosis. Moreover, Bcl-2 produced by IL-6 controls interactions between mitofusins and Bak by inhibiting the dissociation of Bak from Mfn2, which, in turn, prevents the interaction of Bak with Mfn1. These are the two major mitochondrial events that determine cell death. Along with Bcl-2 and Bcl-xL, IL-6 promotes the production of survivin by direct binding of STAT3 to the promoter survivin gene. This helps tumor cells survive. Additionally, the expression of the survivin gene is downregulated, which suppresses STAT3, causing tumor cells to undergo apoptosis. NF-κB, MAPK/extracellular signal-regulated kinase (ERK), and PI3K/Akt signaling pathways are all increased by IL-6 in kidney cancer, hepatocellular carcinoma (HCC), prostate cancer, and multiple myeloma cells, which results in an increase in cyclin A1 expression in these cells. Overall, it seems that interleukin-6 primarily promotes tumor growth by reducing cell death and promoting cell proliferation (Gritsko et al. 2006; Brooks et al. 2007; Waxman and Kolliputi 2009; Mir et al. 2023). In addition to promoting the growth of tumor cells, IL-6 is used to create resistance to anticancer treatments that trigger the body's natural death processes. Direct targets of STAT3 upregulation include the bulk of the genes that control cell differentiation, proliferation, and survival, including p21, Mcl-1, Bcl-xL, Bcl-2, Fas, cyclin E1, and cyclin D1. Moreover, these, including c-Jun, cMyc, and c-Fos, are other transcription factors that promote proliferation and are also targets of STAT3. When STAT3 is activated in tumor cells, p53 expression is transcriptionally repressed, whereas STAT3 inhibition upregulates p53 expression and triggers p53-mediated apoptosis (Hirano et al. 2000; Niu et al. 2005).
5.7 Inflammation Promotes Cancer
Inflammation encourages all phases of carcinogenesis and predisposes to the growth of cancer and promotion of malignant transformation. Many inflammatory mediators like IL-6, TGF-β, IL-10, and tumor necrosis factor-α (TNF-α) have all been implicated in the beginning and progression of cancer. Cancer and inflammation have shown a strong association with cancer prevention, and the use of anti-inflammatory drugs is encouraged. Inflammation related to cancer consists TAMs, white blood cells, and inflammatory mediators such as cytokines (IL-1, IL-6, and TNF-α) and chemokines (CXCL8 and CCL2), which promote tissue remodeling and vasculogenesis (Colotta et al. 2009; Mir et al. 2022a, b, c).
In the tumor microenvironment, interluekin-6 is one of the highly expressed inflammatory mediators, and, in colorectal cancer, IL-6 secretion is associated with STAT3-dependent tumorigenesis and its associated trans-signaling within the tumor milieu induces inflammation. In ulcerative colitis, M2-type macrophages release IL-6 and contribute to the growth of colorectal tumors. These investigations have discovered a connection between IL-6 and inflammation in tumors. Inflammatory cytokines (IL-6, IL-1b, and TNF) as well as transcription factors like NF-κB and STAT3 are the main players in inflammation. Inflammation is heavily regulated by NF-κB, which is dysregulated in many malignancies. IL-6 is a crucial part of the NF-κB/IL-6/STAT3 signaling pathway, which is involved in carcinogenesis, as it is a primary effector molecule of NF-κB that is activated through the JAK/STAT3 signaling pathway. NF-κB activation in tumors must be maintained by STAT3, and IL-6 encourages carcinogenesis by causing inflammation and cell growth. Meanwhile, the stimulation of IL-6-mediated STAT3 signaling promotes inflammation and boosts the progression and development of gastrointestinal cancers, so it is now evident that IL-6, inflammation, and tumor promotion are all strongly associated (Qayoom et al. 2023a).
5.7.1 IL-6 Promotes Invasion, Metastasis, and Angiogenesis
It is common knowledge that inflammation encourages epithelial-to-mesenchymal transition (EMT), which allows cancer cells to spread from the primary tumor site to the nearby tissues and organs. Cancer cells do this by acquiring a mesenchymal phenotype from an epithelial one, which allows them to do so. Tumor cells transform back into epithelial cells after settling into the new organ and multiply to produce metastatic tumors. It is widely known that inflammation encourages EMT. The terms "epithelial-to-mesenchymal transition" (EMT) and "mesenchymal-to-epithelial transition" (MET) refer to the mechanisms by which cells go from having an epithelial phenotype to bearing mesenchymal characteristics (Kalluri and Weinberg 2009; Ricciardi et al. 2015).
Increased levels of IL-6 in the blood are related to the enlarged size of the tumor as well as invasion (EMT), metastasis, and worse survival rate in people with colorectal cancer. According to reported evidence, IL-6 promotes CXCR4 expression through c-Jun and STAT3, which promotes the metastasis of numerous bone tumors, including breast, pulmonary, prostate, neuroblastomas, adenocarcinomas, multiple myeloma, and melanomas (Ricciardi et al. 2015). By boosting the production of growth factors, such as DNA methyltransferase 1 (DNMT1), VEGF, and MMP-9, IL-6 induces hyperactivation of JAK/STAT3, which is involved in EMT, metastasis, and angiogenesis in bladder cancer. It has also been demonstrated that the autocrine production of IL-6 enhances the ability of tumor cells to infiltrate the extracellular matrix in breast cancer. By increasing the expression of several growth factors, including MMP-9, VEGF, tumor-associated macrophages, and basic fibroblast growth factor (bFGF) in tumor-associated endothelial cells and MDSCs, IL-6/JAK/STAT3 signaling promotes angiogenesis in numerous malignancies (Helbig et al. 2003; Kujawski et al. 2008; Tawara et al. 2011; Chen et al. 2013; Sofi et al. 2023).
5.7.2 At Elevated Levels, IL-6 Acts as a Prognostic Marker for Various Cancers
Nearly 1 pg/ml of IL-6 in present in normal blood circulation, under several conditions. An increase in its level is found during acute hyperglycemia, during/after surgery, after a high-fat meal, during a normal menstrual cycle, and during physical activities. IL-6 levels fluctuate throughout the course of pregnancy, peaking at around 128 pg/ml when the baby is delivered and, immediately afterward, dropping by more than twofold (∼58 pg/ml). As mentioned earlier, chronic inflammation promotes carcinogenesis (Sakamoto et al. 1994; Angstwurm et al. 1997; D’Auria et al. 1997; Devaraj et al. 2005; Blackburn et al. 2006; Narbutt et al. 2008; Reihmane and Dela 2014). IL-6 is a potent pro-inflammatory marker, and it exhibits multiple biological activities. Elevated levels of IL-6 in the serum have been reported during cancer; IL-6 acts as a biomarker for various malignant diseases such as gastric cancer, pancreatic cancer, colon cancer, hepatobiliary cancer, prostate cancer, pulmonary cancer, renal cell carcinoma, breast cancer, and ovarian cancer. According to reports, increased IL-6 levels are related to the overall poor prognosis in hepatocellular carcinoma (HCC) patients (Altundag et al. 2004; Groblewska et al. 2008; Culig and Puhr 2012; Waldner et al. 2012; Chang et al. 2013a, b; Dethlefsen et al. 2013; Miura et al. 2015).
5.7.2.1 Colon Cancer
In 32 patients with colorectal cancer, significantly increased levels of IL-6 were seen in comparison to those of normal colorectal tissues, indicating that interluekin-6 plays an important role in tumor development (Komoda et al. 1998). In addition, it has been shown that increased IL-6 levels in the blood are associated with a poorer prognosis in CRC. This is because IL-6 expression is greater in colon rectal cancer tissues than in nonmalignant tissues (Chung and Chang 2003; Zeng et al. 2017). In addition, as compared to cells from a healthy colon epithelium, CRC cells shows high expression of IL-6 and higher levels of STAT3 phosphorylation (Corvinus et al. 2005). Studies have shown that whereas sIL-6R is strongly elevated during carcinogenesis, mbIL-6R is scarcely detectable. This suggests that IL-6 affects CRC through trans-signaling. An increased ADAM17 expression observed in malignant cells in comparison to normal cells serves to authenticate this fact. A sign of this disease's worse prognosis is elevated IL-6 levels (Becker et al. 2005).
5.7.2.2 Breast Cancer
IL-6's effect on breast cancer cell lines has been studied in vitro; the results are inconsistent, displaying tumor-suppressing actions in some models and tumor-promoting activities in others. The results also showed that there was a large concentration of interleukin-6 in Michigan Cancer Foundation-7 (MCF-7)/adriamycin multidrug-resistant breast cancer cell supernatants. However, in parental sensitive cell supernatants, IL-6 was not detectable (Conze et al. 2001). Furthermore, exogenous IL-6 is added to MCF-7 cells, which leads to an increase in their doxorubicin resistance by 8–10-fold, while engineered MCF-7 cells express IL-6, thus showing a 70-fold rise in their sensitivity to various medications. A protein linked to treatment resistance is G96, which was produced by MDA-MB-231 triple-negative breast cancer cells when exogenous interleukin-6 was added to the cells. In line with this, the scientists discovered that IL-6 and G96 protein levels were substantially increased in breast tumors than in normal, noncancerous breast tissues (Haverty et al. 1997; Qayoom et al. 2023).
However, additional research indicates that interluekin-6 may exhibit antitumor activity against a few types of breast cancers. For instance, it has been noted that IL-6 causes MCF-7 breast cancer cells to undergo apoptosis. According to these discoveries, insulin-like growth factor-I (IGF-I), tumor necrosis factor (TNF), and IL-1, along with interluekin-6, are associated with decreased proliferation of MCF-7 cells by suppressing IGF-I, a molecule known to promote DNA synthesis in MCF-7 (Danforth Jr and Sgagias 1993; Chiu et al. 1996). IL-6 can accelerate the migration of breast malignant cells, indicating its function in metastasis, and, despite its contentious role in survival and proliferation, an epithelial-to-mesenchymal transition is induced by IL-6, which then promotes the spread of breast cancer cells. Other findings have shown that IL-6 inhibits the adhesion of breast malignant cells and that this outcome was related to tumor cells' reduced production of E-cadherin (Arihiro et al. 2000).
The IL-6 levels of breast cancer survivors were considerably higher in their blood in comparison to those of healthy women. In patients, elevated blood levels of IL-6 have been linked to a bad prognosis of this disease, despite the fact that different investigations have produced conflicting consequences concerning the usefulness of the predictive value of IL-6 levels in breast tumor tissues (Bachelot et al. 2003; Salgado et al. 2003; Bozcuk et al. 2004). It was also discovered that as compared to individuals with only one metastatic site, patients with extensive metastatic breast cancer had considerably higher amounts of IL-6 in their blood. Additionally, they discovered that patients with higher IL-6 levels had poorer chemo-endocrine treatment response and lower survival rates (Nishimura et al. 2000; Yokoe and Morishita 2000; Bachelot et al. 2003; Salgado et al. 2003; Bozcuk et al. 2004).
5.7.2.3 Ovarian Cancer
Increased blood levels of the pro-inflammatory cytokine IL-6 is associated with ovarian cancer as well, and a compromised immune function is frequently seen in individuals suffering from advanced epithelial ovarian cancer (EOC), which is linked to overexpression of IL-6. Several research studies have revealed the association of interluekin-6 with the progression of EOC, chemotherapy resistance, and poorer prognosis (Macciò et al. 1998; Hagemann et al. 2007). Advanced epithelial ovarian tumors have elevated expression levels of interleukin-6 in their ascites and cystic fluid in comparison to benign ovarian tumors, and these levels are correlated with the growth/size of the tumor and the ascites volume (Paladugu et al. 1985; Nowak et al. 2010). There is evidence that shows that IL-6 may directly contribute to the progression of ovarian tumors. As discussed earlier, IL-6/JAK/STAT3 signaling hyperactivation upregulates the production of antiapoptotic molecules, for instance, Bcl-xL and Bcl-2 (B-cell lymphoma 2), which promotes EOC cell survival In addition, research has revealed that IL-6 in ovarian cancer cells changes the distribution of the cell cycle. IL-6 also stimulates the release of MMP-9 (matrix metalloproteinase-9), and it adds to increase the invasiveness of ovarian cancer cells. MMP-9 levels were lower in anti-IL-6-treated antibody SKOV-3 ovarian cancer cells in comparison to SKOV-3 ovarian cancer cells in control cultures (Rabinovich et al. 2007; Macciò and Madeddu 2013).
In many ovarian cancer models, IL-6 has also been demonstrated to function as a powerful angiogenic agent. Additionally, it has been noted that ovarian cancer cells, particularly SKOV-3 cells, produce more secreted sIL-6R. This finding suggests the potential role of IL-6 trans-signaling in the development of ovarian tumors and tumor angiogenesis (Nilsson et al. 2005; Macciò and Madeddu 2013).
5.7.2.4 Other Types of Cancers
It is not possible to fully summarize the function of interleukin-6 and its signaling in all forms of cancer in a single chapter. Here, an overview has been provided, including how overexpression of IL-6/JAK/STAT signaling is linked to prolonged inflammation, cancer progression, invasion, tumor cell proliferation, metastasis, and inhibition of expression of proapoptotic genes (Fig. 5.7).

IL-6/JAK/STAT3 inhibitors in cancer targeting (ref: https://www.nature.com/articles/nrclinonc.2018.8)
5.8 IL-6/IL-6R Axis Targeting in Cancer
Accumulated findings have shown that increased levels of interleukin-6 are reported in the pathophysiology of several persistent inflammatory conditions and in various malignancies. By inhibiting IL-6 signaling, it can provide beneficial treatment opportunities for diseases like autoimmune diseases and cancer (Nilsson et al. 2005; Mir et al. 2022a, b, c). This can be performed by targeting IL-6, its receptor, or its signaling. There are three main approaches that are currently in use for blocking the signaling mediated by IL-6 at either the ligand level or the receptor level. The first strategy is to directly target the ligand interluekin-6 with antibodies like siltuximab; the second strategy is to directly use antibodies like tocilizumab to target IL-6R; and the third strategy is to directly target the complex of IL-6-soluble interluekin-6 receptor utilizing fusion proteins, including sgp130 that directly prevents trans-signaling. Targeting IL-6 and its receptors prevent traditional signaling and trans-signaling; however, when IL-6/sIL-6R complexes are targeted with sgp130 fusion proteins, only trans-signaling is prevented (Klein et al. 1989, 1991; Puthier et al. 1999; Hideshima et al. 2001; Gislén et al. 2003).
In 2014, after siltuximab underwent effective clinical testing, The US Food and Drug Administration (FDA) authorized it as a means of treating multicentric Castleman illness. Siltuximab is presently the most extensively researched therapeutic drug targeting interleukin-6; it is actually a chimeric antibody of mice and humans (Van Rhee et al. 2010, 2014). However, there are no abundant data showing siltuximab's effectiveness in clinical studies against solid tumors. These data show that inhibiting IL-6 by itself may not have much of an impact on patients with solid tumor prognoses (Angevin et al. 2014; Liu et al. 2016). The development of efficient combination medicines is necessary to address this issue, as is the development of a dependable, strong biomarker that can predict a response (Finkel et al. 2016; Liu et al. 2016; Yanaihara et al. 2016).
A humanized monoclonal antibody called tocilizumab targets the 6R and impairs both trans-signaling and classical signaling. The FDA has approved tocilizumab for use in treating pediatric patients, cytokine release syndrome patients, and patients with systemic juvenile idiopathic arthritis and rheumatoid arthritis. Preclinical research results point to tocilizumab's potential as a treatment for ovarian cancer. A monoclonal antibody sarilumab, which blocks the receptor of IL-6, is presently going through clinical research trails.
Patients with tumors that do not express any or little IL-6R may benefit from selective trans-signaling suppression. Proteins that include the sgp130 sequence bind to interleukin-6 and its receptor there by blocking the construction of the IL-6/IL-6R complex, which is capable of precisely inhibiting trans-signaling (Finkel et al. 2016).
5.9 JAK Inhibitors
The most widely examined JAK inhibitors are tofacitinib, ruxolitinib, and pacritinib with numerous other agents under preclinical development. JAK inhibitors have mostly been utilized to treat myeloproliferative neoplasms and chronic inflammatory diseases; patients with solid tumors are currently receiving less attention when taking these medications.
JAK1 and JAK3 are only selectively inhibited by the oral JAK inhibitor tofacitinib, which has a lesser affinity to JAK2. Rheumatoid arthritis can be treated with tofacitinib, according to the FDA. Other inflammatory disorders being studied clinically for tofacitinib treatment include psoriasis, Crohn's disease, chronic plaque ulcerative colitis, and chronic plaque. Tofacitinib has been FDA-approved for treating individuals at a high or intermediate risk of myelofibrosis and for those with polycythaemia vera that is hydroxyurea-resistant or intolerable. Ruxolitinib is a JAK inhibitor that is oral and selective for JAK1 and JAK2 (Johnson et al. 2018).
5.10 STAT3 Inhibitors
In most cases of human malignancies, STAT3 is hyperactivated and has been shown to have tumor-promoting qualities. It is also frequently linked to a bad prognosis for disease. As a result, the current emphasis is on finding and creating STAT3 inhibitors that can be utilized to treat human malignancies. As an intracellular transcription factor, STAT3 lacks enzymatic activity; it is frequently regarded as an intractable target, and the discovery or development of potential STAT3 inhibitors has proven challenging. Several drugs that block STAT3 expression are now under clinical trials.
The first direct inhibitors of STAT3 were made from peptidomimetics (ISS-610, PM-73G) and tyrosine-phosphorylated peptides (PY*LKTK), which attach to STAT3's SH2 domain and reduce DNA-binding activity (205–208) and STAT3 dimerization. These medications have antitumor and proapoptotic effects on cancer cells that have STAT3 hyperactivation. STA-21, LLL-3, WP1066, STATTIC, and other nonprotein inhibitors of the SH2 domain have also been found, and it has been demonstrated that they can stop the growth of tumors that contain high levels of activated STAT3 (Johnson et al. 2018).
The interactions between the promoter regions of the target genes and STAT3 are competitively inhibited as an alternate strategy for suppressing STAT3 function. It has been demonstrated that a 15-bp, double-stranded oligonucleotide matches the response region of STAT3 and that the FOS promoter competitively reduces DNA binding of STAT3 and inhibits the development of tumors in clinical models of ovary, lung, brain, breast, acute myeloid leukemia (AML), and skin cancer. Antisense oligonucleotide inhibition of STAT3 expression further offers quite a different strategy for reducing STAT3 activity in cells. AZD9150 is a next-generation antisense oligonucleotide inhibitor of STAT3, which outperforms its earlier variants by including ethyl-modified residues with a 2′–4′ constraint (Johnson et al. 2018).
5.11 Conclusions
Interleukin-6 exhibits both anti- and pro-inflammatory activities, upon its interaction with its receptor isoforms via various modes of IL-6 signaling. In addition to aiding in the development of therapeutic resistance, interleukin-6, which is secreted by tumors and a large number of other cells in the tumor milieu, influences and controls almost every characteristic of cancer. This aids in the advancement and maintenance of tumors. Our knowledge of the possible involvement of IL-6 in the interaction between the tumor and its milieu has improved as a result of recent investigations. The majority of IL-6's actions that promote tumor growth appear to be primarily regulated by the JAK/STAT3/IL-6 signaling pathway. Therefore, neutralizing IL-6 or the IL-6 receptor to stop the beginning of signaling or preventing the completion in the end by reducing the action of two additional members, JAK and STAT3, has demonstrated treatment effectiveness in systemic and cellular carcinoma models. The stimulation of interleukin-6/STAT3 signaling occurs via classical signaling or trans-signaling. Typically, prolonged inflammation encourages various malignant diseases. The hyperactivation of JAK/STAT3/IL-6 signaling is classically linked to patients’ overall reduced survival, as it promotes cancer cell survival, angiogenesis, invasiveness, and metastasis, thus severely suppressing the antitumor activity. Employing agents that block interluekin-6 signaling may be potentially therapeutic for cancer.
Further Reading
For further reading, some textbooks mentioned below also provide further understanding of the cytokine and chemokine network.
-
Cytokines and Their Therapeutic Potential by Manzoor Ahmad Mir
-
The Cytokine Handbook by Angus W. Thomson and Michael T. Lotze
The below links for video lectures may also be helpful:
For more insights into the topic, we would suggest detailed findings from the books of Mir et al. (2022a, b, c) https://doi.org/10.1016/C2021-0-02565-7 and https://doi.org/10.1016/C2022-0-00074-X, and Mir (2021) https://doi.org/10.52305/WXJL6770, Mir (2015a, b, c) https://doi.org/10.1016/C2014-0-02898-5, and from the cancer.net website on the following mentioned links:
https://www.cancer.net/cancer-types/breast-cancer/types-treatment
https://discovery.ucl.ac.uk/id/eprint/1472740/
https://www.jmedsciences.com/doi/JMEDS/pdf/10.5005/jp-journals-10045-00138
References
Altundag O et al (2004) Interleukin-6 and C-reactive protein in metastatic renal cell carcinoma. J Clin Oncol 23(5):1044–1044
Angevin E et al (2014) A phase I/II, multiple-dose, dose-escalation study of siltuximab, an anti-interleukin-6 monoclonal antibody, in patients with advanced solid tumors phase I/II study of anti-IL-6 in solid tumors. Clin Cancer Res 20(8):2192–2204
Angstwurm MW et al (1997) Cyclic plasma IL-6 levels during normal menstrual cycle. Cytokine 9(5):370–374
Arihiro K et al (2000) Cytokines facilitate chemotactic motility of breast carcinoma cells. Breast Cancer 7(3):221–230
Bachelot T et al (2003) Prognostic value of serum levels of interleukin 6 and of serum and plasma levels of vascular endothelial growth factor in hormone-refractory metastatic breast cancer patients. Br J Cancer 88(11):1721–1726
Baran P et al (2018) The balance of interleukin (IL)-6, IL-6· soluble IL-6 receptor (sIL-6R), and IL-6· sIL-6R· sgp130 complexes allows simultaneous classic and trans-signaling. J Biol Chem 293(18):6762–6775
Becker C et al (2005) IL-6 signaling promotes tumor growth in colorectal cancer. Cell Cycle 4(2):220–223
Blackburn P et al (2006) Postprandial variations of plasma inflammatory markers in abdominally obese men. Obesity 14(10):1747–1754
Boulanger MJ et al (2003) Hexameric structure and assembly of the interleukin-6/IL-6 α-receptor/gp130 complex. Science 300(5628):2101–2104
Bozcuk H et al (2004) Tumour necrosis factor-alpha, interleukin-6, and fasting serum insulin correlate with clinical outcome in metastatic breast cancer patients treated with chemotherapy. Cytokine 27(2–3):58–65
Brakenhoff J et al (1990) Structure-function analysis of human IL-6. Epitope mapping of neutralizing monoclonal antibodies with amino-and carboxyl-terminal deletion mutants. J Immunol 145(2):561–568
Brakenhoff JP et al (1994) Development of a human interleukin-6 receptor antagonist. J Biol Chem 269(1):86–93
Brocker C et al (2010) Evolutionary divergence and functions of the human interleukin (IL) gene family. Hum Genomics 5(1):1–26
Brooks C et al (2007) Bak regulates mitochondrial morphology and pathology during apoptosis by interacting with mitofusins. Proc Natl Acad Sci 104(28):11649–11654
Buchert M et al (2016) Targeting JAK kinase in solid tumors: emerging opportunities and challenges. Oncogene 35(8):939–951
Chang CH et al (2013a) Circulating interleukin-6 level is a prognostic marker for survival in advanced nonsmall cell lung cancer patients treated with chemotherapy. Int J Cancer 132(9):1977–1985
Chang Q et al (2013b) The IL-6/JAK/Stat3 feed-forward loop drives tumorigenesis and metastasis. Neoplasia 15(7):848–IN845
Chen M-F et al (2013) IL-6 expression regulates tumorigenicity and correlates with prognosis in bladder cancer. PLoS One 8(4):e61901
Chiu JJ et al (1996) Interleukin 6 acts as a paracrine growth factor in human mammary carcinoma cell lines. Clin Cancer Res 2(1):215–221
Chung YC, Chang YF (2003) Serum interleukin-6 levels reflect the disease status of colorectal cancer. J Surg Oncol 83(4):222–226
Colotta F et al (2009) Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis 30(7):1073–1081
Conze D et al (2001) Autocrine production of interleukin 6 causes multidrug resistance in breast cancer cells. Cancer Res 61(24):8851–8858
Corvinus FM et al (2005) Persistent STAT3 activation in colon cancer is associated with enhanced cell proliferation and tumor growth. Neoplasia 7(6):545–555
Culig Z, Puhr M (2012) Interleukin-6: a multifunctional targetable cytokine in human prostate cancer. Mol Cell Endocrinol 360(1–2):52–58
D’Auria L et al (1997) Cytokines in the sera of patients with pemphigus vulgaris: interleukin-6 and tumour necrosis factor-alpha levels are significantly increased as compared to healthy subjects and correlate with disease activity. Eur Cytokine Netw 8(4):383–387
Danforth DN Jr, Sgagias MK (1993) Interleukin-1α and interleukin-6 act additively to inhibit growth of MCF-7 breast cancer cells in vitro. Cancer Res 53(7):1538–1545
Dethlefsen C et al (2013) The role of intratumoral and systemic IL-6 in breast cancer. Breast Cancer Res Treat 138(3):657–664
Devaraj S et al (2005) Hyperglycemia induces monocytic release of interleukin-6 via induction of protein kinase C-α and-β. Diabetes 54(1):85–91
Ehlers M et al (1994) Identification of two novel regions of human IL-6 responsible for receptor binding and signal transduction. J Immunol 153(4):1744–1753
Ferrao R, Lupardus PJ (2017) The Janus kinase (JAK) FERM and SH2 domains: bringing specificity to JAK–receptor interactions. Front Endocrinol 8:71
Finkel KA et al (2016) IL-6 inhibition with MEDI5117 decreases the fraction of head and neck cancer stem cells and prevents tumor recurrence. Neoplasia 18(5):273–281
Fisher DT et al (2014) The two faces of IL-6 in the tumor microenvironment. Semin Immunol 26(1):38–47
Frank SJ et al (1995) Regions of the JAK2 tyrosine kinase required for coupling to the growth hormone receptor. J Biol Chem 270(24):14776–14785
Gandhi NA et al (2016) Targeting key proximal drivers of type 2 inflammation in disease. Nat Rev Drug Discov 15(1):35–50
Gauldie J et al (1987) Interferon beta 2/B-cell stimulatory factor type 2 shares identity with monocyte-derived hepatocyte-stimulating factor and regulates the major acute phase protein response in liver cells. Proc Natl Acad Sci 84(20):7251–7255
Giacinti C, Giordano A (2006) RB and cell cycle progression. Oncogene 25(38):5220–5227
Gislén A et al (2003) Superior underwater vision in a human population of sea gypsies. Curr Biol 13(10):833–836
Gritsko T et al (2006) Persistent activation of stat3 signaling induces survivin gene expression and confers resistance to apoptosis in human breast cancer cells. Clin Cancer Res 12(1):11–19
Groblewska M et al (2008) Serum interleukin 6 (IL-6) and C-reactive protein (CRP) levels in colorectal adenoma and cancer patients. Clin Chem Lab Med 46(10):1423–1428
Grötzinger J et al (1999) IL-6 type cytokine receptor complexes: hexamer, tetramer or both? Biol Chem 380(7–8):803–813
Hagemann T et al (2007) Ovarian cancer cell–derived migration inhibitory factor enhances tumor growth, progression, and angiogenesis. Mol Cancer Ther 6(7):1993–2002
Haverty AA et al (1997) Interleukin-6 upregulates GP96 expression in breast cancer. J Surg Res 69(1):145–149
Heink S et al (2017) Trans-presentation of IL-6 by dendritic cells is required for the priming of pathogenic TH17 cells. Nat Immunol 18(1):74–85
Helbig G et al (2003) NF-kappaB promotes breast cancer cell migration and metastasis by inducing the expression of the chemokine receptor CXCR4. J Biol Chem 278(24):21631–21638
Hibi M et al (1990) Molecular cloning and expression of an IL-6 signal transducer, gp130. Cell 63(6):1149–1157
Hideshima T et al (2001) The role of tumor necrosis factor α in the pathophysiology of human multiple myeloma: therapeutic applications. Oncogene 20(33):4519–4527
Hirano T (2014) Revisiting the 1986 molecular cloning of interleukin 6. Front Immunol 5:456
Hirano T et al (1986) Complementary DNA for a novel human interleukin (BSF-2) that induces B lymphocytes to produce immunoglobulin. Nature 324(6092):73–76
Hirano T et al (1987) Human B-cell differentiation factor defined by an anti-peptide antibody and its possible role in autoantibody production. Proc Natl Acad Sci 84(1):228–231
Hirano T et al (1988) Excessive production of interleukin 6/B cell stimulatory factor-2 in rheumatoid arthritis. Eur J Immunol 18(11):1797–1802
Hirano T et al (2000) Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene 19(21):2548–2556
Hodge DR et al (2005) The role of IL-6 and STAT3 in inflammation and cancer. Eur J Cancer 41(16):2502–2512
Howard M et al (1982) Identification of a T cell-derived b cell growth factor distinct from interleukin 2. J Exp Med 155(3):914–923
Ji C et al (2011) IL-6 induces lipolysis and mitochondrial dysfunction, but does not affect insulin-mediated glucose transport in 3T3-L1 adipocytes. J Bioenerg Biomembr 43(4):367–375
Johnson DE et al (2018) Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol 15(4):234–248
Jostock T et al (2001) Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor transsignaling responses. Eur J Biochem 268(1):160–167
Kalluri R, Weinberg RA (2009) The basics of epithelial-mesenchymal transition. J Clin Invest 119(6):1420–1428
Kishimoto T et al (1978) Induction of IgG production in human B lymphoblastoid cell lines with normal human T cells. Nature 271(5647):756–758
Klein B et al (1989) Paracrine rather than autocrine regulation of myeloma-cell growth and differentiation by interleukin-6. Blood 73(2):517–526
Klein B et al (1991) Murine anti-interleukin-6 monoclonal antibody therapy for a patient with plasma cell leukemia. Blood 78(5):1198–1204
Komoda H et al (1998) Interleukin-6 levels in colorectal cancer tissues. World J Surg 22(8):895–898
Krishnamoorthy N, Paglia M, Ray A, Ray P (2007) A critical role for IL-6 secretion by dendritic cells promoting Th2 and limiting Th1 response (95.24). J Immunol 178(1_Supplement):S181–S181
Kujawski M et al (2008) Stat3 mediates myeloid cell–dependent tumor angiogenesis in mice. J Clin Invest 118(10):3367–3377
Kumari N et al (2016) Role of interleukin-6 in cancer progression and therapeutic resistance. Tumor Biol 37(9):11553–11572
Lai KS et al (1995) A kinase-deficient splice variant of the human JAK3 is expressed in hematopoietic and epithelial cancer cells (∗). J Biol Chem 270(42):25028–25036
Landskron G et al (2014) Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res 2014:149185
Lederle W et al (2011) IL-6 promotes malignant growth of skin SCCs by regulating a network of autocrine and paracrine cytokines. Int J Cancer 128(12):2803–2814
Liu X et al (2016) The biology behind interleukin-6 targeted interventions. Curr Opin Rheumatol 28(2):152–160
Lust JA et al (1992) Isolation of an mRNA encoding a soluble form of the human interleukin-6 receptor. Cytokine 4(2):96–100
Macciò A, Madeddu C (2013) The role of interleukin-6 in the evolution of ovarian cancer: clinical and prognostic implications—a review. J Mol Med 91(12):1355–1368
Macciò A et al (1998) High serum levels of soluble IL-2 receptor, cytokines, and C reactive protein correlate with impairment of T cell response in patients with advanced epithelial ovarian cancer. Gynecol Oncol 69(3):248–252
Mehraj U et al (2021a) Tumor microenvironment promotes breast cancer chemoresistance. Cancer Chemother Pharmacol 87(2):147–158
Mehraj U et al (2021b) The tumor microenvironment as driver of stemness and therapeutic resistance in breast cancer: new challenges and therapeutic opportunities. Cell Oncol 44:1209–1229
Mehraj U et al (2021c) Prognostic significance and targeting tumor-associated macrophages in cancer: new insights and future perspectives. Breast Cancer 28(3):539–555
Mir MA (2015a) Introduction to costimulation and costimulatory molecules. In: Mir MA (ed) Developing costimulatory molecules for immunotherapy of diseases. Academic Press, London, pp 1–43
Mir MA (2015b) Costimulation in lymphomas and cancers. In: Mir MA (ed) Developing costimulatory molecules for immunotherapy of diseases. Academic Press, London, pp 185–254
Mir MA (2015c) T-cell costimulation and its applications in diseases. In: Mir MA (ed) Developing costimulatory molecules for immunotherapy of diseases. Academic Press, London, pp 255–292
Mir MA, Mehraj U (2019) Double-crosser of the immune system: macrophages in tumor progression and metastasis. Curr Immunol Rev 15(2):172–184
Mir MA, Qayoom H, Mehraj U, Nisar S, Bhat B, Wani NA (2020) Targeting different pathways using novel combination therapy in triple negative breast cancer. Curr Cancer Drug Targets 20(8):586–602. https://doi.org/10.2174/1570163817666200518081955. PMID: 32418525
Mir MA et al (2022a) The tumor microenvironment. In: Mir MA (ed) Role of tumor microenvironment in breast cancer and targeted therapies. Academic Press, London, pp 31–58
Mir MA et al (2022b) Role of cancer-associated fibroblasts in tumor microenvironment. In: Mir MA (ed) Role of tumor microenvironment in breast cancer and targeted therapies. Academic Press, London, pp 59–86
Mir MA et al (2022c) Targeting biologically specific molecules in triple negative breast cancer (TNBC). In: Mir MA (ed) Combinational therapy in triple negative breast cancer. Academic Press, London, pp 177–200
Mir WR, Bhat BA, Kumar A, Dhiman R, Alkhanani M, Almilaibary A, Dar MY, Ganie SA, Mir MA (2023) Network pharmacology combined with molecular docking and in vitro verification reveals the therapeutic potential of Delphinium roylei munz constituents on breast carcinoma. Front Pharmacol 14:1135898. https://doi.org/10.3389/fphar.2023.1135898. PMID:37724182; PMCID: PMC10505441
Mitchem JB et al (2013) Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic ResponsesMacrophages regulate tumor-initiating cells. Cancer Res 73(3):1128–1141
Miura T et al (2015) Characterization of patients with advanced pancreatic cancer and high serum interleukin-6 levels. Pancreas 44(5):756–763
Mülberg J et al (1993) The soluble interleukin-6 receptor is generated by shedding. Eur J Immunol 23(2):473–480
Nagasaki T et al (2014) Interleukin-6 released by colon cancer-associated fibroblasts is critical for tumour angiogenesis: anti-interleukin-6 receptor antibody suppressed angiogenesis and inhibited tumour–stroma interaction. Br J Cancer 110(2):469–478
Nakajima T et al (1993) Phosphorylation at threonine-235 by a ras-dependent mitogen-activated protein kinase cascade is essential for transcription factor NF-IL6. Proc Natl Acad Sci 90(6):2207–2211
Narazaki M et al (1993) Soluble forms of the interleukin-6 signal-transducing receptor component gp130 in human serum possessing a potential to inhibit signals through membrane-anchored gp130. Blood 82(4):1120–1126
Narbutt J et al (2008) Serum concentration of interleukin-6 is increased both in active and remission stages of pemphigus vulgaris. Mediators Inflamm 2008:875394
Naugler WE et al (2007) Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 317(5834):121–124
Nevins JR (2001) The Rb/E2F pathway and cancer. Hum Mol Genet 10(7):699–703
Nilsson MB et al (2005) Interleukin-6, secreted by human ovarian carcinoma cells, is a potent proangiogenic cytokine. Cancer Res 65(23):10794–10800
Nishimura R et al (2000) An analysis of serum interleukin-6 levels to predict benefits of medroxyprogesterone acetate in advanced or recurrent breast cancer. Oncology 59(2):166–173
Niu G et al (2005) Role of Stat3 in regulating p53 expression and function. Mol Cell Biol 25(17):7432–7440
Nolen BM et al (2008) Serum biomarker profiles and response to neoadjuvant chemotherapy for locally advanced breast cancer. Breast Cancer Res 10(3):1–9
Nowak M et al (2010) Proinflammatory and immunosuppressive serum, ascites and cyst fluid cytokines in patients with early and advanced ovarian cancer and benign ovarian tumors. Neuroendocrinol Lett 31(3):375–383
Nozawa H et al (2006) Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc Natl Acad Sci 103(33):12493–12498
Oh K et al (2013) A mutual activation loop between breast cancer cells and myeloid-derived suppressor cells facilitates spontaneous metastasis through IL-6 trans-signaling in a murine model. Breast Cancer Res 15(5):1–16
Paladugu RR et al (1985) Bronchopulmonary Kulchitzky cell carcinomas. A new classification scheme for typical and atypical carcinoids. Cancer 55(6):1303–1311
Paonessa G et al (1995) Two distinct and independent sites on IL-6 trigger gp 130 dimer formation and signalling. EMBO J 14(9):1942–1951
Peyser ND et al (2016) Frequent promoter hypermethylation of PTPRT increases STAT3 activation and sensitivity to STAT3 inhibition in head and neck cancer. Oncogene 35(9):1163–1169
Puthier D et al (1999) Mcl-1 and Bcl-xL are co-regulated by IL-6 in human myeloma cells. Br J Haematol 107(2):392–395
Qayoom H et al (2021) An insight into the cancer stem cell survival pathways involved in chemoresistance in triple-negative breast cancer. Future Oncol 17(31):4185–4206
Qayoom H, Sofi S, Mir MA (2023) Targeting tumor microenvironment using tumor-infiltrating lymphocytes as therapeutics against tumorigenesis. Immunol Res. https://doi.org/10.1007/s12026-023-09376-2. Epub ahead of print. PMID: 37004645
Qayoom H, Mehraj U, Sofi S, Aisha S, Almilaibary A, Alkhanani M, Mir MA (2022) Expression patterns and therapeutic implications of CDK4 across multiple carcinomas: a molecular docking and MD simulation study. Med Oncol 39(10):158. https://doi.org/10.1007/s12032-022-01779-9. PMID: 35870089
Qayoom H, Alkhanani M, Almilaibary A, Alsagaby SA, Mir MA (2023a) Mechanistic elucidation of Juglanthraquinone C targeting breast cancer: a network pharmacology-based investigation. Saudi. J Biol Sci 30(7):103705. https://doi.org/10.1016/j.sjbs.2023.103705. Epub 2023 Jun 15. PMID: 37425621; PMCID: PMC10329161
Qayoom H, Alkhanani M, Almilaibary A, Alsagaby SA, Mir MA (2023b) A network pharmacology-based investigation of brugine reveals its multi-target molecular mechanism against breast cancer. Med Oncol 40(7):202. https://doi.org/10.1007/s12032-023-02067-w. PMID: 37308611
Rabinovich A et al (2007) Regulation of ovarian carcinoma SKOV-3 cell proliferation and secretion of MMPs by autocrine IL-6. Anticancer Res 27(1A):267–272
Rane SG, Reddy EP (1994) JAK3: a novel JAK kinase associated with terminal differentiation of hematopoietic cells. Oncogene 9(8):2415–2423
Reihmane D, Dela F (2014) Interleukin-6: possible biological roles during exercise. Eur J Sport Sci 14(3):242–250
Ricciardi M et al (2015) Epithelial-to-mesenchymal transition (EMT) induced by inflammatory priming elicits mesenchymal stromal cell-like immune-modulatory properties in cancer cells. Br J Cancer 112(6):1067–1075
Riethmueller S et al (2017) Proteolytic origin of the soluble human IL-6R in vivo and a decisive role of N-glycosylation. PLoS Biol 15(1):e2000080
Rohleder N et al (2012) Role of interleukin-6 in stress, sleep, and fatigue. Ann N Y Acad Sci 1261(1):88–96
Rose-John S et al (2015) “Family Reunion”–a structured view on the composition of the receptor complexes of interleukin-6-type and interleukin-12-type cytokines. Cytokine Growth Factor Rev 5(26):471–474
Sakamoto K et al (1994) Elevation of circulating interleukin 6 after surgery: factors influencing the serum level. Cytokine 6(2):181–186
Salgado R et al (2003) Circulating interleukin-6 predicts survival in patients with metastatic breast cancer. Int J Cancer 103(5):642–646
Savino R et al (1994a) Rational design of a receptor super-antagonist of human interleukin-6. EMBO J 13(24):5863–5870
Savino R et al (1994b) Generation of interleukin-6 receptor antagonists by molecular-modeling guided mutagenesis of residues important for gp130 activation. EMBO J 13(6):1357–1367
Schafer ZT, Brugge JS (2007) IL-6 involvement in epithelial cancers. J Clin Invest 117(12):3660–3663
Scheller J et al (2011) The pro-and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta 1813(5):878–888
Seif F et al (2017) The role of JAK-STAT signaling pathway and its regulators in the fate of T helper cells. Cell Commun Signal 15(1):1–13
Shabo Y et al (1988) The myeloid blood cell differentiation-inducing protein MGI-2A is interleukin-6. Blood 72(6):2070–2073
Simpson RJ et al (1997) Interleukin-6: structure-function relationships. Protein Sci 6(5):929–955
Sofi S, Mehraj U, Qayoom H, Aisha S, Almilaibary A, Alkhanani M, Mir MA (2022) Targeting cyclin-dependent kinase 1 (CDK1) in cancer: molecular docking and dynamic simulations of potential CDK1 inhibitors. Med Oncol 39(9):133. https://doi.org/10.1007/s12032-022-01748-2. PMID: 35723742; PMCID: PMC9207877
Sofi S, Jan N, Qayoom H, Alkhanani M, Almilaibary A, Ahmad MM (2023) Elucidation of interleukin-19 as a therapeutic target for breast cancer by computational analysis and experimental validation. Saudi J Biol Sci 30(9):103774. https://doi.org/10.1016/j.sjbs.2023.103774. Epub 2023 Aug 11. PMID: 37675062; PMCID: PMC10477739
Sprang SR, Bazan JF (1993) Cytokine structural taxonomy and mechanisms of receptor engagement. Curr Opin Struct Biol 3(6):815–827
Taga T, Kishimoto T (1997) Gp130 and the interleukin-6 family of cytokines. Annu Rev Immunol 15:797
Taga T et al (1989) Interleukin-6 triggers the association of its receptor with a possible signal transducer, gp130. Cell 58(3):573–581
Takahashi T, Shirasaw T (1994) Molecular cloning of rat JAK3, a novel member of the JAK family of protein tyrosine kinases. FEBS Lett 342(2):124–128
Takeuchi O, Akira S (2010) Pattern recognition receptors and inflammation. Cell 140(6):805–820
Tanaka T et al (2014) IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol 6(10):a016295
Taniguchi K et al (2015) A gp130–Src–YAP module links inflammation to epithelial regeneration. Nature 519(7541):57–62
Taniguchi K et al (2017) YAP–IL-6ST autoregulatory loop activated on APC loss controls colonic tumorigenesis. Proc Natl Acad Sci 114(7):1643–1648
Tartaglia M et al (2003) Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet 34(2):148–150
Tawara K et al (2011) Clinical significance of interleukin (IL)-6 in cancer metastasis to bone: potential of anti-IL-6 therapies. Cancer Manag Res 3:177
Urashima M et al (1996) Interleukin-6 promotes multiple myeloma cell growth via phosphorylation of retinoblastoma protein. Blood 88(6):2219–2227
Vaillant AAJ, Qurie A (2021) Interleukin. In: StatPearls. StatPearls Publishing, St. Petersburg, FL
Van Rhee F et al (2010) Siltuximab, a novel anti–interleukin-6 monoclonal antibody, for Castleman’s disease. J Clin Oncol 28(23):3701–3708
Van Rhee F et al (2014) Siltuximab for multicentric Castleman’s disease: a randomised, double-blind, placebo-controlled trial. Lancet Oncol 15(9):966–974
Velazquez L et al (1995) Distinct domains of the protein tyrosine kinase tyk2 required for binding of interferon-α/β and for signal transduction. J Biol Chem 270(7):3327–3334
Verbsky JW et al (1996) Expression of Janus kinase 3 in human endothelial and other non-lymphoid and non-myeloid cells. J Biol Chem 271(24):13976–13980
Waldner MJ et al (2012) Interleukin-6-a key regulator of colorectal cancer development. Int J Biol Sci 8(9):1248
Waxman AB, Kolliputi N (2009) IL-6 protects against hyperoxia-induced mitochondrial damage via Bcl-2–induced Bak interactions with mitofusions. Am J Respir Cell Mol Biol 41(4):385–396
Wei Q et al (2015) Estrogen suppresses hepatocellular carcinoma cells through ERβ-mediated upregulation of the NLRP3 inflammasome. Lab Investig 95(7):804–816
Wunderlich FT et al (2010) Interleukin-6 signaling in liver-parenchymal cells suppresses hepatic inflammation and improves systemic insulin action. Cell Metab 12(3):237–249
Yamasaki K et al (1988) Cloning and expression of the human interleukin-6 (BSF-2/IFNβ 2) receptor. Science 241(4867):825–828
Yanaihara N et al (2016) Antitumor effects of interleukin-6 (IL-6)/interleukin-6 receptor (IL-6R) signaling pathway inhibition in clear cell carcinoma of the ovary. Mol Carcinog 55(5):832–841
Yawata H et al (1993) Structure-function analysis of human IL-6 receptor: dissociation of amino acid residues required for IL-6-binding and for IL-6 signal transduction through gp130. EMBO J 12(4):1705–1712
Yokoe T, Morishita Y (2000) Trends of IL-6 and IL-8 levels in patients with recurrent breast cancer: preliminary report. Breast Cancer 7(3):187–190
Zeng J et al (2017) Clinicopathological significance of overexpression of interleukin-6 in colorectal cancer. World J Gastroenterol 23(10):1780
Zhang X et al (2007) Identification of STAT3 as a substrate of receptor protein tyrosine phosphatase T. Proc Natl Acad Sci 104(10):4060–4064
Zhao W et al (2008) p38α stabilizes Interleukin-6 mRNA via multiple AU-richElements. J Biol Chem 283(4):1778–1785
Zilberstein A et al (1986) Structure and expression of cDNA and genes for human interferon-beta-2, a distinct species inducible by growth-stimulatory cytokines. EMBO J 5(10):2529–2537
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Mir, M.A., Bashir, M., Jan, N. (2023). The Role of Interleukin (IL)-6/IL-6 Receptor Axis in Cancer . In: Mir, M.A. (eds) Cytokine and Chemokine Networks in Cancer. Springer, Singapore. https://doi.org/10.1007/978-981-99-4657-0_5
Download citation
DOI: https://doi.org/10.1007/978-981-99-4657-0_5
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-99-4656-3
Online ISBN: 978-981-99-4657-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)