
Abstract
Esophageal squamous cell carcinoma (ESCC) is one of the most common cancers. In this study, our objective was to identify differentially regulated proteins in ESCC using isobaric tag for relative and absolute quantification (iTRAQ) technique and liquid chromatography–tandem mass spectrometry (LC–MS/MS). We compared the protein expression profiles of ESCC tumor tissues with the corresponding adjacent normal tissue from three patients. It was determined that 72 and 57 unique proteins were significantly up-regulated and down-regulated in all three samples. In addition, there were 431 significantly differentially regulated proteins having at least two biological samples. This subject found some of the differential proteins, such as prolyl 4-hydroxylase subunit alpha-1, prolyl 4-hydroxylase subunit alpha-2, and calponin-2, immunoglobulin superfamily containing leucine-rich repeat protein, and prolyl 3-hydroxylase1, which were few studies about them in ESCC. In order to determine the results, we performed another independent experiment. Our results indicated quantitative proteomics, as a robust discovery tool for the identification, differentially regulated proteins in cancers.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Esophageal squamous cell carcinoma (ESCC) is one of the most common malignant tumors worldwide. Compared with other populations, ESCC, which is the main histological subtype of esophageal cancers, is at a very high frequency rate in Chinese population [1]. Despite significant advances in surgery and other therapeutic strategies, the 5-year survival rate after surgery is being estimated only 17 % [2]. The ample data show that smoking and alcohol abuse play certain roles in the etiology of esophageal cancer, and other environmental influences including nutritional deficiencies, nitrosamines, low socioeconomic status, specific carcinogens, consumption of very hot beverages, and limited intake of vegetables and fruits have also been risk factors of esophageal cancer [3]. ESCC is an unresectable or metastatic disease. Dysphagia, which is the most common symptom of esophageal cancer, occurs on the advanced stage of the tumors. The high mortality rate of ESCC is a major attribute to the lack of early clinical symptoms, effective biomarkers for early detection, and the limited understanding of its carcinogenic mechanisms. Some scholars think that the overall 5-year survival rate may rise to 90 % when the tumor is removed in the early stage of ESCC [4]. Therefore, it is imperative to detect more effective biomarkers of ESCC and study its molecular characterization.
In the postgenomic era, proteomics provides an effective approach to investigate the different protein expressions and provides the basis for discovery of the potential biomarkers and drug targets for cancer. By the two-dimensional electrophoresis (2DE)/matrix-assisted laser desorption/ionization-time-of-flight mass spectrometry (MALDI-TOF MS), it was found that there were plenty of differential proteins and some potential molecules in buccal squamous cell carcinoma, cholangiocarcinoma, prostate cancer, squamous cervical cancer, renal cell carcinoma, lung squamous carcinoma, and gastric carcinoma [5, 6]. In the differential proteins, only a small section was finally identified. In recent years, isobaric tag for relative and absolute quantification (iTRAQ) technique and liquid chromatography–tandem mass spectrometry (LC–MS/MS) have been widely applied to identify tumor-associated proteins, and specific protein markers even provided a broad perspective to understanding the complex biological behaviors of cell [7]. In this study, we are ready to apply iTRAQ technique to analyze three ESCC tumors and paired adjacent normal esophageal squamous epithelium tissues, which were obtained from ESCC patients in Xinjiang, China. The aim of the study was to identify the proteins with significantly different expressions in ESCC. In this study, the identified proteins may be able to illuminate the molecular basis of ESCC progression and provide potential molecular markers for diagnosis or prognosis of ESCC.
Materials and methods
Tissue specimens
Informed consent was signed by all patients, and the study was approved by the Ethical Committee of the First Affiliated Hospital of Xinjiang Medical University. The samples, which were more than 80 % of target cells (cancer cells or normal epithelial cells) without necrosis, would be selected. Tumor tissues and adjacent normal epithelium tissues were obtained from three ESCCs, who underwent esophagectomy at the First Affiliated Hospital of Xinjiang Medical University. All of the samples were histopathologically diagnosed as ESCC by two independent experienced pathologists. All the tissues were frozen at −80 °C until use. The patients come from Xinjiang, where incidence and mortality rates of ESCC were higher than the average level of China.
Protein preparation
Protein extract and quantification
The tissues were ground in liquid nitrogen and extracted with lysis buffer. To obtain the supernatant, the tissues were shaken by 15 min of ultrasound and centrifuged at 25,000 cycles of 4 °C for 20 min. Then, to obtain the precipitates, the supernatant was mixed with ice-cold acetone and centrifuged. The above steps for the precipitates were repeated, and the following steps were recovering the disulfide bonds in proteins and blocking the cysteines. Again, the tissues were ground, shaken by ultrasound, and centrifuged. Finally, the supernatant was transferred to a new tube and quantified, using Bradford Protein Assay Kit.
iTRAQ labeling and SCX fractionation
The protein was digested with Trypsin Gold (Promega, Madison, WI, USA) at 37 °C for 16 h. After trypsin digestion, peptides were dried by vacuum centrifugation. Peptides were reconstituted in 0.5 M TEAB and performed according to manufacturer’s protocol. Strong cation exchange (SCX) chromatography was performed with a LC-20AB HPLC Pump system (Shimadzu, Kyoto, Japan). The iTRAQ-labeled peptide mixtures were reconstituted with 4 mL buffer A. The samples were eluted at a flow rate of 1 mL/min in a SCX column. Elution was monitored at the 214-nm absorbance. In addition, the eluted peptides were divided into 20 segments. Finally, each segment were desalted with a Strata X C18 column (Phenomenex) and vacuum dried.
Mass spectrometry analysis
LC–MS/MS proteomic analysis
The mass spectroscopy analysis was performed by a Triple TOF 5600 (AB SCIEX, Concord, ON), integrated online micro flow LC-20AD nanoHPLC (Shimadzu, Kyoto, Japan) as described before [7]. Data acquisition was performed with a Triple TOF 5600 System comprising a Nanospray III source and a pulled quartz tip as the emitter. When the data was collected, the parameters were respectively 2.5 kV of the ion spray voltage, 30 psi of the curtain gas, 15 psi of the nebulizer gas, and 150 °C of the interface heater temperature. The scan patterns were at a reflective mode at ≥30,000 resolutions. Every ion signal was recorded by the four bins and was translated into the date.
Proteomic data analysis
In the analysis of bioinformatics, only converting into MGF formates can raw data files be used. Mascot search engine, which was regarded as the gold standard of biological mass spectrometry technology, was used to identify the proteins against database. To reduce the probability of false peptide identification, only peptides with significance scores (≥20), which were at the 99 % confidence intervals (CIs) by a Mascot probability analysis, were regarded as identified, and each confident protein identification involved at least one unique peptide. For protein quantification, it was required that a protein contains at least two unique peptides. The quantitative protein ratios were weighted and normalized by the median ratio in Mascot. Differences were indicated when a p value was less than 0.05, and only fold changes more than 1.5 were considered.
Functional analysis
To better understand the annotation and distribution of protein functions, we used the Blast2GO program to obtain Gene Ontology (GO) annotations. GO provides a set of dynamic updating controlled vocabulary to describe genes and gene product attributes in the organism and is an international standardization of gene function classification system. GO has three ontologies which can describe molecular function, cellular component, and biological process, respectively. The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database is a collection of manually drawn graphical diagrams, representing molecular pathways for metabolism, genetic information processing, environmental information processing, other cellular processes, human diseases, and drug development.
Results
Overview of quantitative proteomics
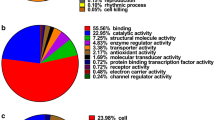
The iTRAQ labeling of peptide tissue specimens derived from three human ESCC tissues and paired adjacent normal tissues and marked by isobaric tags of 6-plex iTRAQ Reagent Multi-Plex kit respectively as follows: cancer tissues, 115/118/121 and paired adjacent normal tissues, 113/116/119. Via the iTRAQ—LC–MS/MS, 342,788 spectra were obtained on the ESCC tissues and normal tissues. After removing low-scoring spectra, there were 60,041 unique spectra remaining, which matched to 4999 unique proteins. According to protein molecular weight (MW) distribution, the majority of the MW was ranged from 10 to 100 kDa. However, it was found that the MW of almost 1000 proteins were ≥100 kDa (Fig. 1a). In addition, most of the proteins were identified with good peptide coverage, of which about 52 % proteins were with more than 10 % of the sequence coverage, and about 38 % proteins were with 20 % of the sequence coverage (Fig. 1b).

MV distribution, coverage, and functional category of proteins identified in this study. a Distribution of protein identified among different molecular weights; b coverage of proteins by the identified peptides; and c functional category coverage of the proteins identified
Functional annotations of all the identified proteins by GO including biological processes, cellular components, and molecular function are shown in Additional file 1. There were 23, 16, and 18 functional groups, respectively. All identified proteins were matched against the Clusters of Orthologous Groups (COG) database and classified into 25 categories. Among the 25 COG function classes, the cluster for “General function prediction” represented the largest group (754), followed by “Posttranslational modification, protein turnover, chaperones”(395), and “Translation, ribosomal structure and biogenesis”(274). According to date, it was easily found that nuclear structure and cell motility were the smallest groups (Fig. 1c). The source of variation differs from one experiment to another, and it was caused by experiment time, manpower, instrument, subject, subject condition, and preparation process. Therefore, these sources of variation (technical, experimental, and biological) must be minimized or identified. The CV was calculated by quantitative value of proteins in repeating data, and the distribution of CV was collected (Fig. 2).

CV distribution. X-axis represents different levels of CV. The left of Y-axis represents quantitative protein of different CV levels, corresponding the cylindrical. The right of Y-axis represents different CV level accumulations that accounted for total quantitative protein ratio, corresponding the line graph
Differentially expressed proteins
According to the standard, differences were indicated statistically significant when p was less than 0.05 and cutoff range was 1.5-fold. Statistics suggested that there were 72 and 57 unique proteins that were significantly up- and down-regulated between ESCC tissues and paired adjacent normal tissues in all three samples. According to the data analysis, there were 190, 172, and 363 especially significantly dysregulated expression in sample 1, sample 2, and sample 3, respectively (Fig. 3). In addition, there were 208 (125 up- and 83 down-regulated) proteins that were significantly dysregulated expressed between sample 1 and sample 2, 245 (156 up- and 89 down-regulated) proteins between sample 2 and sample 3, and 262 (145 up- and 117 down-regulated) proteins between sample 1 and sample 3. The fold difference distribution was obtained from three samples, respectively (Fig. 4). Overall, 431 significantly differentially regulated proteins having at least two biological samples with an acceptable level of confidence were shortlisted for the bioinformatics analysis to retrieve useful biological trends.

Differentially expressed proteins in all three samples: a up-regulated and b down-regulated

Box-whisker plot of fold difference. From the top to down, representing the maximum, upper quartile, median, lower quartile, and minimum, respectively
Functional enrichment analysis
To obtain a comprehensive view of the differential expression proteins, GO and KEGG pathway enrichment analyses were analyzed. The enriched molecular function of up-regulated proteins was mainly including to binding (220 proteins), catalytic activity (127 proteins), and transporter activity (17 proteins), whereas the down-regulated proteins were predominantly involved in binding (121 proteins), catalytic activity (44 proteins), and structural molecule activity (22 proteins). Among the differently expressed proteins in this study, most of them were shown to be associated with various functions, involved in cell proliferation, cell differentiation, cell adhesion, cell migration, and cell apoptosis (Table 1). Moreover, majority of proteins play the roles by signaling pathways, such as p53 signaling pathway (Supplementary figure 1), Wnt signaling pathway (Supplementary figure 2), Jak-STAT signaling pathway (Supplementary figure 3), and NF-kappa B signaling pathway (Supplementary figure 4). These signaling pathways have been well studied in multiple types of cancer. In additional, we also found 12 differential proteins associated with cell cycle. The detailed information of proteins is shown in Table 2.
To investigate protein interactions at the level of Pfam domains, the web resource that provided the analysis of the Protein Data Bank structures was adopted. It was found that RNA recognition motif (RRM) and helicase C, the most prominent two Pfam domains, involved 34 proteins. And the up-regulated and down-regulated proteins involved in the top ten Pfam domains were respectively shown in Fig. 5a, b. In normal tissues, the main function of the spliceosome components was to control cells growing; however, they were inactive in multiple types of cancer. In addition, it was found that 22 spliceosome components were identified as differential proteins (Fig. 6).

Pfam domains analyses of differentially expressed proteins. The top 10 Pfam domains of the up-regulated (a) and down-regulated (b) proteins

Identified differentially expression proteins in spliceosome components. The red box indicates down-regulation
Novel overexpressed proteins identified in this study
The present study identified many novel ESCC-associated differential proteins. Among the novel candidates, prolyl 4-hydroxylase subunit alpha-1 (P4HA1), prolyl 4-hydroxylase subunit alpha-2 (P4HA2), and calponin-2 (CNN2) have not been described previously in the reports about ESCC. So far, immunoglobulin superfamily (ISLR) containing leucine-rich repeat protein and prolyl 3-hydroxylase1 (P3H1/LEPRE1) hardly have been reported in any carcinoma.
Validation of differential expression pattern
In this study, five proteins were chosen which have been reported in carcinoma as novel potential candidates. In order to validate the results from the three human ESCC tissues and paired adjacent normal tissues, another independent same experiment was performed between another three human ESCC tissues and normal tissues. There was no difference between twice experiments. Table 3 showed that the selected five proteins were stable differential expression and that iTRAQ was an effective means for the ESCC proteomic quantification.
Discussion
Clarifying the pathogenesis of ESCC is essential for providing new treatment strategies and take preventive measures. It was the most basic level to detect potential biomarkers of ESCC by analyzing different protein expressions. 2DE/MALDI-TOF MS has been widely used for initial and comprehensive exploration of some cancers, such as buccal squamous cell carcinoma, cholangiocarcinoma, prostate cancer, squamous cervical cancer, renal cell carcinoma, lung squamous carcinoma, and gastric carcinoma. At the same time, plenty differential proteins were screened to be as the basis of the further study. However, there were some disadvantages of 2DE such as difficulties resolving hydrophobic proteins and low-abundance proteins with extreme pI and molecular weights [8]. In recent years, iTRAQ labeling technique combined with 2DE/MALDI-TOF MS has been increasingly used label quantification methods. Because the unaccessible information by 2DE can be obtained using iTRAQ. It is a highly sensitive proteomic platform and is considered as a superior choice due to its high proteome coverage and labeling efficiency. In addition, the iTRAQ has the advantage of analysis capability of multiple samples simultaneously, and the low-abundance protein sensitivity is enough to meet important research, for example, breast cancer, lung cancer, and nasopharyngeal carcinoma [9]. Therefore, to choose the identified proteins, the iTRAQ and 2DE/MALDI-TOF MS were employed in this study.
Only through a subset of residues in any protein can protein interactions be investigated. Finn had established a web resource that can retrieve protein interactions by the Protein Data Bank structures at the level of Pfam domains and amino acid residues [10]. In this study, the web was mainly used to analyze the protein interactions. RRM and helicase C were found as the second outstanding Pfam domains involved in 34 proteins.
Among the Pfam domains, RRMs were the main part of RNA-binding proteins, which were used to pre-messenger RNA (mRNA) splicing. SR protein family, whose members play roles in splicing, can transmit pre-mRNAs to the splicing pathway with overlapping [11]. The present study found six SR differential proteins, including in SRSF1, SRSF2, SRSF5, SRSF9, SRSF10, and SRSF11. SR proteins were composed by one or two N-terminal RRMs and a variable length C-terminal arginine–serine (RS)-rich domain [11]. The RRMs are required for trans-acting factors binding, while the RS domains are required for protein–protein interactions with other components of the splicing machinery and serve as splicing activation domains. SR proteins serve as recruiting components of the general splicing machinery to the nearby intron [11]. Xu provided persuasive evidence of cancer-specific splice variants by 316 human genes [12]. SR proteins control mRNA translocation and translation [13]. RBM25, another splicing factor, was a different expression between tumor tissues and adjacent normal epithelium tissues. There was a family of RNA-binding proteins sharing the N-terminal RRM, ER central region, and C-terminal PWI motif [14]. RBM25 regulated the RNA-binding and directed the alternative splicing of apoptotic factors [15].
There are five novel proteins, P4HA1, P4HA2, CNN2, ISLR, and P3H1/LEPRE1, found in this study. However, the five potential candidates hardly were reported in any carcinoma.
The family of collagen prolyl 4-hydroxylases α isoforms, including in P4HA1, P4HA2, and P4HA3, have been identified in human tissue. P4HA1, as the key enzyme in collagen synthesis, provides the major part of the catalytic site of the active enzyme. Its function is prompting the posttranslational formation of 4-hydroxyproline in -Xaa-Pro-Gly- sequences in collagens and other proteins [16]. When the prolyl 4-hydroxylases activity is inhibited efficiently, cell–collagen interaction will be enhanced, including proliferation, migration, and invasion [17]. It has been demonstrated that the P4HA1 positive was related with breast cancer and prostate cancer [18, 19]. According to Chakravarthi, in vitro studies P4HA1 played a critical role in prostate cancer progression and tumor cell growth. Molecular typing studies about P4HA1 in prostate cancer also suggested that P4HA1 played an important role in the regulation of matrix metalloproteases 1 [19]. P4HA2 is mainly expressed in capillary endothelial cells, chondrocytes, and osteoblasts. High expression of P4HA2 has been observed in breast cancer, papillary thyroid cancer, and oral cavity squamous cell carcinoma [20–22]. Gaofeng Xiong results suggested that collagen deposition caused by P4HA2 promoted the progression of breast cancer. It was conceivable that inhibiting prolyl 4-hydroxylases activity is a new potential therapeutic target to breast cancer [18]. Moreover, Daniele M. Gilkes discovered that the hydroxylase inhibitor, ethyl 3,4-dihydroxybenzoate (DHB), was able to reduce the rate of breast cancer fibrosis and metastasis [18]. However, the main function of P4HA1 and P4HA2 in cancer progression remains to be poorly understood. Therefore, further research was worth doing.
Calponin, a family of actin-associated proteins, was first identified in smooth muscle cells. It is composed of three isoforms: h1-calponin, h2-calponin, and acidic calponin. The actin cytoskeleton as ezrin worked in many cellular processes, including in contraction, cell division, anchorage-independent cell growth, cell migration, and invasion [23]. Kaneko reported that H1-calponin was associated with tumorigenesis, cell migration, and inhibition of cell differentiation [24]. H1-calponin has also been found related with renal angiomyolipoma and colon cancer [25]. Moreover, Tomonori Ogura had demonstrated that the drugs targeting the CNh1 gene were effective against ovarian cancer by in vitro cell study [26]. h2-calponin, encoded by CNN2, can bind actin, tropomyosin, troponin C, and calmodulin. Its function was to direct the structural organization of actin filaments. The interaction between calponin and actin inhibits the actomyosin Mg-ATPase activity. The main function of h2-calponin was to regulate smooth muscle contraction, motor activity, and cell adhesion. Hossain MM suggested that in growing and remodeling tissues, the expression of h2-calponin is at a significant level [27]. Choi also found that h2-calponin was of high expression in cutaneous squamous cell carcinoma and rectal carcinoma [28]. Moreover, Hossain indicated that its overexpression reduced cell proliferation [29]. The gene regulation and function of h2-calponin are largely unknown. Therefore, a large number of studies are eager to be carried out.
Immunoglobulin superfamily (ISLR) containing a leucine-rich repeat (LRR) with conserved flanking sequences and a C2-type immunoglobulin (Ig)-like domain was considered to be a new member of the Ig superfamily [30]. It was known that these domains be critical to protein–protein interaction or cell adhesion. Therefore, the ISLR protein may also interact with other proteins or cells. However, only a little part of proteins is reported including immunoglobulin domain and LRR.
Prolyl 3-hydroxylase1 (encoded by LEPRE1) is a collagen-modifying enzyme, which is composed of carboxyl-terminal dioxygenase domain and amino-terminal domain. The carboxyl-terminal dioxygenase domain is similar to the α-subunit of prolyl 4-hydroxylase and lysyl hydroxylases. The amino-terminal domain contains four cysteine (CXXXC) repeats. Although its function is not clear, it contains multiple tetratricopeptide repeat domains that are important in protein–protein interactions [31]. It has been suggested that the P3H1 complex may function as both an enzyme and a chaperone in collagen molecular assembly, possibly by preventing premature aggregation of collagen chains during protein synthesis [32].
In this study, the sample size was less, but all samples selected were according to the principle of grouping and pairing. Paired design has the advantages of keeping the uniformity of the conditions, higher comparability, higher validity, and less error. So, by comparing ESCC tissues to paired adjacent normal epithelium tissues, the differential proteins were found effectively. On the other hand, the repeated tests could reduce the deviation, screening the shared differential proteins. In a word, the grouping and pairing study can provide more accurate and effective results. The selected differentially expressed proteins were reliable and objective (Fig. 6).
In recent years, there were similar experiments reported about LC–MS-based proteomic analysis in ESCC. And almost all of the experimenters regarded the serum as sample. For example, Harsha Gowda selected 40 ESCC and 10 control serum samples to compare and identify dysregulated proteins or molecular features, and Zhang Li selected 10 ESCC and 10 control serum samples [33, 34]. However, Harsha Gowda and Zhang Li found respectively 652 significantly dysregulated molecular features in serum and 21 expressed proteins, most of which were only about metabolomic. The results of our experiment involved in cell proliferation, cell differentiation, cell adhesion, cell migration, apoptotic, and so on. In addition, the protein level in serum was lower to the protein levels in tissue. Therefore, the accuracy of serum experiments was lower compared to the histological experiments. It was a new way to regard tissue as samples of the protein identification experiment by LC–MS. The advantages of our experiment were to more easily extract proteins, to obtain more dysregulated and more comprehensive proteins, and to keep the accuracy of the results.
Conclusion
By iTRAQ technique and LC–MS/MS, 72 and 57 unique proteins were significantly up-regulated and down-regulated between ESCC tissues and paired adjacent normal epithelium tissues. Among the differential proteins, five novel proteins, including in P4HA1, P4HA2, CNN2, ISLR, and P3H1/LEPRE1, were discovered as the identified proteins, and the further research and the experiment of a larger sample size were worth doing.
References
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90.
Zhang P, Xi M, Li Q-Q, He L-R, Liu S-L, Zhao L, et al. The modified Glasgow prognostic score is an independent prognostic factor in patients with inoperable thoracic esophageal squamous cell carcinoma undergoing chemoradiotherapy. J Cancer. 2014;5(8):689–95.
Keszei AP, Alexandra Goldbohm R, Schouten LJ, Jakszyn P, van den Brandt PA. Dietary N-nitroso compounds, endogenous nitrosation, and the risk of esophageal and gastric cancer subtypes in the Netherlands. Am J Clin Nutr. 2013;97(1):135–46.
D’Journo XB, Thomas PA. Current management of esophageal cancer. J Thorac Dis. 2014;6 Suppl 2:S253–64.
Guo X, Hao Y, Kamilijiang M, Hasimu A, Yuan J, Wu G, et al. Potential predictive plasma biomarkers for cervical cancer by 2D-DIGE proteomics and ingenuity pathway analysis. Tumour Biol. 2015;36(3):1711–20.
Atrih A, Mudaliar MA, Zakikhani P, Lamont DJ, Huang JT, Bray SE, et al. Quantitative proteomics in resected renal cancer tissue for biomarker discovery and profiling. Br J Cancer. 2014;110(6):1622–33.
Gan CS, Chong PK, et al. Technical, experimental, and biological variations in isobaric tags for relatives and absolute quantitation (iTRAQ). J Proteome Res. 2007;6(2):821–7.
Gilmore JM, Washburn MP. Advances in shotgun proteomics and the analysis of membrane proteomes. J Proteomics. 2010;73(11):2078–91.
Xiao Z, Li G, Chen Y, Li M, Peng F, et al. Quantitative proteomic analysis of formalin-fixed and paraffin-embedded nasopharyngeal carcinoma using iTRAQ labeling, two-dimensional liquid chromatography and tandem mass spectrometry. J Histochem Cytochem. 2010;58(6):517–27.
Finn RD, Marshall M, Bateman A. Pfam: visualization of protein–protein interactions in PDB at domain and amino acid resolutions. Bioinformatics. 2005;21(3):410–2.
Saccone G, Louis C, Zhang H, Petrella V, Di Natale M, Perri M, et al. Male-specific phosphorylated SR proteins in adult flies of the Mediterranean fruitfly Ceratitis capitata. BMC Genetics. 2014;15 Suppl 2:S6.
Xu Q, Lee C. Discovery of novel splice forms and functional analysis of cancer-specific alternative splicing in human expressed sequences. Nucleic Acids Res. 2003;31(19):5635–43.
Sanford JR, Gray NK, Beckmann K, Cáceres JF. A novel role for shuttling SR proteins in mRNA translation. Genes Dev. 2004;18(7):755–68.
Fortes P, Longman D, McCracken S, Ip JY, Poot R, Mattaj IW, et al. Identification and characterization of RED120: a conserved PWI domain protein with links to splicing and 3′-end formation. FEBS Lett. 2007;581:3087–97.
Zhou A, Ou AC, Cho A, Benz Jr EJ, Huang SC. Novel splicing factor RBM25 modulates Bcl-x Pre-mRNA 5′ splice site selection. Mol Cell Biol. 2008;28(19):5924–36.
Gilkes DM, Saumendra B, Pallavi C, Denis W, Semenza GL. Hypoxia-inducible factor 1 (HIF-1) promotes extracellular matrix remodeling under hypoxic conditions by inducing P4HA1, P4HA2, and PLOD2 expression in fibroblasts. J Biol Chem. 2013;288(15):10819–29.
Zhang K, Corsa CA, Ponik SM, Prior JL, et al. The collagen receptor discoidin domain receptor 2 stabilizes SNAIL1 to facilitate breast cancer metastasis. Nat Cell Biol. 2013;15(6):677–87.
Gilkes DM, Chaturvedi P. Collagen prolyl hydroxylases are essential for breast cancer metastasis. Cancer Res. 2013;73(11):3285–96.
Chakravarthi BV, Pathi SS, Goswami MT, et al. The miR-124-prolyl hydroxylase P4HA1-MMP1 axis plays a critical role in prostate cancer progression. Oncotarget. 2014;5(16):6654–69.
Pan PW, Zhang Q, Bai F, Hou J, Bai G. Profiling and comparative analysis of glycoproteins in Hs578BST and Hs578T and investigation of prolyl 4-hydroxylase alpha polypeptide II expression and influence in breast cancer cells. Biochemistry (Mosc). 2012;77(5):539–45.
Jarzab B, Wiench M, Fujarewicz K, Simek K, et al. Gene expression profile of papillary thyroid cancer: sources of variability and diagnostic implications. Cancer Res. 2005;65(4):1587–97.
Chang KP, Yu JS, Chien KY, Lee CW, Liang Y, et al. Identification of PRDX4 and P4HA2 as metastasis associated proteins in oral cavity squamous cell carcinoma by comparative tissue proteomics of microdissected specimens using iTRAQ technology. J Proteome Res. 2011;10(11):4935–47.
Ridley AJ. Rho GTPases and actin dynamics in membrane pro-trusions and vesicle trafficking. Trends Cell Biol. 2006;16(10):522–9.
Kaneko M, Takeoka M, Oguchi M, Koganehira Y, et al. Calpon in hl suppresses tumor growth of src-induce d transformed 3Y1 cells in association with a de crease in angiogenesis. Jpn J Cancer Res. 2002;93(8):935–43.
Yanagisawa Y, Takeoka M, Ehara T, Itano N, Miyagawa S, Taniguchi S. Reduction of calponin H1 expression in human colon cancer blood vessels. Eur J Surg Oncol. 2008;34(5):531–7.
Ogura T, Kobayashi H, Ueoka Y, Okugawa K, Kato K, Hirakawa T, et al. Adenovirus-mediated calponin h1 gene therapy directed against peritoneal dissemination of ovarian cancer: bifunctional therapeutic effects on peritoneal cell layer and cancer cells. Clin Cancer Res. 2006;12(17):5216–23.
Hossain MM, Hwang D-Y, Huang Q-Q, Sasaki Y, Jin J-P. Developmentally regulated expression of calponin isoforms and the effect of h2-calponin on cell proliferation. Am J Physiol. 2003;284(1):156–67.
Choi SY, Jang JH, Kim KR. Analysis of differentially expressed genes in human rectal carcinoma using suppression subtractive hybridization. Clin Exp Med. 2011;11(4):219–26.
Hossain MM, Hwang D-Y, Huang Q-Q, Sasaki Y, Jin J-P. Developmentally regulated expression of calponin isoforms and the effect of h2-calponin on cell proliferation. Am J Physiol. 2003;284(1):C156–67.
Nagasawa A, Kubota R, Imamura Y, Nagamine K, et al. Cloning of the cDNA for a new member of the immunoglobulin superfamily (ISLR) containing leucine-rich repeat (LRR). Genomics. 1997;44(3):273–9.
D’Andrea LD, Regan L. TPR proteins: the versatile helix. Trends Biochem Sci. 2003;28(12):655–62.
Ishikawa Y, Wirz J, Vranka JA, Nagata K, Bächinger HP. Biochemical characterization of the prolyl 3-hydroxylase 1.cartilage-associated protein.cyclophilin B complex. J Biol Chem. 2009;284(26):17641–7.
Ahmad Mir S, Pavithra R, Jain AP, Khan AA, Datta KK, Mohan SV, et al. LC–MS-based serum metabolomic analysis reveals dysregulation of phosphatidylcholines in esophageal squamous cell carcinoma. J Proteomics. 2015;S1874-3919(15):00231–6.
Li C, Guo X, Jianqing Z, Yang M, Ge B, Li Z. Serum differential protein identification of Xinjiang Kazakh esophageal cancer patients based on the two-dimensional liquid-phase chromatography and LTQ MS. Mol Biol Rep. 2014;41(5):2893–905.
Conflicts of interest
None
Funding
This study was supported by the grant from the National Natural Science Foundation of China, NO: 81260308, and the Open Subject about Xinjiang Major Diseases of China, NO: SKLIB-XJMDR-2014-12.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Feiyan Deng and Keming Zhou contributed equally to this work.
Rights and permissions
About this article

Cite this article
Deng, F., Zhou, K., Li, Q. et al. iTRAQ-based quantitative proteomic analysis of esophageal squamous cell carcinoma. Tumor Biol. 37, 1909–1918 (2016). https://doi.org/10.1007/s13277-015-3840-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-015-3840-1