
Abstract
The current methods available for screening and detecting cervical squamous cell carcinoma (CSCC) have insufficient sensitivity and specificity. As a result, many patients suffered from erroneous and missed diagnosis. Because CSCC is usually asymptomatic at potentially curative stages, identification of biomarkers is an urgent need for the early detection of CSCC. Comparative proteomics based on two-dimensional differential in-gel electrophoresis (2D-DIGE) was employed to quantitatively analyze plasma proteins of healthy Uyghur women and with early stage cervical carcinoma. The 2D-DIGE image were analyzed statistically using DeCyder™ 2D software. The statistical analysis of proteomic data revealed that 43 protein spots showed significantly different expression (ratio > 1.5, P < 0.01). A further identification of these protein spots by MALDI-TOF-MS found out 16 different proteins. Bioinformatic analysis within the framework of Ingenuity Pathway Analysis (IPA@) showed that 10 plasma proteins as candidate biomarker were screened, mainly including lipid metabolism-related proteins (APOA4, APOA1, APOE), complement (EPPK1, CFHR1), metabolic enzymes (CP, F2, MASP2), glycoprotein (CLU), and immune function-related proteins (IGK@). Networks involved in lipid metabolism, molecular transport, and small molecule biochemistry were dysfunctional in CSCC. Acute phase response signaling and JAK/Stat signaling and IL-4 signaling, etc., were identified as the canonical pathways that are overrepresented in CSCC. Furthermore, the expression of three proteins (APOA1, APOE, CLU) were validated using ELISA in plasma of patients with different stage cervical lesion. With the combined proteomic and bioinformatic approach, this study was successful in identifying biomarker signatures for cervical cancer and might provide new insights into the mechanism of CSCC progression, potentially leading to the design of novel diagnostic and therapeutic strategies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cervical cancer is one of the endemic diseases of high-incidence in Xinjiang, China [1]. The prevalence rate of cervical cancer among ethnic Uyghur women in south part of Xinjiang (590/100,000) is four times of the average in China (138/100,000) [2, 3]. According to clinical data, the proportion of hospitalized patients with cervical cancer reaches up to 20 %, of which Uyghur women account for more than 75 %, and most of them are detected at medium or advanced stages of the cancer, who may lose the opportunity of surgical therapy [4]. Therefore, there is an urgent need to establish specific and sensitive methods for early diagnosis of cervical cancer and its precursor lesions, or prognosis for long-term survival of patients.
Current approaches for the prevention of cervical cancer is mainly the cytologic screening, known as the Pap test, often combined with the detection of high-risk human papillomaviruses (hr-HPVs) [5]. Patients with abnormal Paps undergo colposcopy with directed biopsies. If precursor lesions are identified, the patients receive and cryotherapy or loop electrosurgical excision procedure (LEEP). The treatment is effective in the prevention of cervical cancer, however, it is expensive, cumbersome, and dependent on very good infrastructure and well-trained personnel [6]. Nevertheless, the diagnosis may results in a poor outcome, which lies on the lack of valuable objective indicators for determining cervical chronic inflammation, reactive hyperplasia, and benign or malignant lesions.
The development and survival of tumor cells are just the failure of the body in immune surveillance, and the pathological state associated with dynamic changes in whole body, which is not only manifested in the subtle deregulation of gene expression network within tumor cells but also integrated into plasma proteome as a fine tuning, in the form of plasma (serum) protein markers [7]. In recent years, the identification and application of such molecular markers have already become a cutting edge in the establishment of noninvasive methods in clinical diagnosis [8, 9]. Thus, plasma (serum) proteomics promises a revolution in early diagnosis and therapeutic monitoring as well as revealing the molecular mechanisms of cervical carcinogenesis.
Compared with conventional 2-DE, two-dimensional differential in-gel electrophoresis (2D-DIGE)-based quantitative proteomics has several advantages, such as higher sensitivity, accuracy, and reproducibility, which facilitate spot-to-spot comparisons, precisely because of pre-labelling of protein samples with different fluorescent dyes (Cy2, Cy3, and Cy5) prior to separation by 2-DE [10]. As a result, samples labeled with different dyes are separated in the same 2D gel, in addition to the same internal standard used in all gels to avoid inter-gel variation [11].
In this study, we analyzed the plasma proteomic profile Uygur women with cervical cancer using 2D-DIGE system followed by protein detection using matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF-MS) for identification of differentially expressed proteins as potential plasma biomarkers for noninvasive diagnosis. Additionally, the differentially expressed proteins were analyzed by a bioinformatics approach (IPA@) for functional annotation and enrichment, canonical pathway analysis, and biomarker assessment. Furthermore, candidate plasma protein markers were validated by enzyme-linked immunosorbent assay (ELISA). The findings of this study will contribute to establishing diagnostic methods based on plasma biomarkers for cervical cancer, and revealing the biological nature of cervical pathogenesis and carcinogenesis.
Materials and methods
Instruments and reagents
Enrichment Kit specific to low-abundance proteins (ProteoMiner™ Protein Enrichment Kits), Isoelectric focusing device (protein IEF cell), and Vertical Electrophoresis System were purchased from Bio-Rad Company; fluorescent dye, dithiothreitol (DTT), iodoacetamide (IAA), solid phase pH gradient nonlinear dry strip (IPG dry strip), and IEF buffer (pH 4∼7) were provided by Sweden Amersham Biosciences Company; CHAPS, SDS, Tris, NH4HCO3, mineral oil, glycerol, glycine, and urea were provided by Sigma-Aldrich Company; Scanners (Typhoon 9410) was purchased from America GE Healthcare Company; Image analysis software (DeCyder™ 6.5 image analysis) and Mass Spectrometer (ABI-4800) were provided by America ABI Company; Trypsin was purchased from Promega Company; BCA Protein Assay Kit was provided by Novagen Company; IPA@ online bioinformatics software was provided by America Ingenuity Systems Company.
Plasma sample preparation
This study was approved and monitored by the Ethics Committee of Xinjiang Medical University, Urumqi, China. Signed informed consent was obtained from all patients and healthy individuals, and all data was analyzed anonymously throughout the study. For proteomics analysis, plasma samples from 48 Uyghur women were collected including 26 cases of early stage cervical squamous cell carcinoma (CSCC) (before the stage IIa), and 22 cases of age-matched healthy controls, before any clinical treatment, at the Affiliated Cancer Hospital of Xinjiang Medical University from January 2008 and September 2010. The CSCC samples were obtained from women before the stage IIa for early warning indicators screening because the cervical cancer of before stage IIa is considered as early stage of cervical cancer, and the patients before stage IIa can be treated with surgery. The diagnosis was confirmed by histopathology for all patients, and the cancer stage was established according to the International Federation of Gynecology and Obstetrics (FIGO). Patients’ age ranged from 25 to 65 years, with a mean of 53 ± 3 years. For each person, 3 ml blood was obtained by venipuncture into evacuated blood collection tubes that contained EDTA as anticoagulant, and the plasma was preserved for further use after centrifugation at −80 °C.
Enrichment of low-abundance proteins
Plasma samples from 26 early stage CSCC patients and 22 individuals in the control group were mixed (70 µl each) to form a samples pool for further test by 2D-DIGE. Pooled plasma samples were enriched of low-abundance proteins, by depletion of the most abundant proteins using a prepacked 1-ml affinity LC column provided by the ProteoMiner™ Protein Enrichment Kit (Bio-Rad) according to the recommended manufacturer’s procedure. Simply, the column containing 1 ml settled beads was washed with 1 ml deionized water and 1 mL wash reagent followed by incubation with 1 ml whole plasma sample. With the elimination of high-abundance proteins by saturation limit of the column, low-abundance proteins bound to the column were preserved by subsequent wash procedure and dissolved in 100 µl rehydration elution reagent.
2D-DIGE analysis and in-gel digestion
Low-abundance proteome plasma sample was pre-treated with ProteoMiner Protein Enrichment Kit. We identified the total protein by utilizing BCA reagent (Novagen) with 10, 20, and 30 times series of gradient dilution. After enrichment and quantification of healthy control and early stage CSCC groups of low-abundance protein, took 50 μg of plasma samples in each group, adjusted the pH to 8.5 by 50 mM sodium hydroxide. Then, 50 μg protein from each sample were pooled together as the internal standard. A total of 50 μg protein of each sample were randomly labeled with 400 pmol Cy3 or Cy5, and the internal standard was labeled with Cy2 for 30 min on ice. Samples were then rehydrated into 24 cm IPG strips (pH 3–10) (GE Healthcare, UK) overnight in rehydration buffer (2 M thiourea, 7 M urea, 2 % CHAPS, 0.2 % DTT, and 1 % IPG buffer 3–10, GE Healthcare). Isoelectric focusing was carried out using an IPGphor apparatus (GE Healthcare) and the following procedures are as follows: 12 h at 50 V, 2 h at 100 V, 2 h at 300 V (gradient), 2 h at 600 V (gradient), 2 h at 1,000 V (gradient), 2 h at 3,000 V (gradient), and 2 h at 9,000 V. Strips were equilibrated for 15 min in 50 mM Tris, 6 M urea, 2 % SDS, and 30 % glycerol, containing 1 % DTT, and then for 15 min in the same buffer containing 2.5 % iodoacetamide. Equilibrated IPG strips were transferred onto 12 % SDS-polyacrylamide gels that had been pre-casted in low fluorescence glass plates. The second dimension separation was then carried out at 20 W/gel using Ettan Dalt six electrophoresis system (GE Healthcare, UK). After the 2-DE separation, the gels were scanned using the Typhoon 9410 scanner (GE Healthcare, UK).
The images were then analyzed using the DeCyder™ 2D Differential Analysis Software (DeCyder 2D V8.0) by differential in-gel analysis (DIA) and biological variation analysis (BVA). Protein spots were marked and selected with changes in abundance ratio >1.5-fold, P values <0.01 for protein identification. The gels were subjected to Coomassie blue staining for spot visualization and picking. Spots of interest were excised and destained with 15 mM potassium ferricyanide and 50 mM sodium thiosulphate. The spots were then reduced in 10 mM DTT followed by alkylation in 55 mM IAA (iodoacetamide). After being washed with 50 % acetonitrile (ACN) and dehydrated in 100 % ACN, the gel fragment was dried in a speed vacuum. The dried gel fragment was then digested with 7 ng/μl trypsin (Promega, CA, USA) overnight at 25 °C. The digested proteins were extracted twice with 50 % ACN and followed by 100 % ACN and kept at −80 °C until further analysis by MALDI-TOF-MS/MS.
MALDI-TOF/TOF MS analysis
After enzyme digestion, the tryptic-digested peptides were spotted on a MALDI plate. MALDI-TOF-MS and TOF/TOF tandem MS were performed on an ABI 4800 mass spectrometer (Applied Biosystems, Framingham, MA, USA). TOF/TOF tandem MS fragmentation spectra were acquired for each sample, averaging 4000 laser shots per fragmentation spectrum on each of the 5–10 most abundant ions present in each sample excluding trypsin autolytic peptides and other known background ions. Both the resulting peptide mass and the associated fragmentation spectra were submitted to the MASCOT (Matrix Science, London, UK) search engine in order to search the nonredundant database of SwissProt databases (IPI human 3.62 database). Searches were performed without restricting the protein molecular weight or isoelectric point, with variable carbamidomethylation of cysteine and oxidation of methionine residues and with one missed cleavage allowed in the search parameters. Candidates with either a protein score confidence interval (C.I.%) greater than 95 or Mascot scores greater than 61 were considered significant as positive identifications. The maximum error permitted in the search were MS 0.2 Da and MSMS 0.3 Da.
IPA@ bioinformatics analysis
The differentially expressed proteins were characterized by core analysis and Biomarker® Filter analysis of the online IPA@ software package (Version 8.7) for getting insight into the molecular mechanism of the protein through annotations of biological functions, identification of interaction networks and canonical pathways, and the discovery of potential biomarkers. Set the network size parameters in order to optimize the network visualization and analyze the biologically relevant background. Networks analysis can provide a quick solution to assess the data of interest regulatory networks, metabolic pathways, and physiological processes. Biofunction analysis could aid to understand the interaction of differential protein in the known pathways which have been researched maturely, to distinguish how proteins affect the phenotype and whether those proteins can increase or decrease a biological process.
By IPA Biomarker® Filter, we can optimize the candidate biomarker quickly and determine the most likely candidate biomarkers which are most relevant to experimental data. Those candidate biomarkers could be used as prediction for disease diagnosis and biomarker for disease progression in clinical and/or they can be used as markers for drug efficacy and safety and biomarkers for patient response to treatment.
Validation experiments by ELISA
The expression of candidate plasma biomarkers proteins identified by IPA were further analyzed by enzyme-linked immunosorbent assay (ELISA) according to manufacturer’s instruction in re-collected 82 cases of plasma samples composed of four cervical lesions group, including chronic cervicitis (n = 22); HSIL (high-grade squamous intraepithelial lesion, i.e. cervical intraepithelial lesion II–III, CINII–III) (n = 20); cervical cancer of before stage IIa, (n = 20); cervical cancer of after stage IIa, (n = 20), respectively. For all ELISA experiments, there were three biological replicates. The final data was confirmed by three independent ELISA detections of each plasma protein. The standard curves were created with protein concentration on the y-axis and average absorbance on the x-axis. Then the results of concentration were calculated from the standard curves and multiplied by the dilution factor. For comparison of means, ANOVA tests were used as appropriate (SPSS 17.0 software).
Statistical analysis
The level of protein expression between different groups of ELISA experiment was compared using one-way ANOVA test. All statistical analysis was performed with SPSS17.0 (SPSS Inc, Chicago, IL, USA), and P values ≤0.05 were considered statistically significant.
Result
2D-DIGE map analysis
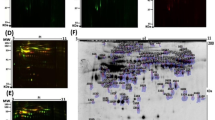
To systematically compare the performance of different Cy staining methods used in differential protein profiling, a DIGE gel was run with an independently prepared proteome from the plasma from a paired chronic cervicitis (control, Ctl) stained with Cy3 (green), from CSCC group stained with Cy5 (red), and from a mixture of all protein samples as the internal standard stained with Cy2 (blue) (Fig. 1). The black and white scanning images of each stained gel were showed at the right (Fig. 2). The digital images were analyzed using DeCyder software. A total of 43 spots were significantly and eventually selected for MALDI-TOF/TOF identification on the basis of Student’s t test P values <0.05 and fold changes >1.5 from images analysis by DeCyder software. Compared with the corresponding samples of normal tissue, 18 spots showed agreater abundance and 25 showed a lower abundance in the CSCC groups.

Representative 2D-DIGE proteome map of CSCC plasma samples vs. benign plasma samples.(a Chronic cervicitis group (control, Ctl) stained with Cy3 (green), b CSCC group stained with Cy5 (red), c a mixture of all protein samples as the internal standard stained with Cy2 (blue), and d overlapping of three groups stained with Cy2, Cy3, and Cy5, respectively

The differential expression profile of low abundant plasma proteome between patients with early stage cervical carcinoma and cervicitis based on the analysis by two-dimensional electrophoresis. Green box was the location of differential expression protein spots, and the digital in the yellow box were the number of differential expression protein spots
Differentially expressed proteins identified by mass spectrometry
The 43 significantly altered spots were selected for further identification by MALDI-TOF/TOF MS analysis. Following a Mascot database search using the acquired MS data, 16 proteins out of 43 spots were identified as differentially expressed in CSCC group as compared to control group. Details of the experimental findings for protein identification, protein score, theoretical pI value, molecular weight, and average relative change are shown in Table 1.
In our parallel study, we have already screened out the 103 proteins (to be published) using two-dimensional liquid chromatography (2D HPLC) and LTQ mass spectrometry (LTQ-MS). Among 103 proteins which coincided with this study were A-I (apolipoprotein A-I, APOA1), APOE (apolipoprotein E, APOE), CLU (clusterin, CLU), and IGK@ (immunoglobulin kappa, IGK@).
Networks analysis of candidate proteins by Ingenuity Pathway Analysis
A total of 16 proteins identified by mass spectrometry were analyzed by IPA@ core analysis in the PANTHER database in terms of their networks function, molecular and cellular function, and canonical signal transduction pathway. For the category of associated network function, the proteins were classified into groups consisting of lipid metabolism, molecular transport, small molecule biochemistry, cellular development, cellular growth and proliferation, and hematological system development and function (Fig. 3). For the category of molecular and cellular function, the proteins were classified into groups consisting of cell-to-cell signaling and interaction, lipid metabolism, molecular transport, small molecule biochemistry, and cell death and survival (Fig. 4). For the category of canonical pathways, the proteins were classified into groups consisting of LXR/RXR activation and acute-phase response pathway (Fig. 5).

Associated network function of differentially expressed proteins in patients with CSCC

Molecular and cellular functions of differentially expressed proteins in patients with CSCC

Canonical pathway analysis of differentially expressed proteins in patients with CSCC
Candidate biomarkers were filtered from the differentially expressed proteins by the terms “cancer” provided in GeneGo diseases ontology using biomarker assessment workflow tool, and the results shown in Table 2 are mainly the following: lipid metabolism-related proteins (APOA4, APOA1, APOE), complement (EPPK1, CFHR1), metabolic enzymes (CP, F2, MASP2), glycoprotein (CLU), and immune function-related proteins (IGK@). It is designed to analyze proteomic data by matching them with biomarkers known for a disease. All relevant information is assembled by GeneGo scientists as a result of the scrupulous hand curation process based on the published peer-reviewed literature.
The validation of ELISA assay
The plasma level of three potential biomarkers APOA1, APOE, and CLU was measured in plasma samples from 82 patients by ELISA (Table 3 and Fig. 6). The results showed gradually altered plasma concentration of the three proteins with pathological progression of uterine cervix from the normal to cervical precursor lesions (HSIL), and early and late stage CSCC. Among them, the plasma level of APOA1 was downregulated, whereas APOE and CLU was upregulated significantly (P < 0.05).

The levels of APOA1, APOE, and CLU are significantly different in HSIL and cervical cancer of before and after stage IIa groups compared to chronic cervicitis by ELISA assay. Comparisons of means (ANOVA) were made among four cervical lesions groups. In HSIL and cervical cancer, there was a significant decrease of APOA1 and increase of APOE and CLU. The plotted were considered as outliers
Discussion
Potential disease biomarkers are often present at low concentration, and high-abundance protein depletion is a formidable technical challenge of such in-depth proteomic analyses due to the complexity and extremely large dynamic range of protein concentrations in plasma. Therefore, it is necessary to reduce sample complexity by either depleting plasma samples of high abundant proteins with specific antibody-based affinity columns or by multidimensional pre-fractionation strategies such as liquid-phase isoelectric focusing (IEF) or chromatography. A novel approach for mining the “unseen proteome” is the bead-based library of combinatorial peptide ligands, ProteoMiner, recently described by Righetti et al. [12].
In our research, ProteoMiner™ protein enrichment kit accomplished through the use of a large, highly diverse bead-based library of combinatorial peptide ligands. So when complex plasma is applied to the beads, the high-abundance proteins saturate their high affinity ligands and excess protein is washed away. In contrast, the medium and low-abundance proteins are concentrated on their specific affinity ligands. As a result of that, high-abundance proteins in our plasma samples had the same concentration, while only low-abundance proteins had various contents. The eluted material will therefore consist of significantly lower amount of total protein representing a higher diversity of species. Sihlbom et al. [13] evaluated the combination of beads with SELDI-TOF-MS and two-dimensional differential gel electrophoresis (2D-DIGE) regarding their efficiency, reproducibility, sensitivity, and compatibility, and the data suggest that integration of the bead technology with 2D-DIGE proteomic technologies will enhance the possibility to deliver new peptide/protein biomarker candidates. It is validated by forward research of many researchers for their effectiveness [13, 14].
Tumor development is accompanied by a complex host systemic response, which reflecting with plasma proteome, which are more likely to contain information of tumor markers [15–17]. Using the proteomic technique to screen and monitor its existence or content change has important implication for early detection and diagnosis, monitoring recurrence or metastasis, guiding treatment and judging prognosis for cervical cancer [18–21]. Our proteomic data revealed 43 spots detected by 2D-DIGE that were altered in plasma of CSCC compared to the benign group. Among these spots, 43 were significantly altered in their abundance (r > 1.5 folds, P < 0.01). Compared to the control, 16 spots were in higher abundance and 27 were at lower abundance in the CSCC group and judged to be valid and selected for further identification. Following a Mascot database search using the acquired MS data, 16 proteins of 43 spots were identified as differentially expressed in CSCC compared to benign group. Based on criteria which included network enrichment, molecular and cellular biofunction, canonical transduction pathway, biomarker filter, 16 proteins identified by mass spectrometry were analyzed by IPA@ online bioinformatics platform.
Several differences of the plasma proteome profile between CSCC and inflammatory states were identified; many are unique candidates for active contributors to the generation of CSCC involved in LXR/RXR activation, acute-phase response pathway and transcriptional signaling pathway, or different adaptor molecules and receptors, cross-talk with other regulatory pathways, and activate or suppress a plethora of kinases that are involved in a multitude of signaling cascades that are important factors in shaping the type, magnitude, and duration of the inflammatory response, leading to cytological anomalies or cancer [21–23]. IPA@ analysis provides a quick solution and assesses the data of interest regulatory networks, metabolic pathways, molecular action networks, and physiological processes.
Owing to the consistency of statistical results obtained from analyses of CSCC and control group (paired t test N, average ratio, Mascot score, multipoint repetitive identification, IPA@ bioinformatics analysis), a total of 10 plasma proteins as candidate biomarker were screened by biomarker filter analysis of the IPA@ [24]. The IPA database consists of proprietary ontology representing 300,000 biologic genes, proteins, and molecular and cellular processes [25]. It can provide a trusted resource, thus explaining and mining the experimental data. Based on this database, we successfully identified several proteins that were definitely involved in cancer, including APOA1, APOE, CP, CLU, FBLN1, MASP2, and TTR. But some proteins investigated in our study were not associated with cancer so far, including APOA4 and F2. In addition, the study about IGK@ protein that has been reported is rare in the recent days, but it has been screened out in a variety of body fluids proteomic studies [26]. This is the first report to confirm that the upregulated expression proteins IGK@ are related to cervical tumorigenesis, which could provide illustration for the following study of this protein. In our previous studies, we applied multiple proteomics strategies and techniques for identification of candidate plasma protein markers. We have already screened out of the 103 proteins (to be published) using 2D HPLC coupled with LTQ-MS. Among 103 proteins which coincided with this study were APOA1, APOE, CLU, and IGK@. We speculated that these four proteins play an important role in some possible mechanism in cervical cancer and the development process and, furthermore, could be used as candidate biomarkers in cervical cancer.
Downregulated expression apolipoprotein A-1 (APOA1) and upregulated expression apolipoprotein E (APOE) are lipid metabolism-related proteins which exist in plasma of Uygur women with cervical cancer in our study. APOA1 is the major protein component of high-density lipoprotein (HDL) in plasma and an established antiatherogenic factor in lipid metabolism [27]. In recent years, many research looked into the association between lipid profiles and cancer risk using HDL and apoA-I and suggested that low HDL and apoA-I levels as well as increased lipid ratios were inversely associated with higher cancer risk in various malignancies, such as prostate cancer, ovarian cancer, breast cancer, and pancreatic cancer and could be one of plasma biomarkers for early detection [28–31]. A study by Muntoni et al. [32] compared 519 patients, with any type of solid tumors, and 928 healthy controls and showed that in the cancer group, the alterations of lower HDL and apoA-I and higher triglycerides could be considered as a specific consequence of the presence of a malignant tumor with a diagnostic and prognostic significance using multivariate analysis.
APOE as a constituent of very-low-density lipoproteins and high-density lipoproteins is a high-risk factor of many diseases such as cardiovascular disease, Alzheimer’s disease, neuromuscular disease, multiple sclerosis, stroke, and diabetes [33] and involved in many functions, including lipid metabolism, cholesterol transport, immune response and regulation as well as cell growth and differentiation. However, the majority of recent studies have examined the significance of APOE as a molecular marker for a variety of cancer [34]. Su et al. [35] found that APOE overexpression promotes cancer proliferation and migration and contributes to an aggressive clinical course in patients with lung adenocarcinoma and malignant pleural effusions (MPE).
Clusterin (CLU) is a heterodimeric disulfide-linked glycoprotein (449 amino acids) and widely distributed in different tissues and highly conserved in species. CLU is implicated in a number of biological processes, including lipid transport, cell adhesion, programmed cell death, and complement cascade, representing a truly multifunctional protein [33]. In recent years, many studies show that elevated levels of CLU, a stress-induced and secreted cytoprotective chaperone, are associated with advanced tumor stage, metastasis, treatment resistance, and adverse outcome in several cancers by modulation of the nuclear factor kappa B pathway, inhibition of the apoptosis, and exerting other protumor activities, etc. [36–38].
A method for serological monitoring of differentially expressed secretory proteins is of great value for tumor screening. In this study, we found that concentrations of APOA1, APOE, and CLU were differential expression in CSCC patients than in benign lesion and were correlated with the histological classification or the processing of the cervical lesion. Therefore, our future study will further investigate the roles of APOA1, APOE, and CLU in cervical tumorigenesis and the levels of APOA1, APOE, and CLU in the plasma of a population at high risk for cervical cancer to establish criteria for medical surveillance.
References
Liu KJ, Liu JW, LI XR, et al. Epidemiology study of risk factors on Uigur and Han cervical cancer in Xinjiang. J Xinjiang Med Univ. 2008;31:1335–8.
Suzuke L, Peng YH, Zhou K, et al. The analysis of pathogenetic tendency of cervical cancer in various ethnic women in Xinjiang. J Xinjiang Med Univ. 2006;29:569–71.
Zhang GQ, LIU KJ, Lai XJ, et al. Distribution of malignant tumor patients in hospital from 1989 to 2002 in the Affiliated Tumor Hospital of Xinjiang Medical University. J Xinjiang Med Univ. 2003;26:393–5.
Zhang SQ, Yakup K, Abliz G, et al. The study of the relationships between HPV multiple infection and cervical cancer of Uygur Women in Xinjiang. J Xinjiang Med Univ. 2009;32:525–8.
Lynge E, Rygaard C, Baillet MV, et al. Cervical cancer screening at crossroads. APMIS. 2014;122(8):667–73.
Kim HS. Correction: primary, secondary, and tertiary prevention of cervical cancer. J Gynecol Oncol. 2014;25(3):261.
Garbis SD, Townsend PA. Proteomics of human prostate cancer biospecimens: the global, systems-wide perspective for protein markers with potential clinical utility. Expert Rev Proteomics. 2013;10(4):337–54.
Nair M, Sandhu SS, Sharma AK. Prognostic and predictive biomarkers in cancer. Curr Cancer Drug Targets. 2014.
Baker ES, Liu T, Petyuk VA, et al. Mass spectrometry for translational proteomics: progress and clinical implications. Genome Med. 2012;4(8):63.
Lihong H, Linlin G, Yiping G, et al. Proteomics approaches for identification of tumor relevant protein targets in pulmonary squamous cell carcinoma by 2D-DIGE-MS. PLoS One. 2014;9(4):e95121.
Pressey JG, Pressey CS, Robinson G, et al. 2D-difference gel electrophoretic proteomic analysis of a cell culture model of alveolar rhabdomyosarcoma. J Proteome Res. 2011;10(2):624–36.
Righetti PG, Boschetti E, Lomas L, Citterio A. Protein Equalizer Technology: the quest for a “democratic proteome”. Proteomics. 2006;6:3980–92.
Sihlbom C, Kanmert I, Bahr HV, Davidsson P. Evaluation of the combination of bead technology with SELDI-TOF-MS and 2-D DIGE for detection of plasma proteins. J Proteome Res. 2008;7(9):4191–8.
Yadav AK, Bhardwaj G, Basak T. A systematic analysis of eluted fraction of plasma post immunoaffinity depletion: implications in biomarker discovery. PLoS ONE. 2011;6(9):e24442.
Hanash S. A call for a fresh new look at the plasma proteome. Proteomics Clin Appl. 2012;6(9–10):443–6.
Matsubara J, Honda K, Ono M, et al. Identification of adipophilin as a potential plasma biomarker for colorectal cancer using label-free quantitative mass spectrometry and protein microarray. Cancer Epidemiol Biomarkers Prev. 2011;20(10):2195–203.
Zhou C, Simpson KL, Lancashire LJ, et al. Statistical considerations of optimal study design for human plasma proteomics and biomarker discovery. J Proteome Res. 2012;11(4):2103–13.
Lin YW, Lai HC, Lin CY, et al. Plasma proteomic profiling for detecting and differentiating in situ and invasive carcinomas of the uterine cervix. Int J Gynecol Cancer. 2006;16(3):1216–24.
Xia T, Zheng ZG, Gao Y, et al. [Application of SELDI-TOF serum proteome profiling in cervical squamous cell carcinoma]. Ai Zheng. 2008;27(3):279–82.
Looi ML, Karsani SA, Rahman MA, et al. Plasma proteome analysis of cervical intraepithelial neoplasia and cervical squamous cell carcinoma. Biosci. 2009;34(6):917–25.
Dae HJ, Hyoung KK, Abd-EI BP, et al. Plasma proteomic analysis of patients with squamous cell carcinoma of the uterine cervix. Gynecol Oncol. 2008;19(3):173–80.
Paradkar PH, Joshi JV, Mertia PN, et al. Role of cytokines in genesis, progression and prognosis of cervical cancer. Asian Pac J Cancer Prev. 2014;15(9):3851–64.
Medina-Martinez I, Barrón V, Roman-Bassaure E, et al. Impact of gene dosage on gene expression, biological processes and survival in cervical cancer: a genome-wide follow-up study. PLoS One. 2014;9(5):e97842.
Zhao Y, Wang H, Gustafsson M, et al. Combined multivariate and pathway analyses show that allergen-induced gene expression changes in CD4+ T cells are reversed by glucocorticoids. PLoS One. 2012;7(6):e39016.
Lamba JK, Crews KR, Pounds SB, et al. Identification of predictive markers of cytarabine response in AML by integrative analysis of gene-expression profiles with multiple phenotypes. Pharmacogenomics. 2011;12(3):327–39.
Wang HY, Tian YF, Chien CC, et al. Differential proteomic characterization between normal peritoneal fluid and diabetic peritoneal dialysate. Nephrol Dial Transplant. 2010;25(6):1955–63.
Bencharif K, Hoareau L, Murumalla RK, et al. Effect of apoA-I on cholesterol release and apoE secretion in human mature adipocytes. Lipids Health Dis. 2010;20(9):75.
Cine N, Baykal AT, Sunnetci D, et al. Identification of ApoA1, HPX and POTEE genes by omic analysis in breast cancer. Oncol Rep. 2014;32(3):1078–86.
His M, Zelek L, Deschasaux M, et al. Prospective associations between serum biomarkers of lipid metabolism and overall, breast and prostate cancer risk. Eur J Epidemiol. 2014;29(2):119–32.
Zamanian-Daryoush M, Lindner D, Tallant TC, et al. The cardioprotective protein apolipoprotein A1 promotes potent anti-tumorigenic effects. J Biol Chem. 2013;288(29):21237–52.
Van Hemelrijck M, Walldius G, Jungner I, et al. Low levels of apolipoprotein A-I and HDL are associated with risk of prostate cancer in the Swedish AMORIS study. Cancer Causes Control. 2011;22(7):1011–9.
Muntoni S, Atzori L, Mereu R, et al. Serum lipoproteins and cancer. Nutr Metab Cardiovasc Dis. 2009;19(3):218–25.
Moore RJ, Chamberlain RM, Khuri FR. Apolipoprotein E and the risk of breast cancer in African-American and non-Hispanic white women. Oncology. 2004;66(2):79–93.
Anand R, Prakash SS, Veeramanikandan R, et al. Association between apolipoprotein E genotype and cancer susceptibility: a meta-analysis. J Cancer Res Clin Oncol. 2014;140(7):1075–85.
Su WP, Chen YT, Lai WW, et al. Apolipoprotein E expression promotes lung adenocarcinoma proliferation and migration and as a potential survival marker in lung cancer. Lung Cancer. 2011;71(1):28–33.
Koltai T. Clusterin: a key player in cancer chemoresistance and its inhibition. Onco Targets Ther. 2014;20(7):447–56.
Lee S, Hong SW, Min BH, et al. Essential role of clusterin in pancreas regeneration. Dev Dyn. 2011;240(3):605–15.
Zoubeidi A, Ettinger S, Beraldi E, et al. Clusterin facilitates COMMD1 and I-kappaB degradation to enhance NF-kappaB activity in prostate cancer cells. Mol Cancer Res. 2010;8(1):119–30.
Conflicts of interest
None
Funding
This study was supported by Natural Science Foundation of China (81060171, 81260380 and 81360321). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Xia Guo and Yi Hao contributed equally to this work.
Rights and permissions
About this article

Cite this article
Guo, X., Hao, Y., Kamilijiang, M. et al. Potential predictive plasma biomarkers for cervical cancer by 2D-DIGE proteomics and Ingenuity Pathway Analysis. Tumor Biol. 36, 1711–1720 (2015). https://doi.org/10.1007/s13277-014-2772-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-014-2772-5