
Abstract
Breast cancer is the most common malignancy among women worldwide. Risk assessment is one of the main services delivered by cancer clinics. Biomarker analysis on different tissues including the peripheral blood can provide crucial information. One of the potential epigenetic biomarkers (epimarkers) is introduced as the peripheral blood DNA methylation pattern. This study was conducted to evaluate the potential value of peripheral blood epimarkers as an accessible tool to predict the risk of breast cancer development. WBC’s DNA was the focus of several case-control studies at both genome wide and candidate gene levels to reveal epigenetic changes accounting for predisposition to breast cancer, leading to suggest that ATM, TITF1, SFRP1, NUP155, NEUROD1, ZNF217, DBC2, DOK7 and ESR1 genes and the LINE1, Alu and Sat2 DNA elements could be considered as the potential epimarkers. To address that by which mechanisms WBC’s DNA methylation patterns could be linked to the propensity to breast cancer, several contemplations have been offered. Constitutional epimutation during embryonic life, and methylation changes secondary to either environmental exposures or tumor-mediated immune response, are the two main mechanisms. One can deduce that epimarkers based on their potential properties or regulatory impacts on cancer-related genes may be employed for risk prediction, prognosis, and survival inferences that are highly required for breast cancer management toward personalized medicine.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Breast cancer, with a lifelong risk of one in nine, is the most common cancer among women [1]. The prognosis of breast cancer, in particular its survival rate, crucially depends on its earlier detection. In terms of any interventions, these points highlight the need for regular life-time screening tests because of the disease frequency, and the feasibility and safety of such tests since they may be repeated many times.
Adopting a comprehensive screening approach including genetic molecular test could be highly recommended for this intervention. In the case of hereditary breast cancer, there are specific blood tests, BRCA1/2 mutation analysis that only covers less than 10 % of breast cancer cases in total, relative to sporadic form of the disease. For the latter form, although some new molecular tests namely particular sets of SNPs [2] have been added to the regular screening methods such as mammography, predicting the risk of breast cancer development in the absence of familial history is still a big challenge. The current screening methods are criticized by both the low sensitivity [3, 4] and overdiagnosis pitfalls [5, 6].
Since each screening molecular test only suggests a relative risk for predisposition to breast cancer, providing a more inclusive set of these tests could collectively infer more precise estimation for risk assessment. In this regard, coupling of epigenetic test to conventional molecular panel may even confer more values, as it could uncover new dimension of molecular architecture relative to cancer susceptibility. It is well known that epigenetic alterations, especially DNA methylation, are involved in tumorigenesis [7]. These alterations include gene-specific hypermethylation and global genomic hypomethylation that are well defined in tumor tissues [8]. However, recent studies have identified some epigenetic changes in the WBC genome among breast cancer patients which propose the predictive value of these changes as new biomarkers. Epigenetic biomarkers (epimarkers) are noteworthy, because of the stability of DNA molecules, dynamic nature of epigenetic changes, and availability of different ways for its measurement.
The purpose of this review is to introduce epimarkers as potential markers in risk prediction, diagnosis, and prognosis of breast cancer.
Peripheral blood epimarkers
In the case of identifying epimarker in the peripheral blood shown in Table 1, we have two alternatives: methylation in DNA of circulating tumor cell and WBC. Since cell-free DNA of circulating tumor cells originates from the tumor mass, it can be regarded as a source of diagnostic rather than predictive information. Presence of circulatory tumor DNA in low amounts and possibility of their contamination with normal cell DNA are serious limitations to consider them as reliable source. However, undergoing techniques may overcome these limitations to catch circulatory tumor cell DNA in good quantity and purity, giving them a momentum to be apply for diagnostic and prognostic purposes [9]. On the contrary, high amount and good quality of WBC DNA and more importantly its predictive potentials and implications concluded by several case-control studies make it a practical source to examine the epigenetic changes of WBCs before and during breast cancer development. Although there is no known mechanistic correlation between WBC epigenomic pattern and tumor development by now, such epigenetic changes may be looked as an independent risk factor quite long before breast cancer development. Since WBC DNA is accessible easily, its epigenetic along with its genetic influences on cancer propensity could be repeatedly evaluated in specified time intervals.
History of WBC DNA methylation as epimarker in cancers
The quest for WBC-based epimarkers was launched in 2001, when Karen et al. [10] showed a relationship between hypomethylation of P53 gene in WBC and risk of lung cancer development. Consequently, methylation alterations in different kind of cancers have been studied, concluding that WBC DNA methylation can be considered as a surrogate biomarker for the risk of cancer development, including breast cancer (Table 2) [27–37].
How methylation of WBC genome was explored in breast cancer?
According to previous studies mentioned in Table 2, there are two ways for discovering epimarkers in WBC: searching for methylation pattern alteration at target gene (candidate gene approach) and at the epigenomic level (whole genome approach). In the candidate gene approach, researchers evaluate genes involved in breast cancer carcinogenesis or genes that are mostly methylated in breast tumors. Although this approach is logical for DNA methylation studies of circulatory tumor cell, in the case of WBCs, because of their unknown role in breast cancer, may not embrace methylation pattern as of the tumor. For this reason, whole genome approach has been more popular, recently. In this regard, a WBCs gene which its methylation alteration pattern is associated with the breast cancer risk will be disclosed, which does not necessarily mean its direct role in carcinogenesis.
Evidences based on candidate gene approach
BRCA1, because of the crucial role of its mutations in hereditary breast cancer, has been considered as a natural choice to conduct a candidate gene analysis. In terms of epigenetic changes, BRCA1 promoter hypermethylation has received good attention as it offers another alternative inactivation mechanism for this critical gene. Some studies have shown an association between WBCs BRCA1 methylation changes and the risk of breast cancer, which was more significant among sporadic breast cancer cases than the hereditary ones [14, 22, 38]. Such association was also stronger among cases with early age at the onset [age ≤40] and high-grade tumors. Therefore, some researchers suggested that in sporadic breast cancer, the first hit may influence somehow BRCA1 methylation according to the Knudson’s two hit theory, while such pivotal role has been given to germ line mutation in hereditary breast cancer [18, 22, 39–42]. However, while that potential value for BRCA1 methylation as a candidate biomarker has not been approved by other studies [15, 17, 43], a very recent investigation on sporadic breast cancer concludes that WBC methylation signature may have such value conditioned to prior presence of BRCA1 mutations [25]. In this study, WBC DNA methylation signature in BRCA1 mutation carriers could predict breast cancer risk (area under curve 0.65). Also, the mentioned signature could predict mortality (area under curve 0.67) in sporadic breast cancer patients.
In addition to BRCA1, other genes which are involved in breast carcinogenesis such as estrogen receptor target genes (ERT), polycomb group target genes (PcGT) and ATM also have been studied [7, 11], which suggest ATM, NUP155, ZNF217, PTGS2, TITF1, NEUROD1 and SFRP1as potential biomarkers for breast cancer. It should be noted that methylation of ATM was in a repetitive element in the gene body and because of notable overlap between methylation in cases and controls; it seems that this marker alone is not sufficient as a diagnostic test (which area under curve of 0.59 shows insufficient specificity as a diagnostic biomarker).
Evidences based on whole genome approach
Since there are uncertainties behind candidate gene(s) selection, and the logic for representative value of peripheral blood for epigenetic changes in diseased breast tissue, whole genome approach could be considered as preferred alternative. Both hypermethylation and hypomethylation changes of WBC at genomic level could accompany with cancer developments.
According to the results of several genomic studies, hypermethylation changes of some genes in WBC genome have been evident in different cancers, including bladder, head and neck, pancreas, and breast cancers [23, 31, 33, 44, 45]. As a result, a number of genes have been nominated as potential epimarkers for epigenetic predisposition to breast cancer with prediction accuracy of 65.8 % [23].
Analysis of global genomic hypomethylation as a well-known epigenetic manifestation in cancer has been referred by several whole genome studies. Recent studies have reported that such genomic hypomethylation continuum can be evident even at WBC DNA level and may detect high-risk women before developing breast cancer suggesting its value as predictive biomarker [26, 30, 32, 36]. In this regard, some researchers have suggested that methylation analysis of repetitive elements such as LINE1, sat2 and Alu in WBC may be a surrogate for genomic hypomethylation. Based on this approach, some studies have shown that these repetitive elements in the leukocytes of healthy women with a family history of breast cancer are hypomethylated [16, 46] as well as breast cancer patients compared to controls [8, 21, 47].
Another way for evaluating global hypomethylation is measurement of 5-methyl cytosine (5mc) amounts in the genome. Xu and Ambrosone in two case-control studies showed that 5mc (not methylation of repetitive element) decreases in the WBC genome in breast cancer patients [12, 48]. The correlation between global 5mc and methylation of repetitive elements was not reported in another study [49], which makes it an unresolved controversy.
Limitation in previous studies
Some recent evidence has indicated that different epigenetic markers in the WBC genome could be considered as potential biomarkers for risk estimation of breast cancer. However, it is necessary to reassess these findings through studies with larger sample sizes. Most of the previous investigations were case-control studies, and the past history of their participants was not provided. It is important to note that many factors such as demographic characteristics, environmental exposures, and even genetic susceptibility can change the leukocyte methylation signatures [50]. Therefore, these factors may confound the interpretation of results and should be taken into account in future studies, which have been properly respected by Brennan et al. study [19], which summoned women from three large cohorts and followed them for 2–4 years and examined their past history along with their blood samples that were taken before cancer development. Another limitation of these studies is choosing the right candidate genes in the WBC genome that may reflect methylation deviation in breast cancer patients. Therefore, it is highly recommended to adhere with prospective and whole genome approaches, meanwhile considering other risk factors which affect the methylation signature in WBCs.
Biological relevance between WBC genome methylation and cancers
In spite of the documented association between WBC genome methylation and breast cancer, there is still a major debate in this issue:“how such association could be explained at mechanistic level?” Here, five explanations for the association between WBC DNA methylation status and breast cancer are overviewed:
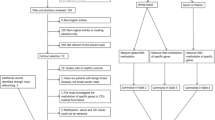
First, there is a constitutional epimutation which means that some epigenetic changes can be observed in normal tissues (blood) as well as in tumor tissues [13, 51–57]. In this case, constitutional epimutation can be considered as a consequence of germ line alteration that is not fully understood. However, it is clear that in germ line development, there are two phases of demethylation and remethylation which any remained errors in each phase can be passed through in the germ line cells [11]. In addition, constitutional epimutation can occur in early developmental stages at intrauterine phase (Fig. 1—rational 2). Second, some evidence have indicated that genetic variation could lead to specific epigenetic signatures [33, 58]; so, genetic susceptibility may lead to epigenetic alteration (Fig. 1—rational 1).

Potential mechanisms for association between WBC DNA methylation and cancers
Third, lifetime exposures to environmental influences could change epigenomic map of somatic cell/stem cell, which in turn is a result of direct or indirect effect by mutation of driver gene that control DNA methylation [44] (Fig. 1—rational 3). Such exposures may lead to the methylation changes in whole body somatic cells or only in specific tissues like hematopoietic stem cells. Considering the fact that DNA methylation is a lengthy process and blood cells have a short life span, their methylation condition may have been acquired from certain subgroups of hematopoietic cells.
The other concerns are connected to the nature of immune system response that is defined by clonal-specific expansion of leukocytes. There are evidences that different cancers evoke immune responses in specific manner mirrored by proliferation of particular subset of these immune cells. So, if one considers that this activation is somehow followed by epigenetic changes at activated clones, inspection of these changes could show new information. Therefore, the point is how we could find the main cellular types/clones to explore their methylation signature patterns according to the existing type of the cancer. However, this assumption could still be sound if we consider that the differential shift or polarization of the immune cells can be also due to cancer-associated environmental exposures in addition to cancer inflammation itself [20, 59] (Fig. 1—rational 4 and 5).
Future applications of peripheral blood epimarkers
Since epimarkers can help to identify high-risk women for breast cancer in population, and as epigenetic changes are reversible [60], it seems that reversing the epigenetic alterations and turning cells back to their normal epigenetic state can be considered as a future modality in prevention of breast cancer. On the other hand, because of potentiality of epimarkers, they can be used for various purposes. Epigenetic alterations, similar to genetic variations, can change the prognosis and survival parameters in cancer. Some studies on the ovaries, stomach, and pancreas cancer patients suggested that hypermethylation of some genes in the WBC genome affects the survival [61–63]. Another study by Zhuang et al. suggested that DNA methylation can be used as a prognostic biomarker [64]. Therefore, in near future, different applications may be defined for epimarkers: therapeutic (e.g., for epidrugs efficacy assessment), prognostic (for patient survival assessment), predictive (to adjust the best treatment options), and recurrence (for estimation of cancer relapse). In this view, new comprehensive studies holding more statistical power and less confounding cracks could pave the way for the era of epigenetic biomarkers in medicine.
Conclusion
Recent investigations have identified some epigenetic alterations in the WBC genome of breast cancer patients that could predict the risk of breast cancer. Although the current findings are quite promising, it seems that application of WBC methylation pattern as a sensible predicting test for breast cancer requires more search and research, for example by designing a prospective and whole genome studies. It is well known that many factors such as the lifestyle and environmental exposures could affect the WBC methylation signature and should be considered in future studies.
References
Radpour R, Barekati Z, Kohler C, Lv Q, Burki N, Diesch C, et al. Hypermethylation of tumor suppressor genes involved in critical regulatory pathways for developing a blood-based test in breast cancer. PLoS One. 2011;6(1):e16080.
Easton DF, Pooley KA, Dunning AM, Pharoah PD, Thompson D, Ballinger DG, et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007;447(7148):1087–93.
Wacholder S, Hartge P, Prentice R, Garcia-Closas M, Feigelson HS, Diver WR, et al. Performance of common genetic variants in breast-cancer risk models. N Engl J Med. 2010;362(11):986–93.
Mukhtar TK, Yeates DR, Goldacre MJ. Breast cancer mortality trends in England and the assessment of the effectiveness of mammography screening: population-based study. J R Soc Med. 2013;106(6):234–42.
Marmot MG, Altman DG, Cameron DA, Dewar JA, Thompson SG, Wilcox M. The benefits and harms of breast cancer screening: an independent review. Br J Cancer. 2013;108(11):2205–40.
Bell RJ. Screening mammography - early detection or over-diagnosis? Contribution from Australian data. Climacteric. 2014;2:1–7.
Widschwendter M, Apostolidou S, Raum E, Rothenbacher D, Fiegl H, Menon U, et al. Epigenotyping in peripheral blood cell DNA and breast cancer risk: a proof of principle study. PLoS One. 2008;3(7):e2656.
Cho YH, Yazici H, Wu HC, Terry MB, Gonzalez K, Qu M, et al. Aberrant promoter hypermethylation and genomic hypomethylation in tumor, adjacent normal tissues and blood from breast cancer patients. Anticancer Res. 2010;30(7):2489–96.
Sonnenberg A, Marciniak JY, Skowronski EA, Manouchehri S, Rassenti L, Ghia EM, et al. Dielectrophoretic isolation and detection of cancer-related circulating cell-free DNA biomarkers from blood and plasma. Electrophoresis. 2014;35(12–13):1828–36.
Woodson K, Mason J, Choi SW, Hartman T, Tangrea J, Virtamo J, et al. Hypomethylation of p53 in peripheral blood DNA is associated with the development of lung cancer. Cancer Epidemiol Biomarkers Prev. 2001;10(1):69–74.
Flanagan JM, Munoz-Alegre M, Henderson S, Tang T, Sun P, Johnson N, et al. Gene-body hypermethylation of ATM in peripheral blood DNA of bilateral breast cancer patients. Hum Mol Genet. 2009;18(7):1332–42.
Choi J-Y, James SR, Link PA, McCann SE, Hong C-C, Davis W, et al. Association between global DNA hypomethylation in leukocytes and risk of breast cancer. Carcinogenesis. 2009;30(11):1889–97.
Wong EM, Southey MC, Fox SB, Brown MA, Dowty JG, Jenkins MA, et al. Constitutional methylation of the BRCA1 promoter is specifically associated with BRCA1 mutation-associated pathology in early-onset breast cancer. Cancer Prev Res. 2011;4(1):23–33.
Iwamoto T, Yamamoto N, Taguchi T, Tamaki Y, Noguchi S. BRCA1 promoter methylation in peripheral blood cells is associated with increased risk of breast cancer with BRCA1 promoter methylation. Breast Cancer Res Treat. 2011;129(1):69–77.
Wojdacz TK, Thestrup BB, Overgaard J, Hansen LL. Methylation of cancer related genes in tumor and peripheral blood DNA from the same breast cancer patient as two independent events. Diagn Pathol. 2011;6:116.
Wu HC, John EM, Ferris JS, Keegan TH, Chung WK, Andrulis I, et al. Global DNA methylation levels in girls with and without a family history of breast cancer. Epigenetics. 2011;6(1):29–33.
Bosviel R, Garcia S, Lavediaux G, Michard E, Dravers M, Kwiatkowski F, et al. BRCA1 promoter methylation in peripheral blood DNA was identified in sporadic breast cancer and controls. Cancer Epidemiol. 2012;36(3):e177–82.
Pang D, Zhao Y, Xue W, Shan M, Chen Y, Zhang Y, et al. Methylation profiles of the BRCA1 promoter in hereditary and sporadic breast cancer among Han Chinese. Med Oncol. 2012;29(3):1561–8.
Brennan K, Garcia-Closas M, Orr N, Fletcher O, Jones M, Ashworth A, et al. Intragenic ATM methylation in peripheral blood DNA as a biomarker of breast cancer risk. Cancer Res. 2012;72(9):2304–13.
Wu HC, Delgado-Cruzata L, Flom JD, Perrin M, Liao Y, Ferris JS, et al. Repetitive element DNA methylation levels in white blood cell DNA from sisters discordant for breast cancer from the New York site of the Breast Cancer Family Registry. Carcinogenesis. 2012;33(10):1946–52.
Kitkumthorn N, Tuangsintanakul T, Rattanatanyong P, Tiwawech D, Mutirangura A. LINE-1 methylation in the peripheral blood mononuclear cells of cancer patients. Clin Chim Acta. 2012;413(9–10):869–74.
Hansmann T, Pliushch G, Leubner M, Kroll P, Endt D, Gehrig A, et al. Constitutive promoter methylation of BRCA1 and RAD51C in patients with familial ovarian cancer and early-onset sporadic breast cancer. Hum Mol Genet. 2012;21(21):4669–79.
Xu Z, Bolick SC, DeRoo LA, Weinberg CR, Sandler DP, Taylor JA. Epigenome-wide association study of breast cancer using prospectively collected sister study samples. J Natl Cancer Inst. 2013;105(10):694–700.
Heyn H, Carmona FJ, Gomez A, Ferreira HJ, Bell JT, Sayols S, et al. DNA methylation profiling in breast cancer discordant identical twins identifies DOK7 as novel epigenetic biomarker. Carcinogenesis. 2013;34(1):102–8.
Anjum S, Fourkala EO, Zikan M, Wong A, Gentry-Maharaj A, Jones A, et al. A BRCA1-mutation associated DNA methylation signature in blood cells predicts sporadic breast cancer incidence and survival. Genome Med. 2014;6(6):47.
Kuchiba A, Iwasaki M, Ono H, Kasuga Y, Yokoyama S, Onuma H, et al. Global methylation levels in peripheral blood leukocyte DNA by LUMA and breast cancer: a case–control study in Japanese women. Br J Cancer. 2014;110(11):2765–71.
Cash HL, Tao L, Yuan JM, Marsit CJ, Houseman EA, Xiang YB, et al. LINE-1 hypomethylation is associated with bladder cancer risk among nonsmoking Chinese. Int J Cancer. 2012;130(5):1151–9.
Hou L, Wang H, Sartori S, Gawron A, Lissowska J, Bollati V, et al. Blood leukocyte DNA hypomethylation and gastric cancer risk in a high-risk Polish population. Int J Cancer. 2010;127(8):1866–74.
Hsiung DT, Marsit CJ, Houseman EA, Eddy K, Furniss CS, McClean MD, et al. Global DNA methylation level in whole blood as a biomarker in head and neck squamous cell carcinoma. Cancer Epidemiol Biomarkers Prev. 2007;16(1):108–14.
Lim U, Flood A, Choi SW, Albanes D, Cross AJ, Schatzkin A, et al. Genomic methylation of leukocyte DNA in relation to colorectal adenoma among asymptomatic women. Gastroenterology. 2008;134(1):47–55.
Marsit CJ, Koestler DC, Christensen BC, Karagas MR, Houseman EA, Kelsey KT. DNA methylation array analysis identifies profiles of blood-derived DNA methylation associated with bladder cancer. J Clin Oncol. 2011;29(9):1133–9.
Moore LE, Pfeiffer RM, Poscablo C, Real FX, Kogevinas M, Silverman D, et al. Genomic DNA hypomethylation as a biomarker for bladder cancer susceptibility in the Spanish Bladder Cancer Study: a casecontrol study. Lancet Oncol. 2008;9(4):359–66.
Pedersen KS, Bamlet WR, Oberg AL, de Andrade M, Matsumoto ME, Tang H, et al. Leukocyte DNA methylation signature differentiates pancreatic cancer patients from healthy controls. PLoS One. 2011;6(3):e2656.
Teschendorff AE, Menon U, Gentry-Maharaj A, Ramus SJ, Gayther SA, Apostolidou S, et al. An epigenetic signature in peripheral blood predicts active ovarian cancer. PLoS One. 2009;4(12):e8274.
Wilhelm CS, Kelsey KT, Butler R, Plaza S, Gagne L, Zens MS, et al. Implications of LINE1 methylation for bladder cancer risk in women. Clin Cancer Res. 2010;16(5):1682–9.
Pufulete M, Al-Ghnaniem R, Leather AJ, Appleby P, Gout S, Terry C, et al. Folate status, genomic DNA hypomethylation, and risk of colorectal adenoma and cancer: a case control study. Gastroenterology. 2003;124(5):1240–8.
Terry MB, Delgado-Cruzata L, Vin-Raviv N, Wu HC, Santella RM. DNA methylation in white blood cells: association with risk factors in epidemiologic studies. Epigenetics. 2011;6(7):828–37.
Snell C, Krypuy M, Wong EM, Loughrey MB, Dobrovic A. BRCA1 promoter methylation in peripheral blood DNA of mutation negative familial breast cancer patients with a BRCA1 tumour phenotype. Breast Cancer Res. 2008;10(1):R12.
Suijkerbuijk KP, Fackler MJ, Sukumar S, van Gils CH, van Laar T, van der Wall E, et al. Methylation is less abundant in BRCA1-associated compared with sporadic breast cancer. Ann Oncol. 2008;19(11):1870–4.
Birgisdottir V, Stefansson OA, Bodvarsdottir SK, Hilmarsdottir H, Jonasson JG, Eyfjord JE. Epigenetic silencing and deletion of the BRCA1 gene in sporadic breast cancer. Breast Cancer Res. 2006;8(4):R38.
Butcher DT, Rodenhiser DI. Epigenetic inactivation of BRCA1 is associated with aberrant expression of CTCF and DNA methyltransferase (DNMT3B) in some sporadic breast tumours. Eur J Cancer. 2007;43(1):210–9.
Al-Moghrabi N, Al-Qasem AJ, Aboussekhra A. Methylation-related mutations in the BRCA1 promoter in peripheral blood cells from cancer-free women. Int J Oncol. 2011;39(1):129–35.
Kontorovich T, Cohen Y, Nir U, Friedman E. Promoter methylation patterns of ATM, ATR, BRCA1, BRCA2 and p53 as putative cancer risk modifiers in Jewish BRCA1/BRCA2 mutation carriers. Breast Cancer Res Treat. 2009;116(1):195–200.
Marsit CJ, Christensen BC, Houseman EA, Karagas MR, Wrensch MR, Yeh RF, et al. Epigenetic profiling reveals etiologically distinct patterns of DNA methylation in head and neck squamous cell carcinoma. Carcinogenesis. 2009;30(3):416–22.
Langevin SM, Koestler DC, Christensen BC, Butler RA, Wiencke JK, Nelson HH, et al. Peripheral blood DNA methylation profiles are indicative of head and neck squamous cell carcinoma: an epigenome-wide association study. Epigenetics. 2012;7(3):291–9.
Delgado-Cruzata L, Wu HC, Liao Y, Santella RM, Terry MB. Differences in DNA methylation by extent of breast cancer family history in unaffected women. Epigenetics. 2014;9(2):243–8.
Delgado-Cruzata L, Wu HC, Perrin M, Liao Y, Kappil MA, Ferris JS, et al. Global DNA methylation levels in white blood cell DNA from sisters discordant for breast cancer from the New York site of the Breast Cancer Family Registry. Epigenetics. 2012;7(8):868–74.
Xu X, Gammon MD, Hernandez-Vargas H, Herceg Z, Wetmur JG, Teitelbaum SL, et al. DNA methylation in peripheral blood measured by LUMA is associated with breast cancer in a population-based study. FASEB J. 2012;26(6):2657–66.
Wu HC, Delgado-Cruzata L, Flom JD, Kappil M, Ferris JS, Liao Y, et al. Global methylation profiles in DNA from different blood cell types. Epigenetics. 2011;6(1):76–85.
Li L, Choi JY, Lee KM, Sung H, Park SK, Oze I, et al. DNA methylation in peripheral blood: a potential biomarker for cancer molecular epidemiology. J Epidemiol. 2012;22(5):384–94.
Ally MS, Al-Ghnaniem R, Pufulete M. The relationship between gene-specific DNA methylation in leukocytes and normal colorectal mucosa in subjects with and without colorectal tumors. Cancer Epidemiol Biomarkers Prev. 2009;18(3):922–8.
Chan TL, Yuen ST, Kong CK, Chan YW, Chan AS, Ng WF, et al. Heritable germline epimutation of MSH2 in a family with hereditary nonpolyposis colorectal cancer. Nat Genet. 2006;38(10):1178–83.
Hajikhan Mirzaei M, Noruzinia M, Karbassian H, Shafeghati Y, Keyhanee M, Bidmeshki-Pour A. Evaluation of Methylation Status in the 5′UTR Promoter Region of the DBC2 Gene as a Biomarker in Sporadic Breast Cancer. Cell J. 2012;14(1):19–24.
Hitchins MP, Ward RL. Constitutional (germline) MLH1 epimutation as an aetiological mechanism for hereditary non-polyposis colorectal cancer. J Med Genet. 2009;46(12):793–802.
Ligtenberg MJ, Kuiper RP, Chan TL, Goossens M, Hebeda KM, Voorendt M, et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3′ exons of TACSTD1. Nat Genet. 2009;41(1):112–7.
Raval A, Tanner SM, Byrd JC, Angerman EB, Perko JD, Chen SS, et al. Downregulation of deathassociated protein kinase 1 (DAPK1) in chronic lymphocytic leukemia. Cell. 2007;129(5):879–90.
Hitchins MP, Rapkins RW, Kwok CT, Srivastava S, Wong JJ, Khachigian LM, et al. Dominantly inherited constitutional epigenetic silencing of MLH1 in a cancer-affected family is linked to a single nucleotide variant within the 5′UTR. Cancer Cell. 2011;20(2):200–13.
Boks MP, Derks EM, Weisenberger DJ, Strengman E, Janson E, Sommer IE, et al. The relationship of DNA methylation with age, gender and genotype in twins and healthy controls. PLoS One. 2009;4(8):e6767.
Koestler DC, Marsit CJ, Christensen BC, Accomando W, Langevin SM, Houseman EA, et al. Peripheral blood immune cell methylation profiles are associated with nonhematopoietic cancers. Cancer Epidemiol Biomarkers Prev. 2012;21(8):1293–302.
Agrawal A, Murphy RF, Agrawal DK. DNA methylation in breast and colorectal cancers. Mod Pathol. 2007;20(7):711–21.
Al-Moundhri MS, Al-Nabhani M, Tarantini L, Baccarelli A, Rusiecki JA. The prognostic significance of whole blood global and specific DNA methylation levels in gastric adenocarcinoma. PLoS One. 2010;5(12):e15585.
Flanagan JM, Wilhelm-Benartzi CS, Metcalf M, Kaye SB, Brown R. Association of somatic DNA methylation variability with progression-free survival and toxicity in ovarian cancer patients. Ann Oncol. 2013;24(11):2813–8.
Dauksa A, Gulbinas A, Barauskas G, Pundzius J, Oldenburg J, El-Maarri O. Whole blood DNA aberrant methylation in pancreatic adenocarcinoma shows association with the course of the disease: a pilot study. PLoS One. 2012;7(5):e37509.
Zhuang J, Jones A, Lee SH, Ng E, Fiegl H, Zikan M, et al. The dynamics and prognostic potential of DNA methylation changes at stem cell gene loci in women’s cancer. PLoS Genet. 2012;8(2):e1002517.
Conflicts of interest
None
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Khakpour, G., Pooladi, A., Izadi, P. et al. DNA methylation as a promising landscape: A simple blood test for breast cancer prediction. Tumor Biol. 36, 4905–4912 (2015). https://doi.org/10.1007/s13277-015-3567-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-015-3567-z