
Abstract
Benzo[a]pyrene (BaP) is one of the most studied targets among polycyclic aromatic hydrocarbons (PAHs). Because of the complexity of the toxicity mechanism in BaP, little is known about the molecular mechanism at the level of transcription of BaP in marine fishes. The primary objective of this study was to investigate the molecular basis of the effects of BaP on marine fish, using Mugilogobius chulae (Smith 1932) as the model. A closed colony of M. chulae was used for the BaP toxicity test. Two fish liver samples per replicate from each group were excised and blended into one sample by pooling an equal amount of liver tissue. Total RNA of all samples was extracted separately. Equal quantities of total RNA from the three replicates of the two groups were pooled for sequencing. The sequencing cDNA libraries were sequenced using Illumina HiSeq 2000 system. Differentially expressed genes were detected with the DEGSeq R package. In total, 52,364,032 and 53,771,748 clean nucleotide reads were obtained in the control and BaP-exposed libraries, respectively, with N50 lengths of 1277 and 1288 bp, respectively. Gene Ontology and Kyoto Encyclopedia of Genes and Genomes pathway analyses revealed a significant enrichment of genes related to detoxification, transportation, and lipid metabolism. We also identified, for the first time, an association between endoplasmic reticulum dysfunction and lipid metabolism resulting from BaP exposure. Using quantitative real-time PCR, some effective molecular biomarkers for monitoring of BaP-polluted seawater were identified. The results demonstrate that BaP enhanced the expression of genes involved in detoxification in M. chulae and inhibited that of genes related to lipid metabolism, possibly by suppressing the expression of numerous ER-related genes involved in fat digestion and absorption.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Polycyclic aromatic hydrocarbons (PAHs) are a broad class of organic compounds comprising two or more fused aromatic rings that mainly originate from the inefficient combustion of organic matter (Kim et al. 2013). These compounds are considered as persistent organic pollutants, and they have received much attention because of their toxicity and mutagenicity (Long et al. 2016). With the continued consumption of fossil fuels, the accumulation of PAH pollutants in marine environments has increased worldwide (Sinaei and Mashinchian 2014; Squadrone et al. 2014; Wang et al. 2014). In the China Sea, the concentrations of PAHs in sediments reach 287.05 ng/g and 1606.89 ng/g in southern and northern regions, respectively (Lu et al. 2012; Men et al. 2009). In Northern China, the concentration in seawater is as high as 1717.87 ng/L (Men et al. 2009), which is higher than the maximum allowed for drinking water (200 ng total PAHs/L) regarding human health protection (Eisler 1987).
Benzo[a]pyrene (BaP) is a typical PAH ubiquitous in marine ecosystems. Many countries and regions include BaP in their endocrine-disrupting chemical screening programs. A previous study on BaP pollution in several marine environments in China reported BaP concentrations in seawater and sediment up to 4799 ng/L and 2640 ng/g dry weight, respectively (Liu et al. 2015). In addition, the mean concentration of BaP in aquatic products from China has been reported to vary between 0.0008 and 343.97 ng/g (Li et al. 2010; Tian et al. 2003; Wan et al. 2007). Thus, BaP pollution is a critical issue in aquatic environments.
It has been suggested that BaP can cause damage to aquatic organisms because of its mutagenicity and carcinogenicity (Denissenko et al. 1996; Labib et al. 2013). Silicon metabolism of Thalassiosira pseudonana, a critical diatom of marine ecosystems, is disturbed by BaP (Carvalho et al. 2011). Disruption of sex steroidogenesis and endocrine were observed in crustaceans (Tian et al. 2013; Wen and Pan 2016). Moreover, in the two most widely used freshwater fishes, zebrafish (Danio rerio) and Japanese medaka (Oryzias latipes), visual system developmental defects (Huang et al. 2014) and bone formation dysfunction (Seemann et al. 2015) were identified, respectively. BaP also causes liver damage in amphibians (Xenopus tropicalis; Regnault et al. 2014). As the most representative species of higher vertebrate marine animals, less information is available on marine fishes. Currently, most other studies focus on freshwater animals or marine crustaceans. Additionally, because of the complexity of the toxicity mechanism in BaP, little is known about the molecular mechanism at the level of transcription of BaP in marine fishes.
The yellow stripe goby (Mugilogobius chulae, Smith, 1932) is an important small bottom-dwelling marine fish distributed mainly in western Pacific region, including Japan, China, Vietnam, Indonesia, Philippines and Thailand (Larson 2001). Previous reproductive biology studies have demonstrated that M. chulae is an ideal marine fish for marine ecotoxicology study. The maximum total length of M. chulae is less than 4 cm, the reproductive cycle is just 2 weeks, the fecundity is higher (approximately 3000 eggs/batch), and artificial breeding in the laboratory is easy to achieve; more importantly, M. chulae is sensitive to pollutants (Cai et al. 2016; Guo et al. 2017; Li et al. 2014). Inbred strain and closed colony of M. chulae have been maintained in laboratory conditions for more than 10 years (Cai et al. 2015), and a quality control standard has also been established for M. chulae (draft national standard for Approval No. 20091329-T-469), which is considered a potential model organism for estuarine and marine environmental toxicology research.
The present study investigated the molecular basis of BaP effects on marine fish using transcriptional sequencing. Obtained data suggest that BaP activates the detoxification system of M. chulae and disrupts lipid metabolism in the liver, specifically targeting the endoplasmic reticulum (ER). These results from standardized laboratory marine fish may provide a useful reference for future research.
Materials and methods
Chemicals
BaP (> 99% purity) was purchased from Sigma-Aldrich (St. Louis, MO, USA). Acetone (99% purity, Sangon, China) was used as the cosolvent for BaP. All other chemicals were obtained from commercial sources.
Animals and BaP exposure
A closed colony of M. chulae (13 months old) at the fifteenth generation was used for the BaP toxicity test. All fishes were raised at Guangdong Laboratory Animals Monitoring Institute, China, and acclimated for 2 weeks before the exposure test. The temperature, salinity, and pH of seawater were maintained at 26 ± 1 °C, 20‰, and 8.1, respectively. One-third of the seawater in the tank was renewed every day. M. chulae were fed dried food once daily.
The exposure test was performed in 5 L round glass tanks, each containing eight healthy fishes and 4 L of exposure medium. The experimental conditions were the same as those used for acclimation. In total, 48 fishes were randomly divided into two groups, with three replicates each. One group was exposed to BaP (6 µg/L) for 24 h, and another group served as control. The concentration of acetone in each group was maintained at 0.001%. The exposure concentration of BaP was determined by the maximum solubility of BaP in water. At the exposure duration of 6, 12, and 24 h, two fish liver samples per replicate from each group were excised and blended into one sample by pooling an equal amount of liver tissue. The samples were frozen in liquid nitrogen immediately.
RNA extraction, cDNA library preparation and sequencing
Total RNA of all samples was extracted separately. Equal quantities of total RNA (1 µg) from the three replicates of the two groups were pooled for sequencing and RT-QPCR validation (10 genes). The remaining total RNA of each sample was frozen at − 80 °C for further assays of temporal expression profiles of CYP2K3, MRP1, PAP, and FABP7. Total RNA was extracted with TRIzol reagent (Thermo, USA) following the manufacturer’s protocol. An Agilent RNA 6000 Nano Reagents kit and Agilent 2100 Bioanalyzer system (Agilent Technologies, Santa Clara, CA, USA) were used to measure the RNA purity and concentration. A sequencing cDNA library was generated using the TruSeq Stranded mRNA Sample Prep kit (Illumina, San Diego, CA, USA). The libraries were sequenced using Illumina HiSeq 2000 system.
Assembly of transcripts and sequence annotation
The low-quality sequences of raw reads from the two samples were removed before assembly, and then the clean, high-quality reads were assembled into contigs and unigenes using Trinity software (Grabherr et al. 2011). Annotation of unigenes was aligned to the protein database (Nr, SwissProt, Kyoto Encyclopedia of Genes and Genomes (KEGG), and Gene Ontology (GO) databases) using BLASTx (E-value < 0.00001), and to the Nt database with the BLASTN program (E-value < 0.00001). The candidate coding regions were identified by TransDecoder software (https://github.com/TransDecoder/TransDecoder). The assembly and annotation methods have been fully described in Cai et al. (2018).
Quantification and differential expression analysis
A robust and efficient stack memory management method was used to count the reads mapped to each unigene (Li and Dewey 2011). The reads per kilobase per million mapped reads (RPKM) value was measured based on transcript length and read counts mapped to the transcript (Marioni et al. 2008).
Differentially expressed genes were detected with the DEGSeq R package (Wang et al. 2009). The results were corrected by the false discovery rate (FDR). A unigene was considered as differentially expressed if FDR was ≤ 0.001 and the absolute value of the log2 ratio was ≥ 1. GO functional and KEGG pathway enrichment analyses were performed using the hypergeometric test.
Identification and validation of differentially expressed genes by quantitative real-time PCR
RNA was reverse-transcribed using the PrimeScript RT Reagent kit (Takara Bio, Dalian, China). Real-time quantitative PCR (qPCR) amplification was performed on an ABI7500 system (Applied Biosystems, Foster City, CA, USA) using a SYBR Premix Ex Taq kit (Takara Bio). Actin was used as an internal reference. The reaction conditions were 95 °C for 30 s followed by 40 cycles of 95 °C for 5 s and 60 °C for 34 s. The primer sequences are shown in Table 1. The relative expression levels of target genes were calculated by the \({{\text{2}}^ - }^{{\Delta \Delta {C_{\text{t}}}}}\) method (Livak and Schmittgen 2001). Statistical analysis was performed by one-way ANOVA followed by the Dunnett’s test using SPSS 17.0 software (SPSS Inc.). A value of p < 0.05 was used to indicate a significant difference.
Results
Sequencing and de novo assembly
The acute BaP toxicity test was performed over 24 h. None of the fishes died during this period. There were no differences in the length (cm) and mass (g) of fishes or in the physicochemical parameters of the water between experimental and control groups (p < 0.05) (Table 2).
Sequencing with the HiSeq 2000 platform generated a total of 56,309,262 and 54,769,792 raw nucleotide reads for the libraries of BaP-exposed and control groups, respectively. After removal of low-quality reads, the total number of clean reads was 53,771,748 (95.49%) and 52,364,032 (95.61%), respectively. The Q20 percentages were 98.80% and 98.85%, respectively, and the GC percentages of clean reads were 47.93% and 47.67%, respectively. The number of contigs assembled from reads for the two groups were 95,521 and 95,804, respectively. The number of unigenes assembled from these contigs was 58,998 and 59,175, respectively, with N50 lengths of 1288 and 1277 bp, respectively. The quality of the assembled transcriptomes of M. chulae was good. Length distribution of all unigenes is shown in Fig. 1.

Length distribution of all unigenes
Raw reads and Transcriptome Shotgun Assembly project are deposited in GenBank under project number SRP115439 and GFVA00000000, respectively.
Functional annotation and classification
In total, 28,044 unigenes were aligned to the Nr, Nt, SwissProt, KEGG, and GO databases (Table 3). For the Nr database, 82% of the annotated sequences had an E-value threshold < 1e−5, with a threshold of 1e−100 constituting the second-largest group (15.7%). In the priority order of Nr, SwissProt, and KEGG, all unigenes were aligned using BLASTX to predicted coding regions; 25,361 unigenes were mapped to the protein database and 1269 were predicted by ESTScan (Iseli et al. 1999).
Based on sequence homology, 16,048 transcripts were categorized into 59 functional groups in the GO analysis. Cell killing was the least dominant group within the biological process category; virion and virion parts constituted the smallest proportion in the cellular components category. For molecular functions, chemoattractant activity, chemorepellent activity, metallochaperone activity protein tag, and translation regulator activity were the least dominant groups. In total, 22,912 unigenes were mapped to reference canonical pathways in the KEGG database, yielding 258 predicted biosynthesis pathways of which metabolic pathway was the most highly represented (12.66%). These pathways and enriched genes are likely to be important for the study of molecular toxicity mechanisms of BaP.
Expression patterns of genes related to BaP exposure
To investigate the significance of the gene expression patterns observed in response to BaP exposure, we identified genes that were differentially expressed between the treatment and control groups and analyzed their functions. There were 846 differentially expressed genes and 27,189 genes with similar expression. Among the 846 differentially expressed genes, 364 and 482 genes were up- and downregulated, respectively. A functional analysis of BaP-regulated genes revealed 390 that encoded proteins with unknown functions, including 180 and 210 that were up- and downregulated, respectively (Table 4). Differentially expressed genes were mapped to terms in the GO database (Fig. 2). In the biological process category, “cellular process”, “metabolic process”, and “response to stimulus” were predominant with 103, 91, and 56 genes, respectively. For molecular functions, “catalytic activity” was the second largest group with 82 genes, whereas 26 genes were enriched in the function of “oxidoreductase activity”. For cellular components, ER-associated terms were significantly enriched, including “ER part”, “ER membrane”, and “nuclear outer membrane–ER membrane network”. The ER is an important organelle for detoxification and lipid metabolism; we found here that the expression of ER-related genes was significantly altered (up- and downregulated) by BaP exposure.

Gene ontology classification of unigenes. a Biological processes, b cellular components, c molecular function
Differentially expressed genes related to xenobiotic metabolism
The annotations only indicated the general functions of transcripts without revealing the biochemical pathways in which the encoded proteins are involved. The KEGG pathway analysis of M. chulae transcripts revealed six pathways related to xenobiotic metabolism, including several detoxification pathways such as “Xenobiotics by cytochrome P450 (CYP)”, “Glutathione metabolism”, and “Metabolism of xenobiotics by CYP”. In total, 28 differentially expressed genes were identified, of which five are known to be involved in BaP biodegradation (CYP1C1, CYP1B1, CYP2K1, CYP2K3, and glutathione S transferase Rho [GSTR]). All these genes except for GSTR were upregulated. Other enzymes involved in xenobiotic detoxification were also identified, including CYP2J6 and reduced glutathione and S-adenosylmethionine mitochondrial carrier protein. Another pathway related to xenobiotic catabolism was “ABC transporters”, with eight and two genes that were up- and downregulated, respectively. These results suggest that BaP exposure promotes the transport of allogeneic substances within cells.
Effect of BaP on lipid metabolism
In addition to transcripts associated with xenobiotic metabolism, 11 pathways related to lipid metabolism were enriched upon BaP exposure. “Peroxisome proliferator-activated receptor [PPAR] signaling” and “fat digestion and absorption”, the two predominant enriched pathways, both play important roles in lipid metabolism. In total, 40 differentially expressed genes were identified; among them, 12 were upregulated, whereas 28 were downregulated, including key genes involved in fatty acid metabolism, such as fatty acid-binding protein (FABP)7, adiponectin (ADPN), apolipoprotein (APO), and phosphatidate phosphatase (PAP). These results suggest that BaP causes dysregulation of lipid metabolism in the liver of M. chulae.
Remarkably, “Sphingolipid metabolism” and “Ether lipid metabolism” pathways were also enriched. Sphingolipids and ether lipids are important components of biomembranes, including the ER. This is consistent with our observation that most genes that were differentially expressed between BaP-treated and untreated groups were associated with the ER and the ER membrane, and all these genes were downregulated. The term “Protein processing in ER”, another pathway related to ER function, was identified in our analysis; nine of ten genes in this pathway were downregulated. The inhibition of the function of the ER, which is the major lipid factory of the cell, may underlie the dysregulation of lipid metabolism caused by BaP.
Quantitative real-time PCR validation of differentially expressed genes
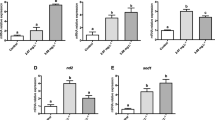
To validate the RNA-seq transcriptome data, ten differentially expressed genes (four downregulated and six upregulated) were analyzed by qPCR. Of the ten genes, six were randomly selected; the other four were non-randomly selected (CYP2K3, multidrug resistance protein 1 (MRP1), PAP and FABP7). Concordance between RNA-seq and qPCR results was observed for all ten genes (Fig. 3).

Quantitative real-time PCR validation of ten genes identified by RNA-sEq. Abbreviations are defined in Table 1
We also examined the temporal expression profiles of CYP2K3, MRP1, PAP, and FABP7 in response to BaP exposure (Fig. 4). CYP2K3 and MRP1 transcripts were significantly upregulated at 6, 12, and 24 h, with maximal expression levels relative to the control group observed at 6 and 12 h. PAP expression initially increased at 6 h before decreasing thereafter (at 12 and 24 h). FABP7 expression was higher than that in the control at 6 and 24 h but showed a slight decline at 12 h.

Time-dependent transcriptional pattern of M. chulae. Bars represent mean ± SD (n = 3). Means not sharing a common letter are significantly different at p < 0.05, as assessed using one-way ANOVA followed by the Dunnett’s test
Discussion
Gobiidae (Perciformes) constitute one of the largest families of marine fish, with more than 1950 species identified to date (Rickborn and Buston 2015). However, the genome and transcriptome of most of these species, including M. chulae, have yet to be described. In the present study, we obtained high-quality transcriptome data regarding the response of M. chulae to BaP-induced stress. We found that short-term exposure to BaP can adversely affect detoxification and lipid metabolism in the liver. In addition, we report, for the first time, that ER dysfunction is related to lipid metabolism in M. chulae following exposure to BaP.
To minimize the effects that sample origin might cause, we used the fifteenth generation of the closed colony of M. chulae for transcriptome sequencing, thereby minimizing the influence of genetic background on the results. We also used 13-month-old fish, which are at the optimal growth period (6–18 months) and pooled equal quantities of total RNA from the three replicates of the two groups to reduce the errors associated with individual differences. For the complexity and difficulty for the test species to be laboratory animals, most studies similar to the present one were carried out with wild or farmed fish (Andersen et al. 2015; Miao et al. 2018; Xu et al. 2017), which might be affected by background unconformities. The reliability, repeatability, and accuracy of toxicity test results from laboratory fish might be better, especially when compared with wild fish.
We sequenced two libraries and obtained 47,979 unigenes with an N50 length of 1658 bp, which was similar to that of Haliotis tuberculata (1529 bp; Harney et al. 2016) but longer than those of Schizothorax richardsonii (1274 bp; Barat et al. 2016), Synechogobius hasta (1298 bp; Chen et al. 2016), Oryzias melastigma (565 bp; Huang et al. 2012), and Larimichthys crocea (627 bp; Qian and Xue 2016). In total, 28,041 (58.4%) unigenes were annotated according to nucleotide and protein reference databases; this number was similar to that for Synechogobius hasta (62.7%), another gobiid fish. There were 26,630 coding sequences that were mapped or predicted. GO categories were assigned to M. chulae transcripts; 9558 were associated with metabolic processes, which can include detoxification. In total, 19,099 transcripts were grouped into 259 pathways, which was more than that reported in Paramisgurnus dabryanus (218; Li et al. 2015) and Gymnocypris przewalskii (252; Tong et al. 2015). These annotations provide valuable information for investigating specific processes and pathways in M. chulae.
A BLASTX search of M. chulae transcripts against the Nr database revealed a high similarity to Nile tilapia (Oreochromis niloticus) (49.27%), followed by Japanese medaka (Oryzias latipes) (15.44%), Japanese puffer (Fugu rubripes) (12.28%), green spotted puffer (Tetraodon nigroviridis) (5.35%), and zebrafish (Danio rerio) (3.30%), in accordance with the known phylogenetic relationships among these species (i.e., M. chulae and O. niloticus both belong to the order Perciformes). However, 9.09% of the unigenes matched genes of unknown species, implying that some lineage-specific genes are represented in M. chulae.
BaP is listed as a Group 1 genotoxic carcinogen by the International Agency for Research on Cancer (IARC 2017); therefore, it is necessary to define the relationship between gene expression and BaP exposure. In the present study, we found that exposure to BaP caused changes in the expression of a variety of genes in M. chulae, including those related to detoxification and xenobiotics and drug metabolism. BaP is first oxidized by CYP to BaP 7,8-oxide, then to BaP-7,8-diol and to benzopyrene diol epoxide, a carcinogenic BaP degradation product. Following BaP exposure, five major genes involved in the biodegradation of BaP were disturbed, likely to protect liver cells from BaP-induced oxidative stress injury. Of these genes, CYP1C1, CYP1B1, CYP2K1, and CYP2K3 levels were increased whereas that of GSTR was decreased. CYP1C1 and CYP1B1 are known to be involved in drug metabolism (Hassanin et al. 2012; Uno et al. 2006) and are upregulated in the presence of BaP; however, to the best of our knowledge, the present study is the first to describe an increase in CYP2K1 and CYP2K3 expression in response to BaP exposure. High CYP2K1 levels are related to catalysis and immune response (Yang et al. 2000), whereas relatively little is known about the function and mechanism of action of CYP2K3. Clarifying this aspect may provide new insight into the effects of BaP on metabolism. GSTR is a phase II enzyme that promotes the detoxification of xenobiotics and contributes to antioxidant defense (Jakoby 1978). GSTR levels were slightly decreased (log2 ratio of − 1.07) at 24 h of BaP exposure. A similar trend was observed in Venerupis philippinarum exposed to 5 µg/L BaP for 24 h (Zhang et al. 2012). However, a high concentration of BaP (50 µg/L) only induced GSTR after 48 h, suggesting that BaP concentration and exposure time are important for GSTR expression. In addition, reduced glutathione (GSH), another key factor in phase II metabolism of xenobiotics (Wu et al. 2004), was upregulated, whereas S-adenosylmethionine mitochondrial carrier protein, which is responsible for the transport of the GSH precursor S-adenosylmethionine and protects against alcohol-induced hepatotoxicity (Hu et al. 2015), was downregulated.
The ATP-binding cassette (ABC) transporter is associated with phase III of xenobiotic metabolism, which mainly involves transmembrane or membrane-associated proteins (Wakabayashi et al. 2006). We observed that 80% of the M. chulae genes in the ABC transporter pathway were upregulated in response to BaP exposure, including multidrug resistance protein 1, ABC-type multidrug transport system, and ABC sub-family A member 1-like. An increase in the expression of ABC transporter genes may be required to for the efficient removal of toxic BaP metabolites.
The liver, which is known to be the major organ for fat digestion, has been linked to lipid metabolism disorders following BaP exposure in animals. In the present study, we observed an abundance of genes in the enriched KEGG pathways of PPAR signaling, fat digestion and absorption, and fatty acid, ether lipid, sphingolipid, glycerolipid, and glycerophospholipid metabolism. Among these genes, four genes related to lipid metabolism were linked to at least two of these pathways, i.e., adiponectin (ADPN), fatty acid-binding protein 7 (FABP7), apolipoprotein (APO), and phosphatidate phosphatase (PAP); all except FABP7 were downregulated. FABP7 overexpression has been shown to impact fatty acid uptake (Ahn et al. 2012), low ADPN levels can lead to severe lipoatrophy (Ye et al. 2014), and downregulation of APO affects fatty acid transport between extra- and intracellular membranes (Weisiger 2002). Low expression of PAP, a key enzyme in intracellular lipid metabolism, can affect the synthesis of lipid triacylglycerol (Carman and Han 2006). The lipid metabolism disorder induced by BaP was observed in the liver of M. chulae.
The ER plays a key role in lipid metabolism. Various ER-related genes were differentially expressed in response to BaP exposure. Most of the differentially expressed genes were associated with the ER membrane or lipid metabolism, including five membrane structure genes, three channel genes, three lipid metabolism genes, and three ER stress-related genes. Disruption of the ER membrane can compromise ER function, including lipid metabolism; additionally, the aberrant expression of lipid-metabolizing genes in the ER induced by BaP supports the idea that functional disorder of the ER may be another key ingredient in the dysregulation of liver lipid metabolism.
Four genes were selected for further examination of temporal expression profiles. Of the four genes, CYP2K3 and MRP1 were involved in detoxification, FABP7 and PAP were related to lipid metabolism. Once inside the body, xenobiotics initiate the process of detoxification by inducing the expression of phase I metabolism enzymes such as CYP (Anzenbacher and Anzenbacherová 2001; Xu et al. 2005). The endogenous substrates of most CYP450 enzymes identified to date are unknown (Anzenbacher and Anzenbacherová 2001). In the present study, CYP2K3 was found to be a BaP-related enzyme. The fact that CYP2K3 mRNA expression reached a peak at an early time point, i.e., after 6 h of BaP exposure, suggests that this enzyme may be involved in phase I metabolism of xenobiotics. The multidrug resistance system, such as that mediated by the MRP1 gene, is another cellular mechanism that counters drug cytotoxicity (Higgins 2007). MRP1 encodes a transporter that protects cells against xenobiotics (Leslie et al. 2001). The change in MRP1 levels upon exposure to BaP was time-dependent, with the highest expression observed after 12 h. Thus, CYP2K3 and MRP1 might be useful biomarkers for monitoring marine pollution.
Although FABP7 is normally expressed in the ventricular zone and in radial glia of the vertebrate brain (Takaoka et al. 2011), recent research has linked it to liver injury (Miyazaki et al. 2014), retinal vasculature maintenance (Su et al. 2016), and tumor cell proliferation (Takaoka et al. 2017). In the present study, we found that FABP7 was upregulated in the liver of BaP-treated M. chulae by both RNA-seq and qPCR analyses, suggesting that BaP may affect lipid metabolism via induction of FABP7. As an important enzyme of the ER membrane, PAP is normally considered to regulate glycerolipid synthesis by converting phosphatidate to diacylglycerol (Brindley et al. 2009; Eastmond et al. 2010). In the present study, PAP was downregulated in response to BaP exposure, with the lowest expression observed after 12 h. Downregulation of PAP can adversely affect ER membrane growth in liver cells (Pascual and Carman 2013). In turn, this structural instability decreases several ER functions, including lipid metabolism.
Conclusion
Our results demonstrate that BaP enhanced the expression of genes involved in detoxification in M. chulae and inhibited that of genes related to lipid metabolism, possibly by suppressing the expression of numerous ER-related genes involved in fat digestion and absorption. These data provide a basis for future studies on the molecular mechanisms of BaP toxicity in marine organisms.
References
Ahn J, Lee H, Jung CH, Ha T (2012) Lycopene inhibits hepatic steatosis via microRNA-21-induced downregulation of fatty acid-binding protein 7 in mice fed a high- fat diet. Mol Nutr Food Res 56(11):1665–1674
Andersen Ø, Frantzen M, Rosland M, Timmerhaus G, Skugor A, Krasnov A (2015) Effects of crude oil exposure and elevated temperature on the liver transcriptome of polar cod (Boreogadus saida). Aquat Toxicol 165:9–18
Anzenbacher P, Anzenbacherová E (2001) Cytochromes P450 and metabolism of xenobiotics. Cell Mol Life Sci 58(5):737–747
Barat A, Sahoo PK, Kumar R, Goel C, Singh AK (2016) Transcriptional response to heat shock in liver of snow trout (Schizothorax richardsonii)-a vulnerable Himalayan Cyprinid fish. Funct Integr Genomics 16(2):203–213
Brindley DN, Pilquil C, Sariahmetoglu M, Reue K (2009) Phosphatidate degradation: phosphatidate phosphatases (lipins) and lipid phosphate phosphatases. Biochim Biophys Acta 1791(9), 956–961
Cai L, Chen XQ, Zheng WQ, Li JJ (2015) Isolation and characterization of polymorphic microsatellitemarkers in Mugilogobius chulae. Acta Lab Anim Sci Sin 23(1):57–62
Cai L, Huang R, Yu LJ, Li JJ (2016) Complete mitochondrial genome of Mugilogobius chulae (Perciformes: Gobiidae). Mitochondrial DNA A 27(6):4054–4055
Cai L, Li JJ, Yu LJ, Wei YZ, Miao ZY, Chen ML, Huang R (2018) De novo transcriptome assembly of the new marine fish model of goby, Mugilogobius chulae. Mar Genomics. https://doi.org/10.1016/j.margen.2018.02.001
Carman GM, Han GS (2006) Roles of phosphatidate phosphatase enzymes in lipid metabolism. Trends Biochem Sci 31(12):694–699
Carvalho RN, Bopp SK, Lettieri T (2011) Transcriptomics responses in marine diatom Thalassiosira pseudonana exposed to the polycyclic aromatic hydrocarbon benzo[a]pyrene. PLoS ONE 6(11):e26985
Chen QL, Luo Z, Huang C, Pan YX, Wu K (2016) De novo characterization of the liver transcriptome of javelin goby Synechogobius hasta and analysis of its transcriptomic profile following waterborne copper exposure. Fish Physiol Biochem 42(3):979–994. https://doi.org/10.1007/s10695-015-0190-2
Denissenko MF, Pao A, Tang MS, Pfeifer GP (1996) Preferential formation of benzo [a] pyrene adducts at lung cancer mutational hotspots in P53. Science 274:430–432
Eastmond PJ, Quettier AL, Kroon JT, Craddock C, Adams N, Slabas AR (2010) PHOSPHATIDIC ACID PHOSPHOHYDROLASE1 and 2 regulate phospholipid synthesis at the endoplasmic reticulum in Arabidopsis. Plant Cell 22(8):2796–2811
Eisler R (1987) Polycyclic aromatic hydrocarbon hazards to fish, wildlife, and invertebrates: a synoptic review (No. PB-87-189825/XAB; BIOLOGICAL-85 (1.11)). Patuxent Wildlife Research Center, Laurel. https://www.pwrc.usgs.gov/eisler/CHR_11_PAHs.pdf. Accessed 14 June 2018
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29(7):644–652
Guo Z, Gao N, Wu Y, Zhang L (2017) The simultaneous uptake of dietary and waterborne Cd in gastrointestinal tracts of marine yellowstripe goby Mugilogobius chulae. Environ Pollut 223:31–41
Harney E, Dubief B, Boudry P, Basuyaux O, Schilhabel MB, Huchette S, Paillard C, Nunes FL (2016) De novo assembly and annotation of the European abalone Haliotis tuberculata transcriptome. Mar Genomics 28:11–16
Hassanin AA, Kaminishi Y, Funahashi A, Itakura T (2012) Cytochrome P450 1C1 complementary DNA cloning, sequence analysis and constitutive expression induced by benzo-a-pyrene in Nile tilapia (Oreochromis niloticus). Aquat Toxicol 109:17–24
Higgins CF (2007) Multiple molecular mechanisms for multidrug resistance transporters. Nature 446(12):749–757
Hu F, Pan L, Cai Y, Liu T, Jin Q (2015) Deep sequencing of the scallop Chlamys farreri transcriptome response to tetrabromobisphenol A (TBBPA) stress. Mar Genomics 19:31–38
Huang Q, Dong S, Fang C, Wu X, Ye T, Lin Y (2012) Deep sequencing-based transcriptome profiling analysis of Oryzias melastigma exposed to PFOS. Aquat Toxicol 120:54–58
Huang L, Zuo Z, Zhang Y, Wu M, Lin JJ, Wang C (2014) Use of toxicogenomics to predict the potential toxic effect of benzo(a)pyrene on zebrafish embryos: ocular developmental toxicity. Chemosphere 108:55–61
International Agency for Research on Cancer (2017) Agents classified by the IARC monographs. http://monographs.iarc.fr/ENG/Classification/latest_classif.php/. Accessed 24 Feb 2018
Iseli C, Jongeneel CV, Bucher P (1999) ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. In ISMB 99:138–148
Jakoby WB (1978) The glutathione S-transferases: a group of multifunctional detoxification proteins. Adv Enzymol Relat Areas Mol Biol 46:383–414
Kim KH, Jahan SA, Kabir E, Brown RJ (2013) A review of airborne polycyclic aromatic hydrocarbons (PAHs) and their human health effects. Environ Int 60:71–80
Labib S, Guo CH, Williams A, Yauk CL, White PA, Halappanavar S (2013) Toxicogenomic outcomes predictive of forestomach carcinogenesis following exposure to benzo(a)pyrene: relevance to human cancer risk. Toxicol Appl Pharmacol 273(2):269–280
Larson HK (2001) A revision of the gobiid fish genus Mugilogobius (Teleostei: Gobioidei), and its systematic placement. Western Australian Museum, Perth
Leslie EM, Deeley RG, Cole SP (2001) Toxicological relevance of the multidrug resistance protein 1, MRP1 (ABCC1) and related transporters. Toxicology 167(1):3–23
Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform 12:323
Li Q, Zhang X, Yan C (2010) Polycyclic aromatic hydrocarbon contamination of recent sediments and marine organisms from Xiamen Bay, China. Arch Environ Contam Toxicol 58(3):711–721
Li JJ, Lin ZT, Chen XQ, Huang R (2014) Single and joint toxicity of four heavy metal ions on Mugilogobius chulae. Mar Environ Sci 33(2):236–241
Li C, Ling Q, Ge C, Ye Z, Han X (2015) Transcriptome characterization and SSR discovery in large-scale loach Paramisgurnus dabryanus (Cobitidae, Cypriniformes). Gene 557(2):201–208
Liu T, Pan L, Jin Q, Cai Y (2015) Differential gene expression analysis of benzo(a) pyrene toxicity in the clam, Ruditapes philippinarum. Ecotoxicol Environ Saf 115:126–136
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−∆∆CT method. Methods 25(4):402–408
Long AS, Lemieux CL, Arlt VM, White PA (2016) Tissue-specific in vivo genetic toxicity of nine polycyclic aromatic hydrocarbons assessed using the Muta™ Mouse transgenic rodent assay. Toxicol Appl Pharmacol 290:31–42
Lu TT, Lin Q, Ke CL, Sun RX (2012) Polycyclic aromatic hydrocarbons and risk assessment in the surface sediments from Lingdingyang, Pearl River Estuary. J Fish Sci China 19(2):336–347
Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y (2008) RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res 18(9):1509–1517
Men B, He M, Tan L, Lin C, Quan X (2009) Distributions of polycyclic aromatic hydrocarbons in the Daliao River Estuary of Liaodong Bay, Bohai Sea (China). Mar Pollut Bull 58(6):818–826
Miao LH, Lin Y, Pan WJ, Huang X, Ge XP, Zhou QL, Liu B, Ren MC, Zhang WX, Liang HL, Yu H (2018) Comparative transcriptome analysis reveals the gene expression profiling in bighead carp (Aristichthys nobilis) in response to acute nitrite toxicity. Fish Shellfish Immunol 79:244–255
Miyazaki H, Sawada T, Kiyohira M, Yu Z, Nakamura K, Yasumoto Y, Kagawa Y, Ebrahimi M, Islam A, Sharifi K, Kawamura S (2014) Fatty acid binding protein 7 regulates phagocytosis and cytokine production in kupffer cells during liver injury. Am J Pathol 184(9):2505–2515
Pascual F, Carman GM (2013) Phosphatidate phosphatase, a key regulator of lipid homeostasis. Biochim Biophys Acta 1831(3):514–522
Qian B, Xue L (2016) Liver transcriptome sequencing and de novo annotation of the large yellow croaker (Larimichthy crocea) under heat and cold stress. Mar Genomics 25:95–102
Regnault C, Worms IA, Oger-Desfeux C, MelodeLima C, Veyrenc S, Bayle ML, Combourieu B, Bonin A, Renaud J, Raveton M, Reynaud S (2014) Impaired liver function in Xenopus tropicalis exposed to benzo[a]pyrene: transcriptomic and metabolic evidence. BMC Genomics 15:666
Rickborn AJ, Buston PM (2015) Life-history transitions of the coral-reef fish Elacatinus lori. J Fish Biol 86(2):637–650
Seemann F, Peterson DR, Witten PE, Guo BS, Shanthanagouda AH, Rui RY, Zhang G, Au DW (2015) Insight into the transgenerational effect of benzo[a]pyrene on bone formation in a teleost fish (Oryzias latipes). Comp Biochem Physiol C 178:60–67
Sinaei M, Mashinchian A (2014) Polycyclic aromatic hydrocarbons in the coastal sea water, the surface sediment and Mudskipper Boleophthalmus dussumieri from coastal areas of the Persian Gulf: source investigation, composition pattern and spatial distribution. J Environ Health Sci Eng 12:59
Squadrone S, Favaro L, Abete MC, Vivaldi B, Prearo M (2014) Polycyclic aromatic hydrocarbon levels in European catfish from the upper Po River basin. Environ Monit Assess 186(4):2313–2320
Su X, Tan QS, Parikh BH, Tan A, Mehta MN, Wey YS, Tun SB, Li LJ, Han XY, Wong TY, Hunziker W (2016) Characterization of fatty acid binding protein 7 (FABP7) in the murine retina characterization of FABP7 in the murine retina. Invest Ophthalmol Vis Sci 57(7):3397–3408
Takaoka N, Takayama T, Teratani T, Sugiyama T, Mugiya S, Ozono S (2011) Analysis of the regulation of fatty acid binding protein 7 expression in human renal carcinoma cell lines. BMC Mol Biol 12:31
Takaoka N, Takayama T, Ozono S (2017) Functional analysis of fatty acid binding protein 7 and its effect on fatty acid of renal cell carcinoma cell lines. BMC Cancer 17:192
Tian Y, Zheng T, Wang X, Luo Y (2003) Contamination characteristics of polycyclic aromatic hydrocarbons in Maluan Bay mariculture area of Xiamen. Mar Environ Sci 22(1):29–33
Tian S, Pan L, Sun X (2013) An investigation of endocrine disrupting effects and toxic mechanisms modulated by benzo[a]pyrene in female scallop Chlamys farreri. Aquat Toxicol 144:162–171
Tong C, Zhang C, Zhang R, Zhao K (2015) Transcriptome profiling analysis of naked carp (Gymnocypris przewalskii) provides insights into the immune-related genes in highland fish. Fish Shellfish Immunol 46(2):366–377
Uno S, Dalton TP, Dragin N, Curran CP, Derkenne S, Miller ML, Shertzer HG, Gonzalez FJ, Nebert DW (2006) Oral benzo[a]pyrene in Cyp1 knockout mouse lines: CYP1A1 important in detoxication, CYP1B1 metabolism required for immune damage independent of total-body burden and clearance rate. Mol Pharmacol 69(4):1103–1114
Wakabayashi K, Tamura A, Saito H, Onishi Y, Ishikawa T (2006) Human ABC transporter ABCG2 in xenobiotic protection and redox biology. Drug Metab Rev 38(3):371–391
Wan Y, Jin X, Hu J, Jin F (2007) Trophic dilution of polycyclic aromatic hydrocarbons (PAHs) in a marine food web from Bohai Bay, North China. Environ Sci Technol 41(9):3109–3114
Wang L, Feng Z, Wang X, Wang X, Zhang X (2009) DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26(1):136–138
Wang Z, Liu Z, Xu K, Mayer LM, Zhang Z, Kolker AS, Wu W (2014) Concentrations and sources of polycyclic aromatic hydrocarbons in surface coastal sediments of the northern Gulf of Mexico. Geochem Trans 15:2
Weisiger RA (2002) Cytosolic fatty acid binding proteins catalyze two distinct steps in intracellular transport of their ligands. Mol Cell Biochem 239(1):35–43
Wen J, Pan L (2016) Short-term exposure to benzo[a]pyrene causes oxidative damage and affects haemolymph steroid levels in female crab Portunus trituberculatus. Environ Pollut 208:486–494
Wu G, Fang YZ, Yang S, Lupton JR, Turner ND (2004) Glutathione metabolism and its implications for health. J Nutr 134(3):489–492
Xu C, Li CYT, Kong ANT (2005) Induction of phase I, II and III drug metabolism/transport by xenobiotics. Arch Pharm Res 28(3):249–268
Xu EG, Mager EM, Grosell M, Hazard ES, Hardiman G, Schlenk D (2017) Novel transcriptome assembly and comparative toxicity pathway analysis in mahi-mahi (Coryphaena hippurus) embryos and larvae exposed to Deepwater Horizon oil. Sci Rep 7:44546
Yang YH, Miranda CL, Henderson MC, Wang-Buhler JL, Buhler DR (2000) Heterologous expression of Cyp2k1 and identification of the expressed protein (bv-Cyp2k1) as lauric acid (ω-1)-hydroxylase and Aflatoxin B1exo-epoxidase. Drug Metab Dispos 28(11):1279–1283
Ye R, Holland WL, Gordillo R, Wang M, Wang QA, Shao M, Morley TS, Gupta RK, Stahl A, Scherer PE (2014) Adiponectin is essential for lipid homeostasis and survival under insulin deficiency and promotes β-cell regeneration. Elife 3:e03851
Zhang L, Qiu L, Wu H, Liu X, You L, Pei D, Chen L, Wang Q, Zhao J (2012) Expression profiles of seven glutathione S-transferase (GST) genes from Venerupis philippinarum exposed to heavy metals and benzo[a]pyrene. Comp Biochem Physiol C 155(3):517–527
Acknowledgements
This work was supported by the National Key Technologies R & D Program of China (Grant No. 2015BAI09B05).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors would like to declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Cai, L., Li, J., Yu, L. et al. Characterization of transcriptional responses mediated by benzo[a]pyrene stress in a new marine fish model of goby, Mugilogobius chulae. Genes Genom 41, 113–123 (2019). https://doi.org/10.1007/s13258-018-0743-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13258-018-0743-8