
Abstract
Previous studies have investigated the physiological responses to chronic copper (Cu) exposure in the liver of Synechogobius hasta; however, little information is available on the underlying molecular mechanisms. In an effort to better understand the mechanisms of Cu toxicity and to illuminate global gene expression patterns modulated by Cu exposure, we obtained the liver transcriptome information of S. hasta by RNA sequencing (RNA-seq) technology and also investigated the differential expression of genes following waterborne Cu exposure. Using the Illumina sequencing platform, as many as 60,217 unigenes were generated, with 815 bp of average length and 1298 bp of unigene N50 after filtering and assembly. For functional annotation analysis, 34,860, 31,526, 31,576, 25,808, 11,542, and 21,721 unigenes were annotated to the NR, NT, Swiss-Prot, KEGG, COG, and GO databases, respectively, and total annotation unigenes were 37,764. After 30 days of exposure to 55 μg Cu/l, a total of 292 and 1076 genes were significantly up- and down-regulated, respectively. By KEGG analysis, 660 had a specific pathway annotation. Subsequent bioinformatics analysis revealed that the differentially expressed genes were mainly related to lipid metabolism, immune system, apoptosis, and signal transduction, suggesting that these signaling pathways may be regulated by Cu exposure. The present study provides comprehensive sequence information for subsequent gene expression studies regarding S. hasta, and the transcriptome profiling after Cu exposure is also expected to improve our understanding of the molecular toxicology of Cu.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Nowadays, the contamination of the aquatic environment by heavy metals severely threatens the sustainable and healthy development of aquaculture. Among the metals, copper (Cu) is of particular concern because of the increased discharge through industrial effluents and wide use in aquaculture to control algae and pathogens (Robinson et al. 2013). As an essential micronutrient required by all living organisms, Cu acts as a cofactor for many enzymes. However, it can be potentially toxic to aquatic organisms when available at elevated concentrations (Chen et al. 2013a; Giacomin et al. 2014). At present, although Cu toxicity has extensively been studied in fish, including histological alterations, oxidative stresses, and physiological modifications (Dethloff et al. 1999; Liu et al. 2010; Chen et al. 2012), still the comprehensive understanding of the mechanisms that are responsible for the toxicity of chronic Cu exposure at low, environmentally realistic concentrations is lacking.
Exposure to environmental Cu can evoke complex responses within an organism, making it difficult to explain the effects observed at the organ and organism levels based on some or a few regulatory entities. System biology approaches in toxicology offer an unprecedented potential to unravel the complex effect of Cu on the health of organisms (Santos et al. 2010). Because of this, an integrated functional genomics approach to study Cu toxicity is appropriate. It is commonly accepted that changes in gene expression are an integral part of the cellular response to Cu exposure. With the advent of next-generation sequencing technologies (NGS), a more comprehensive and accurate transcriptome analysis has become feasible and affordable for relevant species. High-throughput RNA sequencing (RNA-seq), a version of NGS, and progresses in bioinformatics, especially the development of de novo assembly tools, provide a powerful platform for characterizing the transcriptome of various non-model species without a reference genome (Mu et al. 2014), which have been proven to be a superior method for measuring mRNA expression and identifying differentially expressed genes (Marioni et al. 2008; Wang et al. 2009; Ekblom and Galindo 2011; Ali et al. 2014). Hence, large-scaled analysis of gene expression by RNA-seq technology can help to identify specific expression patterns after toxicant exposure, and this information can be used to monitor the effects pollution.
Javelin goby Synechogobius hasta (Perciformes: Gobiidae), a species of carnivorous fish, is widely distributed over the southern coast of Liaoning Province, China. Previous studies in our laboratory have shown that waterborne Cu exposure could influence hepatic lipid deposition and metabolism in this fish (Liu et al. 2010; Chen et al. 2013a, b; Huang et al. 2014). However, due to the lack of genomic resources such as genome and transcriptome sequences, these studies were limited to few candidate genes, and a global understanding of the transcriptome profiling of S. hasta is required. The purpose of the present study is to obtain the liver transcriptome information of S. hasta by RNA-seq technology and also investigate the differential expression of genes following waterborne Cu exposure. Corresponding analysis associated with the change in lipid deposition and metabolism in S. hasta subjected to Cu exposure is presented in a parallel paper (Huang et al. 2014). To our knowledge, this is the first study on the transcriptome profiling of S. hasta, and also the first time differential expression of its genes has been analyzed. Because this study systematically and quantitatively explores the effects of Cu exposure on the genome-wide gene expression, it extends our understanding of how Cu regulates or disrupts the gene expression network. Meanwhile, the transcriptome obtained for S. hasta will become essential for future analyses and also provide useful information for future functional genomic studies in other fish.
Materials and methods
The study was carried out in strict accordance with the recommendation of the Guide for the Care and Use of Laboratory Animals of Huazhong Agricultural University. The protocol was approved by the Committee on the Ethics of Animal Experiments of the University. All efforts were made to minimize suffering in animals.
Fish and Cu exposure
The fish and experimental procedures used here have been described in our recent study (Huang et al. 2014). Briefly, healthy S. hasta (without any bacterial or other parasitic infections) were obtained from a local marine water pond (Panjin, China) and transferred to indoor cylindrical fiberglass tanks (90 cm height, 80 cm diameter) for 2 weeks of acclimation. Afterwards, 144 uniform-sized juvenile fish (initial mean weight 19.2 ± 0.3 g, mean ± SEM, with gland of stage II) were randomly assigned to 6 fiberglass tanks with 24 fish per tank. They were exposed to two nominal Cu concentrations of zero (control, without extra Cu addition) and 57 μg/l (7.5 % of the 96-h LC50 of Cu for S. hasta, Liu et al. 2010), respectively, with triplicates for each concentration. According to Zhan (2005), the highest Cu concentration detected in Panjin (China), where this experiment was conducted, was 105 μg/l. Cu was added as CuSO4·5H2O (AR, Shanghai Sinopharm Group Corporation, Shanghai, China). It was dissolved in distilled water for stock concentrations. Individual test solutions during the experiment were obtained by adding the appropriate volume of the primary stock to the dilution. Cu concentrations in the test tanks were monitored twice every week by graphite furnace atomic absorption spectrometry (GFAAS) following the method described in our recent study (Chen et al. 2013a, b). The measured Cu concentrations for two treatments were 2 ± 1 and 55 ± 3 μg Cu/l, respectively.
The experiment was performed in a static aquarium system with continuous aeration to maintain dissolved oxygen near saturation. During the acclimation and experimental periods, the fish were fed 6 % of their biomass daily (two meals per day) with minced trash fish (Cu content: 2.37 ± 0.18 mg/kg live weight basis). After 15 min, the remaining uneaten food was removed from the tanks. The amount of feed consumed by the fish in each tank was recorded daily, and the results showed that Cu exposure concentration here did not affect the feeding rate (data not shown). Meantime, to ensure good water quality and maintain waterborne Cu levels, water was renewed twice daily. The experiment continued for 30 days.
The experiment was conducted at ambient temperature and subjected to natural photoperiod (approximately 12 h of light and 12 h of darkness). Water quality parameters were monitored twice a week in the morning. The parameters were as follows: water temperature 22.4 ± 3.3 °C; pH 8.4 ± 0.1; dissolved oxygen 6.9 ± 0.7 mg/l; salinity 19.8 ± 0.6 ‰; dissolved organic carbon 5.6 ± 1.2 mg/l; total hardness 74.4 ± 2.0 mM; and total alkalinity 6.4 ± 0.6 mM.
Sampling and total RNA extraction
At the end of 30-day exposure, fish were starved for 24 h before sampling. After euthanized with MS-222 (10 mg/l), six fish (two fish each tank, and three tanks per group) were randomly selected from each group. Their liver samples were excised and immediately frozen in liquid nitrogen and stored at −80 °C until RNA extraction. In the present study, livers do not derive from non-goby sources. Total RNA was isolated from each sample using Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. RNA quality and quantity were measured using the NanoDrop 2000 (Thermo Scientific, Wilmington, DE, USA) and an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). RNA samples with RIN (RNA integrity number) >8.0 were considered acceptable for further processing. All the samples were standardized to 500 ng/μl, and equal volumes of total RNA from six individuals in the same group were combined into one pool for transcriptome analysis.
cDNA library preparation and Illumina sequencing
The library construction and sequencing were performed by Beijing Genomics Institute (Shenzhen, China). Briefly, total RNA was treated by DNase I, and then, magnetic beads with Oligo (dT) were used to isolate mRNA. The mRNA was mixed with the fragmentation buffer and fragmented into short fragments, which were used as templates for cDNA synthesis. The short cDNA fragments were purified and resolved with EB buffer for end reparation and single nucleotide A (adenine) addition, and then, they were connected with adapters. After that, the suitable fragments were selected for the PCR amplification as templates. During the QC steps, an Agilent 2100 Bioanalyzer and an ABI StepOnePlus Real-Time PCR System were used to quantify and qualify the sample library. Finally, the library was sequenced using Illumina HiSeq™ 2000 sequencing platform (San Diego, CA, USA).
De novo assembly and function annotation
Before assembly, dirty raw reads were filtered as follows: (1) removed reads with adaptors; (2) removed reads with unknown nucleotides (N) larger than 5 %; (3) removed low-quality reads (the rate of bases with a quality value ≤10 is more than 20 % of the entire read); and (4) clean reads were obtained. Transcriptome de novo assembly was carried out with Trinity software, a short reads assembling program (Grabherr et al. 2011). The resulting sequences obtained from Trinity were called unigenes. Separate cDNA libraries were constructed from control and Cu-treated fish, and unigenes from each library were taken into further process of sequence splicing and redundancy removing with sequence clustering software to acquire non-redundant unigenes (All-Unigene) as long as possible. These All-Unigenes were submitted to databases for homolog and annotation comparison by BLASTx algorithm (e-value <10−5), including NR (non-redundant protein sequence), non-redundant nucleotide (Nt), Swiss-Prot, Kyoto Encyclopedia of Genes and Genomes (KEGG), and Clusters of Orthologous Groups (COG). Functional annotation by Gene Ontology terms (GO; http://www.geneontology.org) was accomplished with Blast2GO software (Conesa et al. 2005; Gotz et al. 2008). After obtaining GO annotation for each unigene, we used WEGO software (Ye et al. 2006) to perform GO functional classification for All-Unigenes.
Identification of differentially expressed genes
Gene expression levels were calculated using the Reads Per kb per Million reads (RPKM) method (Mortazavi et al. 2008). A rigorous algorithm was developed to identify differentially expressed genes between the two samples (control and Cu exposure) based on a previously described method (Audic and Claverie 1997). The false discovery rate (FDR) was controlled to determine differentially expressed genes (Benjamini and Yekutieli 2001). In this study, FDR ≤ 0.001 and the absolute value of log2 ratio ≥1 (fold-change ≥2) were used as the threshold to judge the significance of gene expression differences. For pathway and GO enrichment analysis, all differentially expressed genes were mapped to terms in GO and the KEGG database.
Real-time quantitative PCR (qPCR) validation
Twenty-five candidate genes related to lipid metabolism, immune responses, and signal transduction were selected for validation by qPCR. Total RNA was extracted from the liver of S. hasta in control and Cu-exposed groups (n = 6 for each group) using the method mentioned above. Total RNA was quantified spectrophotometrically and integrity checked by agarose formaldehyde gel electrophoresis, and first-strand cDNA was synthesized using PrimeScriptTM RT reagent Kit with gDNA Eraser (TaKaRa, Dalian, China). qPCR assays were carried out in a quantitative thermal cycler (MyiQ™ 2 Two-Color Real-Time PCR Detection System, Bio-Rad, Hercules, CA, USA) with a 20-μl reaction volume containing 2 × SYBR Premix Ex Taq™ (TaKaRa, Dalian, China) 10 μl, 10 mM each of forward and reverse primers 0.4, 1 μl diluted cDNA template (tenfold), and 8.2 μl double-distilled H2O. Primer set was designed based on each identified gene sequence of transcriptome library by Primer Premier 5.0 (Supplementary Table S1). The qPCR parameters consisted of initial denaturation at 95 °C for 30 s, followed by 40 cycles at 95 °C for 5 s, 57 °C for 30 s, and 72 °C for 30 s. All reactions were performed in duplicate, and each reaction was verified to contain a single product of the correct size by agarose gel electrophoresis. A non-template control and dissociation curve were performed to ensure that only one PCR product was amplified and that stock solutions were not contaminated. Standard curves were constructed for each gene using serial dilutions of stock cDNA. The amplification efficiencies of all genes were approximately equal and ranged from 96 to 105 %. Gene expression levels were quantified relative to the expression of β-actin using the optimized comparative Ct (2−ΔΔCt) value method (Livak and Schmittgen 2001). Prior to the analysis, we have carried out the trial and proved β-actin expression levels were stable under the experimental conditions (Huang et al. 2014).
Statistical analysis
All data for qPCR result were presented as mean ± standard error of means (SEM). Differences between the control and Cu-exposed group were analyzed by Student’s t test for independent samples. Analysis was performed using the SPSS 17.0 software, and the minimum significant level was set at 0.05.
Results
Illumina sequencing and de novo transcriptome assembly
To obtain an overview of gene expression profile in S. hasta, cDNA libraries were constructed from the liver of control and Cu-exposed fish and were subjected to Illumina sequencing platform, resulting in 60,022,948 and 58,613,188 reads, respectively (Table 1). After quality trimming and adapter clipping, a total of 54,490,824 and 53,012,788 clean reads with 4,904,174,160 and 4,771,150,920 nucleotides (nt) remained, respectively. The percentages of Q20, GC content, and unknown bases (N) were 97.24, 49.92, and 0.01 %, respectively, for the control sample, and 97.15, 50.38, and 0.01 %, respectively, for the Cu-exposed sample (Table 1). These reads were assembled, resulting in 128,888, and 118,146 contigs, respectively. After splicing and redundancy removing, these contigs were further assembled, and sequence data from the two libraries were combined, yielding 60,217 unigenes (All-Unigene), with average length of 815 bp (N50 = 1298 bp) (Table 2). The length distribution of All-Unigene is shown in Fig. 1. This Transcriptome Shotgun Assembly project has been deposited at DDBJ/EMBL/GenBank under the accession GCNT00000000. The version described in this paper is the first version, GCNT01000000.

The length distribution of All-Unigene
Function annotation and classification of unigenes
In order to validate and annotate the assembled unigenes, a BLASTx (e-value <10−5) search using public protein and nucleotide databases (NR, NT, GO, Swiss-Prot, KEGG, and COG) was performed with sequences from 60,217 unigenes. The results showed that 37,764 unigenes (62.71 % of All-Unigene) were aligned function by BLASTx, and 22,453 (37.29 %) assembled sequences could not be matched to any known protein (Table 3). A total of 34,860 (57.89 %) and 31,576 (52.44 %) unigenes were unambiguous alignments to the reference when BLASTx against NR and Swiss-Prot databases, respectively, while BLASTn against NT database returned 31,526 (52.35 %) hits. The e-value distribution of the top hits in the NR database showed that 32.2 % of the mapped sequences have strong homology (<1e−100), whereas 67.8 % of the homolog sequences ranged between 1e−5 and 1e−100 (Fig. 2a). The similarity distribution showed that 43.4 % of the sequences had a similarity >80 % and the remaining 56.6 % of the sequences had a similarity ranging from 17 to 80 % (Fig. 2b). For species distribution, 49.5 % of the matched unigenes showed similarities with Oreochromis niloticus, followed by Oryzias latipes (15.7 %), Fugu rubripes (12.7 %), and Tetraodon nigroviridis (5.6 %) (Fig. 2c).

Homology analysis of S. hasta transcriptome. All distinct gene sequences that had BLAST annotations within the NR database with a cutoff e-value ≤10−5 were analyzed for e-value distribution (a), similarity distribution (b), and species distribution (c)
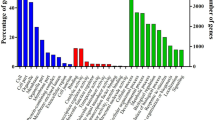
Based on the NR annotation, GO analysis was performed. A total of 21,721 (36.07 %) GO terms were obtained (Table 3), with 53.06 % for biological processes, 33.57 % for cellular components, and 13.37 % for molecular functions. The three main GO categories were classified into 60 subcategories (Fig. 3). Within the biological processes, proteins related to cellular processes were the most enriched. In the category of cellular components, most of the corresponding genes were involved in cell, cell part, and organelle. Under the molecular functions category, binding and catalytic activity were the most highly represented GO terms. Other categories such as “antioxidant activity” and “receptor regulator activity” are also present, but at a lower percentage. In addition, the COG assignments were performed to predict and classify possible functions of unigenes. A total of 11,542 (19.17 %) sequences were functionally classified into 25 COG categories (Table 3; Fig. 4). Among these categories, most enriched terms were “General function of prediction only,” followed by “Replication, recombination and repair,” “Translation, ribosomal structure and biogenesis,” and “Transcription” (Fig. 4). Besides the GO and COG analysis, KEGG pathway parsing was also carried out. A total of 25,808 (42.86 %) unigenes were mapped into 259 signaling pathways including metabolic pathway (12 %), MAPK signaling pathway (3.01 %), chemokine signaling pathway (2.55 %), calcium signaling pathway (1.52 %), and fatty acid metabolism (0.6 %).

GO classification analysis of All-Unigene. GO functions are shown in X axis. The right Y axis shows the numbers of genes which have the GO function, and the left Y axis shows the percentage

COG function classification of All-Unigene. The horizontal coordinates are functional classes of COG, and the vertical coordinates are numbers of unigenes in one class. The notation on the right is the full name of the functions in X axis
Identification of differentially expressed genes
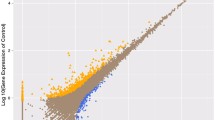
There were a total of 1368 genes identified as differentially expressed, of which 1076 were down-regulated and 292 were up-regulated in the liver from the control compared with those from Cu-exposed fish (Fig. 5, Supplementary Table S2). For the GO analysis, 381, 422, and 419 differentially expressed genes were grouped in cellular component, molecular function, and biological process categories, respectively. Most of the differentially expressed genes were classified into cellular process, metabolic process, cell, cell part, and binding categories (Fig. 6).

a Scatter plots showed gene expression profiles; b histogram showed numbers of differentially expressed genes in the liver of S. hasta from the control and Cu-exposed groups. Differentially expressed genes are indicated in red (increased expression) and green (decreased expression). Blue indicates genes that were not differentially expressed. (Color figure online)

GO classification of differentially expressed unigenes. Unigenes were assigned to three main categories: biological process, cellular components, and molecular function. Values are displayed for each term as the percentage of the total number of genes as well as the number of genes
To characterize the functional consequences of gene expression changes associated with Cu exposure, we performed pathway analysis based on the KEGG database. Of the 1368 genes differentially expressed in the liver from control compared with those from Cu-exposed fish, 660 had a specific KEGG pathway annotation, which clearly indicated the involvement of different biological pathways in the response toward Cu exposure and accumulation, such as stress and detoxification-related processes, cell growth and death, immune response, lipid metabolism, and signal transduction.
Differentially expressed genes involved in lipid metabolism
The differentially expressed genes were identified and involved in a wide aspect of lipid metabolism, including fatty acid degradation, steroid biosynthesis, fatty acid elongation, and glycerophospholipid metabolism. Specially, genes encoding for proteins involved in fatty acid β-oxidation were down-regulated in response to Cu exposure, such as long-chain acyl-CoA synthetase (acsl), carnitine O-palmitoyltransferase 1 (cpt 1), enoyl-CoA hydratase (ech), and acetyl-CoA acyltransferase (acaa) (Fig. 7).

Differentially expressed genes involved in fatty acid β-oxidation. The map was described based on KEGG databases and studies by Kobayashi et al. (2010) and Collins et al. (2010). Genes with green background indicated that the mRNA expression levels of Cu-exposed fish were significantly lower than those in the control (FDR ≤ 0.001, the absolute value of log2[Ratio] ≥1). (Color figure online)
Differentially expressed genes involved in immune system
Pathway enrichment analysis based on Cu exposure responsive unigenes identified significantly immune-related pathways. Among these pathways, “chemokine signaling pathway,” “B cell receptor signaling pathway,” and “complement and coagulation cascades” were the most frequently represented pathways. The vast majority of genes involved in “chemokine signaling pathway” (Supplementary Fig. S1) and “B cell receptor signaling pathway” (Supplementary Fig. S2) were down-regulated following Cu exposure, such as chemokine, Src, PI3K, and IKK. In complement and coagulation cascades pathway, Cu exposure increased the transcription level of TFPI, A2M, KNG, HF1, BF, DF, MCP, and C1Q, while decreased the mRNA levels of C1R, C7, and C9 (Table 4).
Differentially expressed genes involved in apoptosis
As shown in Fig. 8, eight differentially expressed genes involved in apoptosis were found between the control and Cu-exposed fish, with seven down-regulated genes (CASP3, DFF45, DFF40, IKK, IκBα, NFκB, and PI3K) and only one up-regulated gene (calpain).

Differentially expressed genes involved in apoptosis. The map was described based on KEGG databases. Genes with green or red background indicated that the mRNA expression levels of Cu-exposed fish were significantly lower or higher than those in the control, respectively (FDR ≤ 0.001, the absolute value of log2[Ratio] ≥1). (Color figure online)
Differentially expressed genes involved in signal transduction
Analysis of differentially expressed genes involved in signal transduction revealed that MAPK signaling pathway was the largest category, including some up-regulated genes (such as FGF, FGFR, cPLA2, and c-fos) and down-regulated genes (such as CACN, GRB2, and IKK). Moreover, calcium signaling pathway and NFκB signaling pathway were enriched as well. Compared with the control, Cu exposure down-regulated the expression of genes involved in these two signaling pathways (Table 5).
Validation of differential gene expression by qPCR
The expression levels of 25 genes (5 up-regulated and 20 down-regulated genes) from RNA-seq were validated by qPCR analysis. The results showed 24 genes exhibited a concordant direction both in RNA-seq and in qPCR analysis except DFF45 gene (Supplementary Fig. S3). The correlation coefficient between RNA-seq and qPCR results was 0.931 (p < 0.001).
Discussion
To date, because genome and transcriptome resources for most fish species have not been obtained, including S. hasta, molecular studies on the metabolic regulation, signal transduction, and immune response to Cu exposure in fish are still scarce. In this study, we analyzed the liver transcriptome in S. hasta using RNA-seq technology and obtained large amounts of sequence information. Meanwhile, by profiling the transcriptomes of the control and Cu-exposed fish, we identified differentially expressed genes involved in lipid metabolism, immune system, cell growth and death, and signal transduction.
Liver transcriptome of S. hasta generated by RNA-seq
In the present study, we obtained a total of 60,217 unigenes (≥200 bp), with average length 815 bp (N50 = 1298 bp), which was much longer than those documented in other organisms, such as Oryzias melastigma (N50 = 565 bp) (Huang et al. 2012), mandarin fish (mean length = 506 bp, N50 = 611 bp) (He et al. 2013), Oncorhynchus kisutch (mean length = 599 bp) (Harding et al. 2013), and O. niloticus (mean length = 618 bp, N50 = 852 bp) (Zhang et al. 2013). A total of 37,764 (62.71 %) unigenes were annotated with nucleotide and protein reference databases. This annotation ratio was higher than those reported in mandarin fish (41.6 %) (He et al. 2013) and Litopenaeus vannamei (55.58 %) (Guo et al. 2013), probably due to the length of unigenes obtained from the present study. Our results showed that 57.89 % of unigenes were aligned to NR database, which was comparable with these studies using the same method in other fish species, such as Oncorhynchus mykiss (61.17 %) (Ali et al. 2014) and Misgurnus anguillicaudatus (43.76 %) (Long et al. 2013). The species distribution for the best match from each sequence showed the highest homology to O. niloticus, followed by O. latipe, F. rubripes, T. nigroviridis, and other fish species, indicating the high level of phylogenetic conservation between S. hasta and other fish species. The COG database is an attempt to phylogenetically classify proteins, while the GO database is used to analyze gene functions and gene products in organisms (Gui et al. 2013). In addition, KEGG provides useful information for predicting the functional profiles of genes (Kanehisa and Goto 2000). All the multiple annotations for transcriptome provide the predictable information for these important unique transcripts, and each database has its own advantage for facilitating future studies.
Differentially expressed genes involved in lipid metabolism
In fish, lipids act as a major energy source and support various physiological, developmental, and reproductive processes (Tocher 2003). Lipid metabolism is a complex physiological process that includes lipid absorption, transportation, deposition, and mobilization (Sheridan 1988). Our recent studies reported that waterborne Cu exposure influenced lipid deposition (Chen et al. 2013a; Huang et al. 2014) and disturbed the normal processes of lipid metabolism (Chen et al. 2013b; Huang et al. 2014). Meantime, studies indicated that there were differences between species, exposure routes, and tissues of fish subjected to copper exposure (Chen et al. 2013b; Huang et al. 2014; Chen et al. 2015). However, due to the lack of transcriptomic and genomic resources, these studies only investigated the mRNA levels of very limited genes affected by Cu and lacked the understanding of the pathways involved in lipid metabolism following Cu exposure. In this study, transcriptome analyses showed that Cu exposure was associated with profound changes in the expression of genes involved in lipid metabolism, helping to explain the molecular background to the known effect of Cu on lipid profiles. Of these pathways, the genes (such as cpt 1) involved in fatty acid β-oxidation were down-regulated by Cu administration. Mitochondrial fatty acid β-oxidation is a major source of energy in organisms. It has been proved that mitochondrial damage and inhibition of fatty acid β-oxidation is a mechanism of hepatotoxicity (Fromenty and Pessayre 1995). Therefore, it seems that Cu induced hepatotoxicity through, at least in part, impairment of mitochondrial β-oxidation.
Differentially expressed genes involved in immune system
The immune system is a complex set of cellular, chemical, and soluble protein components which interact with each other in a sequential, regulated manner to protect the body against infectious agents. Identifying chemical-induced changes in transcriptional profiling would greatly extend our understanding of the molecular mechanisms of immunotoxicity and would allow for an evaluation of the immune system within the confines of traditional toxicology researches (Frawley et al. 2011). In the present study, following Cu exposure, some differentially expressed genes were associated with “chemokine signaling pathway,” “B cell receptor signaling pathway,” and “complement and coagulation cascades.” In general, the most important function of chemokines and their receptors is the recruitment of immune and non-immune cells into the inflamed sites (Borroni et al. 2010; Seki and Schwabe 2015). In the liver, chemokines play a crucial role in the coordination of the multicellular wound healing response (Seki and Schwabe 2015). B-lymphocytes are involved in antigen-specific defense and activated by binding of antigen to B cell receptors (Ali et al. 2014). Furthermore, the complement system is a proteolytic cascade in blood plasma that acts on the interface of innate and adaptive immunity and is an important component for triggering many immunoregulatory mechanisms (Hovhannisyan et al. 2010). In the present study, many genes involved in these pathways were down-regulated by Cu exposure, including chemokine, chemokine R, AC, GRB2, Src, PI3K, FOXO, IKK, NFκB, IκB, P-Rex1, WASP, Paxillin, FAK, CD22, LYN, BLNK, Bam32, GRB2, BCAP, SHIP, IKKγ, C1R, C7, and C9, indicating that these genes participated in S. hasta defense response after Cu exposure. Similarly, Pierron et al. (2011) found that the transcription levels of many genes (such as complement C3, complement regulatory plasma protein, and complement C9) involved in the immune system were down-regulated in response of yellow perch to metal exposure (Cd and Cu). In the present study, both the complement and coagulation system and the chemokine signaling pathway participated in the Cu process, suggesting that inflammation and immune reactions were involved in Cu-induced stress.
Differentially expressed genes involved in apoptosis
Apoptosis has been clarified as a physiological mechanism regulating tissue mass and architecture during normal tissue development (Ellis et al. 1991). However, the occurrence of deregulated apoptosis has also been shown to be involved in the pathogenesis of various diseases. In the present study, Cu exposure changed the expression levels of several genes involved in apoptosis, such as calpain and caspase-3 (CASP3), suggesting that these genes may be important factors of the apoptotic response to Cu in S. hasta. Calpain and caspase-3 are cysteine proteases, which have been reported to play a critical role in mediating cellular apoptosis (Tassabehji et al. 2005). Calpain is a Ca2+-dependent papain-like neutral cysteine protease, and an elevation of cytoplasmic-free Ca2+ concentration induces calpain activation, which gives rise to the cleavage of various proteins and culminates in cell death (Pietrobon et al. 1990). Among the caspase members, caspase-3 in particular is an essential apoptotic effector (Bratton and Cohen 2001; Tassabehji et al. 2005). Our study showed that the expression of calpain was up-regulated, while caspase-3 was down-regulated by Cu exposure. Similarly, Arnal et al. (2014) found that the activities of caspase-3 and calpain exhibited a differential response to Cu administration. Indeed, activation of one of these two proteases may cause inactivation of another (Rami et al. 2000; Arnal et al. 2014). Furthermore, Sun et al. (2008) reported that caspase-3 inhibition reduced the calpain activities in the rat model of experimental stroke.
Differentially expressed genes involved in signal transduction
Cu has the potential to mediate several biological processes through multiple signal transduction pathways (Mattie et al. 2008). Mitogen-activated protein kinases (MAPKs) are serine–threonine kinases, which phosphorylate specific serine and threonine of target protein substrates and regulate many diverse physiological processes, including survival, development, growth, proliferation, stress responses, and apoptosis (Caffrey et al. 1999; Kim and Choi 2010). A plethora of evidence has shown that the activation of MAPK signaling cascades play important roles in Cu exposure (Nawaz et al. 2006; Mattie et al. 2008). Exposure of rainbow trout hepatocytes to Cu triggered a time- and dose-dependent increase in phosphorylated extracellular signal-regulated kinase (pERK) and thus induced apoptosis (Ebner et al. 2007). In the present study, Cu exposure up-regulated several genes related to MAPK signaling pathway such as c-fos, which was consistent with the in vitro study by Mattie et al. (2008). Cu2+ could induce intracellular reactive oxygen species (ROS) generation, which activated MAPK signaling transduction pathway and contributed to elevation of phosphorylation levels (Mattie et al. 2008).
NFκB is considered as an important redox-sensitive transcriptional factor that regulates a large number of genes controlling immune and inflammatory responses (Collins et al. 1995; Ramesh et al. 1999). Our study showed that Cu exposure down-regulated the expression of NFκB as well as the genes involved in NFκB signaling pathway. At present, the effect of Cu on NFκB signaling pathway is controversial. Some reports showed that Cu activated NFκB, while other researches demonstrated that Cu inhibited or had no effect on NFκB activity (reviewed by McElwee et al. 2009). Besides, studies suggested that the ability of Cu to affect NFκB activity was cell type specific (Lewis et al. 2005; McElwee et al. 2009). Therefore, further experiments are required to determine the effect of Cu on NFκB signaling pathway.
Intracellular calcium is a universal messenger regulating many physiological and pathological processes. Particularly, calcium signals in the liver are able to modulate diverse and specialized functions, such as bile secretion, glucose metabolism, cell proliferation, and apoptosis (Amaya and Nathanson 2013). Generally, exogenous divalent heavy metals including Cu2+ are known to affect cellular calcium homeostasis and calcium signaling (Stohs and Bagchi 1995; Schafer et al. 2009; Song et al. 2016). Disruption of cellular Ca2+ signaling appears to mediate the Cu toxicity (Nielsen et al. 2003). In the present study, Cu exposure down-regulated genes related to calcium signaling pathway, probably reflecting variations in the concentration of calcium in cells. The understanding of how defects in the Ca2+ signaling machinery and in Ca2+ signaling patterns may lead to pathological conditions and help to elucidate the Cu toxicity.
In conclusion, for the first time, the transcriptome information of S. hasta was constructed by de novo assembly of two libraries from the liver of normal and Cu-exposed fish using the Illumina paired-end sequencing. Based on the assembled de novo transcriptome, the differentially expressed genes involved in lipid metabolism, immune system, apoptosis, and signal transduction in response to Cu exposure were identified, which would contribute to a deeper understanding of the molecular mechanisms of Cu-induced hepatic toxicity in fish, and the identified Cu-sensitive genes may serve as biomarkers to monitor aquatic environmental pollution. Further studies are required to elucidate the exact regulatory mechanisms by which Cu causes these coordinated responses and the implications for the health of exposed organisms.
References
Ali A, Rexroad CE, Thorgaard GH, Yao J, Salem M (2014) Characterization of the rainbow trout spleen transcriptome and identification of immune-related genes. Front Genet 5:348
Amaya MJ, Nathanson MH (2013) Calcium signaling in the liver. Compr Physiol 3:515–539
Arnal N, Dominici L, De Tacconi MJ, Marra CA (2014) Copper-induced alterations in rat brain depends on route of overload and basal copper levels. Nutrition 30:96–106
Audic S, Claverie JM (1997) The significance of digital gene expression profiles. Genome Res 7:986–995
Benjamini Y, Yekutieli D (2001) The control of the false discovery rate in multiple testing under dependency. Ann Stat 29:1165–1188
Borroni EM, Mantovani A, Locati M, Bonecchi R (2010) Chemokine receptors intracellular trafficking. Pharmacol Ther 127:1–8
Bratton SB, Cohen GM (2001) Apoptotic death sensor: an organelle’s alter ego? Trends Pharmacol Sci 22:306–315
Caffrey DR, O’neill LA, Shields DC (1999) The evolution of the MAP kinase pathways: coduplication of interacting proteins leads to new signaling cascades. J Mol Evol 49:567–582
Chen QL, Luo Z, Zheng JL, Li XD, Liu CX, Zhao YH, Gong Y (2012) Protective effects of calcium on copper toxicity in Pelteobagrus fulvidraco: copper accumulation, enzymatic activities, histology. Ecotoxicol Environ Saf 76:126–134
Chen QL, Luo Z, Liu X, Song YF, Liu CX, Zheng JL, Zhao YH (2013a) Effects of waterborne chronic copper exposure on hepatic lipid metabolism and metal-element composition in Synechogobius hasta. Arch Environ Contam Toxicol 64:301–315
Chen QL, Luo Z, Pan YX, Zheng JL, Zhu QL, Sun LD, Zhuo MQ, Hu W (2013b) Differential induction of enzymes and genes involved in lipid metabolism in liver and visceral adipose tissue of juvenile yellow catfish Pelteobagrus fulvidraco exposed to copper. Aquat Toxicol 136:72–78
Chen QL, Luo Z, Wu K, Huang C, Zhuo MQ, Song YF, Hu W (2015) Differential effects of dietary copper deficiency and excess on lipid metabolism in yellow catfish Pelteobagrus fulvidraco. Comp Biochem Physiol 184B:19–28
Collins T, Read M, Neish A, Whitley M, Thanos D, Maniatis T (1995) Transcriptional regulation of endothelial cell adhesion molecules: NF-kappa B and cytokine-inducible enhancers. FASEB J 9:899–909
Collins SA, Sinclair G, Mcintosh S, Bamforth F, Thompson R, Sobol I, Osborne G, Corriveau A, Santos M, Hanley B (2010) Carnitine palmitoyltransferase 1A (CPT1A) P479L prevalence in live newborns in Yukon, Northwest Territories, and Nunavut. Mol Genet Metab 101:200–204
Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3676
Dethloff GM, Schlenk D, Hamm JT, Bailey HC (1999) Alterations in physiological parameters of rainbow trout (Oncorhynchus mykiss) with exposure to copper and copper/zinc mixtures. Ecotoxicol Environ Saf 42:253–264
Ebner HL, Blatzer M, Nawaz M, Krumschnabel G (2007) Activation and nuclear translocation of ERK in response to ligand-dependent and-independent stimuli in liver and gill cells from rainbow trout. J Exp Biol 210:1036–1045
Ekblom R, Galindo J (2011) Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity 107:1–15
Ellis RE, Yuan J, Horvitz H (1991) Mechanisms and functions of cell death. Annu Rev Cell Biol 7:663–698
Frawley R, White K, Brown R, Musgrove D, Walker N, Germolec D (2011) Gene expression alterations in immune system pathways in the thymus after exposure to immunosuppressive chemicals. Environ Health Perspect 119:371–376
Fromenty B, Pessayre D (1995) Inhibition of mitochondrial beta-oxidation as a mechanism of hepatotoxicity. Pharmacol Ther 67:101–154
Giacomin M, Jorge MB, Bianchini A (2014) Effects of copper exposure on the energy metabolism in juveniles of the marine clam Mesodesma mactroides. Aquat Toxicol 152:30–37
Gotz S, Garcia-Gomez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, Robles M, Talon M, Dopazo J, Conesa A (2008) High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res 36:3420–3435
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q (2011) Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat Biotechnol 29:644–652
Gui D, Jia K, Xia J, Yang L, Chen J, Wu Y, Yi M (2013) De novo assembly of the indo-pacific humpback dolphin leucocyte transcriptome to identify putative genes involved in the aquatic adaptation and immune response. PLoS One 8:e72417
Guo H, Ye CX, Wang AL, Xian JA, Liao SA, Miao YT, Zhang SP (2013) Transcriptome analysis of the Pacific white shrimp Litopenaeus vannamei exposed to nitrite by RNA-seq. Fish Shellfish Immunol 35:2008–2016
Harding LB, Schultz IR, Goetz GW, Luckenbach JA, Young G, Goetz FW, Swanson P (2013) High-throughput sequencing and pathway analysis reveal alteration of the pituitary transcriptome by 17α-ethynylestradiol (EE2) in female coho salmon, Oncorhynchus kisutch. Aquat Toxicol 142:146–163
He S, Liang XF, Sun J, Li L, Yu Y, Huang W, Qu CM, Cao L, Bai XL, Tao YX (2013) Insights into food preference in hybrid F1 of Siniperca chuatsi (♀)× Siniperca scherzeri (♂) mandarin fish through transcriptome analysis. BMC Genom 14:601
Hovhannisyan LP, Mkrtchyan GM, Sukiasian SH, Boyajyan AS (2010) Alterations in the complement cascade in post-traumatic stress disorder. Allergy Asthma Clin Immunol 6:3
Huang Q, Dong S, Fang C, Wu X, Ye T, Lin Y (2012) Deep sequencing-based transcriptome profiling analysis of Oryzias melastigma exposed to PFOS. Aquat Toxicol 120:54–58
Huang C, Chen QL, Luo Z, Shi X, Pan YX, Song YF, Zhuo MQ, Wu K (2014) Time-dependent effects of waterborne copper exposure influencing hepatic lipid deposition and metabolism in javelin goby Synechogobius hasta and their mechanism. Aquat Toxicol 155:291–300
Kanehisa M, Goto S (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28:27–30
Kim EK, Choi EJ (2010) Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta 1802:396–405
Kobayashi Y, Miyazawa M, Kamei A, Abe K, Kojima T (2010) Ameliorative effects of mulberry (Morus alba L.) leaves on hyperlipidemia in rats fed a high-fat diet: induction of fatty acid oxidation, inhibition of lipogenesis, and suppression of oxidative stress. Biosci Biotechnol Biochem 74:2385–2395
Lewis J, Wataha J, Mccloud V, Lockwood P, Messer R, Tseng WY (2005) Au (III), Pd (II), Ni (II), and Hg(II) alter NFκB signaling in THP1 monocytic cells. J Biomed Mater Res A 74:474–481
Liu X, Luo Z, Xiong B, Liu X, Zhao Y, Hu G, Lv G (2010) Effect of waterborne copper exposure on growth, hepatic enzymatic activities and histology in Synechogobius hasta. Ecotoxicol Environ Saf 73:1286–1291
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−∆∆CT method. Methods 25:402–408
Long Y, Li Q, Zhou B, Song G, Li T, Cui Z (2013) De novo assembly of mud loach (Misgurnus anguillicaudatus) skin transcriptome to identify putative genes involved in immunity and epidermal mucus secretion. PLoS One 8:e56998
Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y (2008) RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res 18:1509–1517
Mattie MD, McElwee MK, Freedman JH (2008) Mechanism of copper-activated transcription: activation of AP-1, and the JNK/SAPK and p38 signal transduction pathways. J Mol Biol 383:1008–1018
McElwee MK, Song MO, Freedman JH (2009) Copper activation of NF-κB signaling in HepG2 cells. J Mol Biol 393:1013–1021
Mortazavi A, Williams BA, Mccue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-seq. Nat Methods 5:621–628
Mu Y, Li M, Ding F, Ding Y, Ao J, Hu S, Chen X (2014) De novo characterization of the spleen transcriptome of the large yellow croaker (Pseudosciaena crocea) and analysis of the immune relevant genes and pathways involved in the antiviral response. PLoS One 9:e97471
Nawaz M, Manzl C, Lacher V, Krumschnabel G (2006) Copper-induced stimulation of extracellular signal-regulated kinase in trout hepatocytes: the role of reactive oxygen species, Ca2+, and cell energetics and the impact of extracellular signal-regulated kinase signaling on apoptosis and necrosis. Toxicol Sci 92:464–475
Nielsen HD, Brown MT, Brownlee C (2003) Cellular responses of developing Fucus serratus embryos exposed to elevated concentrations of Cu2+. Plant, Cell Environ 26:1737–1747
Pierron F, Normandeau E, Defo MA, Campbell PG, Bernatchez L, Couture P (2011) Effects of chronic metal exposure on wild fish populations revealed by high-throughput cDNA sequencing. Ecotoxicology 20:1388–1399
Pietrobon D, Virgilio F, Pozzan T (1990) Structural and functional aspects of calcium homeostasis in eukaryotic cells. Eur J Biochem 193:599–622
Ramesh GT, Manna SK, Aggarwal BB, Jadhav AL (1999) Lead activates nuclear transcription factor-κB, activator protein-1, and amino-terminal c-Jun kinase in pheochromocytoma cells. Toxicol Appl Pharmacol 155:280–286
Rami A, Agarwal R, Botez G, Winckler J (2000) μ-Calpain activation, DNA fragmentation, and synergistic effects of caspase and calpain inhibitors in protecting hippocampal neurons from ischemic damage. Brain Res 866:299–312
Robinson CB, Wills PS, Riche MA, Straus DL (2013) Tissue-specific copper concentrations in red drum after long-term exposure to sublethal levels of waterborne copper and a 21-day withdrawal. N Am J Aquac 75:1–6
Santos EM, Ball JS, Williams TD, Wu H, Ortega F, Van Aerle R, Katsiadaki I, Falciani F, Viant MR, Chipman JK (2010) Identifying health impacts of exposure to copper using transcriptomics and metabolomics in a fish model. Environ Sci Technol 44:820–826
Schafer S, Bickmeyer U, Koehler A (2009) Measuring Ca2+-signalling at fertilization in the sea urchin Psammechinus miliaris: alterations of this Ca2+-signal by copper and 2, 4, 6-tribromophenol. Comp Biochem Physiol C 150:261–269
Seki E, Schwabe RF (2015) Hepatic inflammation and fibrosis: functional links and key pathways. Hepatology 61:1066–1079
Sheridan MA (1988) Lipid dynamics in fish: aspects of absorption, transportation, deposition and mobilization. Comp Biochem Physiol B 90:679–690
Song YF, Luo Z, Zhang LH, Hogstrand C, Pan YX (2016) Endoplasmic reticulum stress and disturbed calcium homeostasis are involved in copper-induced alteration in hepatic lipid metabolism in yellow catfish Pelteobagrus fulvidraco. Chemosphere 144:2443–2453
Stohs S, Bagchi D (1995) Oxidative mechanisms in the toxicity of metal ions. Free Radic Biol Med 18:321–336
Sun M, Zhao Y, Xu C (2008) Cross-talk between calpain and caspase-3 in penumbra and core during focal cerebral ischemia-reperfusion. Cell Mol Neurobiol 28:71–85
Tassabehji NM, Vanlandingham JW, Levenson CW (2005) Copper alters the conformation and transcriptional activity of the tumor suppressor protein p53 in human Hep G2 cells. Exp Biol Med 230:699–708
Tocher DR (2003) Metabolism and functions of lipids and fatty acids in teleost fish. Rev Fish Sci 11:107–184
Wang Z, Gerstein M, Snyder M (2009) RNA-seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10:57–63
Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, Wang J, Li S, Li R, Bolund L (2006) WEGO: a web tool for plotting GO annotations. Nucleic Acids Res 34:W293–W297
Zhan YJ (2005) The status quo of heavy metal pollution in Bohai sea and the abecedarian estimate of the effect of heavy metals on the growth of typical phytoplankton. Doctoral dissertation. Ocean University of China, Qingdao, China
Zhang R, Zhang LL, Ye X, Tian YY, Sun CF, Lu MX, Bai JJ (2013) Transcriptome profiling and digital gene expression analysis of Nile tilapia (Oreochromis niloticus) infected by Streptococcus agalactiae. Mol Biol Rep 40:5657–5668
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grants Nos. 31372547, 31422056, 31072226, and 30800850) and the Fundamental Research Funds for the Central Universities (Grant Nos. 2662015PY017, 2013PY073). We thank Beijing Genomics Institute (Shenzhen, China, http://english.big.cas.cn/) for cDNA library construction and Illumina sequencing.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have declared no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Chen, QL., Luo, Z., Huang, C. et al. De novo characterization of the liver transcriptome of javelin goby Synechogobius hasta and analysis of its transcriptomic profile following waterborne copper exposure. Fish Physiol Biochem 42, 979–994 (2016). https://doi.org/10.1007/s10695-015-0190-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10695-015-0190-2