
Abstract
Bacterial transcriptome profiling in the presence of plant fluids or extracts during microbial growth may provide relevant information on plant–bacteria interactions. Here, RNA sequencing (RNA-Seq) was used to determine the transcriptomic profile of Herbaspirillum seropedicae strain HRC54 at the early stages of response to sugarcane apoplastic fluid. Differentially expressed gene (DEG) analysis was performed using the DESeq2 and edgeR packages, followed by functional annotation using Blast2GO and gene ontology enrichment analysis using the COG and KEGG databases. After 2 h of sugarcane apoplastic fluid addition to the H. seropedicae HRC54 culture, respectively, 44 and 45 genes were upregulated and downregulated. These genes were enriched in bacterial metabolism (e.g., oxidoreductase and transferase), ABC transporters, motility, secretion systems, and signal transduction. RNA-Seq expression profiles of 12 genes identified in data analyses were verified by RT-qPCR. The results suggested that H. seropedicae HRC54 recognized sugarcane apoplastic fluid as the host signal, and some DEGs were closely involved at the early stages of the establishment of plant–bacteria interactions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sugarcane is an important crop in Brazilian agribusiness because of the high value of its products in various industries, including bioenergy production (Bordonal et al. 2018). Over the last decades, sugarcane yield has decreased slightly but significantly, which is typically attributed to low soil fertility and climate change (Pereira et al. 2013; CONAB de CNA 2020). In this light, need for the application of fertilizers, particularly nitrogen, has increased (Schultz et al. 2015), which can be harmful to the environment and increase agricultural costs (Gomes et al. 2008). An alternative to reduce fertilizer application is the use of inoculants with diazotrophic endophytic bacteria (DEB). DEB can convert N2 to NH3 through biological nitrogen fixation (BNF) and can act as plant growth-promoting bacteria (PGPB) (Baldani and Baldani 2005; Oliveira et al. 2006; de Souza et al. 2015; da Fonseca Breda et al. 2019; dos Santos et al. 2020).
Previous studies have shown the positive effects of DEB inoculation on plant growth via BNF, hormone production, defense response, nutrient acquisition, and abiotic and biotic stress tolerance (Oliveira et al. 2002, 2006, 2009; de Souza et al. 2015; Kandel et al. 2017). In sugarcane, the positive effects of inoculating five DEB strains, including Gluconacetobacter diazotrophicus PAL5T (BR11281), Herbaspirillum rubrisubalbicans HCC103 (BR11504), H. seropedicae HRC54 (BR11335), Nitrospirillum (formerly Azospirillum) amazonense CBAmC (BR11145), and Paraburkholderia (formerly Burkholderia) tropica PPe8T (BR11366), have been previously reported (Oliveira et al. 2006; Reis et al. 2009; Renan et al. 2016; dos Santos et al. 2020; Martins et al. 2020; Ramos et al. 2020). However, the beneficial effects attributed to inoculation are highly variable, and their underlying mechanisms remain unclear. Therefore, additional information on the fundamental aspects of plant–microorganism interaction is imperative (Monteiro et al. 2008).
H. seropedicae is a diazotrophic endophytic β-proteobacteria, isolated from crops such as rice, maize, and sugarcane (Baldani et al. 1986; Olivares et al. 1996), and it has also been used as a sugarcane inoculant. This bacterium has been shown to increase plant biomass (Guimarães et al. 2003) through acting on phytohormone signaling (Amaral et al. 2014; Tadra-Sfeir et al. 2015), plant defense responses (Brusamarello-Santos et al. 2012), nitrogen metabolism (Breda et al. 2018), siderophore production, and polyhydroxybutyrate synthesis (Tirapelle et al. 2013; Brader et al. 2014). Furthermore, diverse protein secretion systems help bacteria to successfully interact with plants (Monteiro et al. 2008; Pedrosa et al. 2011). H. seropedicae strain HRC54 is a diazotrophic entophyte isolated from sugarcane roots (Baldani et al. 1996), with potential to serve as a PGPB, particularly for gramineuos plants (Junior et al. 2008; Alves et al. 2014; Martins et al. 2020). However, no study has reported the molecular overview of the interaction between HRC54 and sugarcane.
In 2011, the complete genome of H. seropedicae strain SmR1 was compiled, which enabled the identification of genes involved in several pathways such as auxin biosynthesis, BNF, siderophore production, protein secretion, and plant–bacteria interactions (e.g., chemotaxis proteins, secretion systems, and flagellar biosynthesis) (Pedrosa et al. 2011). These genes allow the bacteria to recognize plant signals and modulate plant gene expression for endophytic colonization and plant growth promotion (Monteiro et al. 2012). However, the molecular pathways involved at the initial stages of the bacterial endophytic lifestyle have not been well studied.
The aboveground parts of host plants, specifically intercellular spaces, are frequently occupied by endophytic bacteria, and the apoplastic space is a niche for bacterial development, providing nutrients such as sugar, ammonium, nitrite, nitrate, amino acids, and proteins, which are essential for bacterial growth (Asis et al. 2003; Haslam et al. 2003; Tejera et al. 2006; Sattelmacher and Horst 2007; Pechanova et al. 2010). An in vitro system to grow microorganisms in the presence of specific plant fluids or tissues can mimic the endophytic environment and help study the initial stages of plant–microbe interactions and provide information on the expression profiles of genes involved in these processes (Dong et al. 1994). Therefore, the exposure of a bacterial culture to apoplastic fluid during a certain period can provide insight into the signaling cascades that activate the bacterial genes necessary for metabolic adaptations to live within the plant.
Transcriptomics can be applied in such experiments, allowing for the evaluation of microbial gene expression profiles under different scenarios, such as abiotic stress, or in specific pathways and interactions (Vacheron et al. 2013; Chandra et al. 2019; Raju et al. 2020). However, in most plant-associated bacterial transcriptomic studies conducted thus far, bacteria were cultured separately from the host plant to obtain sufficient bacterial mRNA transcripts (Levy et al. 2018). We previously used sugarcane apoplastic fluid in in vitro experiments on P. tropica PPe8T and N. amazonense CBAmC and found this approach to be appropriate for evaluating bacterial responses to plant signals (Silva et al. 2018; Terra et al. 2020). The short exposure time to apoplastic fluid set in those experiments allowed us to elucidate initial gene expression changes and their importance at the early stages of plant–bacteria interactions (Pinski et al. 2019; Taulé et al. 2021). To this end, in the present study, we sought to identify the differentially expressed genes (DEGs) of H. seropedicae HRC54 in response to the short-term exposure of its culture to sugarcane (RB867515 variety) apoplastic fluid.
Materials and methods
Sugarcane apoplastic fluid collection
The sugarcane variety RB867515 was sampled from the Embrapa Agrobiologia Experimental Field Station. This commercial variety is characterized by its ability to adapt to low-fertility soils, and it is responsive to PGPB inoculation (Schultz et al. 2014; dos Santos et al. 2020). After superficial washing, the stems were peeled, and the internodes were removed and disinfected by flaming (Silva et al. 2018). Sugarcane apoplastic fluid was obtained by centrifuging the stems in 50 mL tubes at 3,000 × g for 20 min at 4 °C (Dong et al. 1994). The extracted apoplastic fluid was passed through a 0.22 µm filter (diameter, 47 mm) and stored at − 70 °C until use.
H. seropedicae HRC54 growth in the presence of sugarcane apoplastic fluid
A pre-inoculum of H. seropedicae HRC54 was grown in 5 mL of DYGS medium (Baldani et al. 2014) for 12 h at 30 °C while shaking at 100 rpm. Next, the bacteria (105 cells mL−1) were inoculated in 100 mL of semi-selective JNFB liquid medium containing nitrogen (Baldani et al. 2014). After 16 h of growth under the same conditions mentioned above, the culture was divided in half and subjected to the following treatment: addition of sugarcane apoplastic fluid or water to the bacterial culture (at 1:1 proportion). The experiment was performed with three biological replicates for each treatment, resulting in six samples. The treatment lasted for 2 h at 30 °C, while shaking at 100 rpm. From each replicate of both treatments, 5 mL of the culture was collected and centrifuged at 2300×g for 10 min (Cordeiro et al. 2013), and the residual medium was discarded.
Cellular viability of H. seropedicae HRC54 in the presence of apoplastic fluid
To evaluate the effects of sugarcane apoplastic fluid on H. seropedicae HRC54, a LIVE/DEAD cell viability assay was performed. At the time of the start of the treatment (T0) and after 2 h (T2), samples (1 mL) were collected and centrifuged at 10,000×g for 10 min; the pellet was resuspended in 1 mL of 0.85% saline solution (NaCl, w/v). Then, 1.5 µL each of reagents A and B from the LIVE/DEAD BacLight Bacterial Viability Kit (Invitrogen™) was added, and the samples were incubated in the dark for 15 min. An aliquot of 10 µL of the incubated sample was used for microscopy using LSM700 AxioObserver (Zeiss) equipped with the Plan-Apochromat 40x/1.3 Oil DIC M27 and 63x/1.4 Oil DIC M27 objective lenses. For SYTO 9 staining, the excitation wavelength was 555 nm and detection wavelength was 578–800 nm (green fluorescence, live cells). For propidium iodide staining, the excitation wavelength was 488 nm and detection wavelength was 300–578 nm (red fluorescence, dead cells). Transmitted light was used with a T-PMT detector. To confirm the results of microscopy, bacterial colony-forming units were counted (CFU·mL−1) using selective media for Herbaspirillum sp. JNFb (Baldani et al. 2014). Briefly, 1 mL of sample from each biological replicate was centrifuged at 10,000×g for 10 min, and the resulting pellet was resuspended in 1 mL of 0.9% saline solution (NaCl, w/v). The suspensions were serially diluted to a density of 10–10 cells mL−1; then, 10 µL of the diluted suspension was spread on JNFb solid medium containing a nitrogen source and cultured for 24 h at 30 °C. Statistical analyses were performed using R (R Foundation for Statistical Computing, Vienna, Austria; http://www.R-project.org/). The results were compared using Student’s t test, considering 0.05 as the level of significance (p).
Total RNA extraction, mRNA enrichment, and RNA Sequencing (RNA-Seq)
Total RNA was extracted using the TRIZOL™ reagent (Invitrogen), followed by DNase I (Epicenter) treatment to completely remove genomic DNA. Both procedures were performed according to the manufacturer’s protocols. mRNA enrichment was performed using the MICROBExpress™ Bacterial mRNA Enrichment Kit (Ambion), following the manufacturer’s instructions. Six cDNA libraries were constructed using the Ion PITM Sequencing 200 Kit and sequenced using an ion proton semiconductor sequencer (Life).
Transcriptomic data analysis
The quality of raw reads generated from RNA-Seq was analyzed using FastQC (Andrews 2010). The reads were trimmed using FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit) and mapped to the genome of H. seropedicae HRC54 (unpublished). Read mapping data were used to calculate gene expression levels as reads per kilobase of transcript per million mapped reads (RPKM) using EDGE-pro (Magoc et al. 2013). Only reads aligned at least once with the reference genome of H. seropedicae HRC54 were used for the subsequent analyses. The list of DEGs with associated fold changes and significance estimates was generated using the R packages DESeq2 (Love et al. 2014) and edgeR (Robinson et al. 2010), with a cut-off p value of 0.05 and a log2 fold change of 1.5 (fold change of ≤ 1.5 indicated downregulated genes, and fold change of ≥ 1.5 indicates upregulated genes). The results from both packages were used for DEGS analyses to ensure the reliability of our final data. In the edgeR package, the false discovery rate (FDR) was analyzed at a cut-off of ≤ 0.05. The fold change was calculated as the difference between the strain + apoplast (apoplast) and strain + water (water) treatments. Only genes that were differentially expressed in both packages were selected for functional annotation with Blast2GO (Conesa et al. 2005) using the Kyoto Encyclopedia of Genes and Genomes (KEGG) and Clusters of Orthologous Groups (COG) databases (Kanehisa and Goto 2000; Tatusov et al. 2000).
RT-qPCR
To validate the results of RNA-Seq analysis, 12 DEGs were randomly selected for RT-qPCR. Primers were designed using Primer3Plus (Untergasser et al. 2007) according to the following criteria: sequence length of 19–22 nucleotides; annealing temperature of 58–62 °C; GC content of 50–80%; and amplicon size of 100–180 base pairs. The primers designed were also checked for dimer and hairpin formation using Oligo Explorer (http://www.uku.fi/˜kuulasma/OligoSoftware/) (Table S2). For this assay, a new experiment was designed using the same conditions as above and the same total RNA extraction and DNAse I treatment protocols, followed by cDNA synthesis using SuperScript™ III Reverse Transcriptase (Invitrogen). RT-qPCR was performed using the 7500 Fast Real-Time PCR System and QuantiTect SYBR® Green PCR (Qiagen). The raw fluorescence data were converted to cycle threshold (Ct) using Miner (Zhao and Fernald 2005), and relative expression was calculated using the delta–delta Ct method (2−ΔΔCT) and qBase v1.3.5 (Livak and Schmittgen 2001; Hellemans et al. 2007). rpoA and groEL were used as the reference genes (Pessoa et al. 2016). Statistical analyses were performed as described above (see section Cellular viability of H. seropedicae HRC54 in the presence of apoplastic fluid).
Results and discussion
Apoplastic fluid does not negatively affect the viability of H. seropedicae HRC54 cells
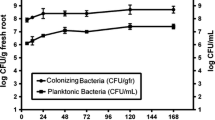
Confocal microscopy showed that the addition of sugarcane apoplastic fluid to the culture did not negatively affect the viability of H. seropedicae HRC54 cells (Fig. 1). However, the result observed in CFU counts was different; as such, the number of cells was not higher following the addition of apoplastic fluid (Fig. 2). Indeed, bacterial growth following the addition of water was higher than that after the addition of apoplastic fluid; however, this analysis was focused on the viability of HRC54 cell following exposure to sugarcane apoplastic fluid. In micrographs D, H, M, and Q, the bacterial morphology was not altered following the addition of sugarcane apoplastic fluid.

Confocal microscopy of H. seropedicae HRC54 in the presence of sugarcane apoplastic fluid using LIVE/DEAD BacLight Bacterial Viability Kits (Invitrogen™). Micrographs A, B, C, and D are from sample Apoplast; and micrographs E, F, G and H are from sample Water in the moment of the addition of the treatments (T0). Micrographs I, J, L, and M are from sample Apoplast; and micrographs N, O, P and Q are from sample Water after the two hours exposure of the treatments (T0). Red fluorescence represents dead cells and green fluorescence represents the live cells. Micrographs D, H, M and Q are using the 63 × lent, presenting the cell morphology. Scale bars represents 20 µm

Count of the UFC/mL from H. seropedicae HRC54 from the experiment with apoplastic fluid from sugarcane. Were evaluate for this assay, the moment from addition of the treatments (T0) and after 2 h of exposure (T2). The asterisks represent a significant difference in t test (p < 0.05), and NS represents a non-significant difference in t test (p > 0.05)
Transcriptomic analysis revealed 89 DEGs in H. seropedicae HRC54 in response to sugarcane apoplastic fluid
After raw data quality analysis, the trimmed reads were mapped against the H. seropedicae HRC54 genome, and the RPKM values were calculated (Table 1). Analyses using DESeq2 and edgeR with a p value of 0.05 revealed that the expression patterns of 1009 genes were altered. Using a cut-off of log2 fold change of ± 1.5, 89 genes were identified as differentially expressed under the experimental conditions, of which, respectively, 44 and 45 were upregulated and downregulated (Table 2). Of these DEGs, 18% encoded proteins or domains with unknown functions (DUFFs), 16.9% were related to oxidoreductase activity, and 13.5% were involved in transport and transmembrane transport (Tables S4 and S5). In addition, some DEGs encoded proteins related to signal transduction, transferase activity, secretion systems, cell division, and motility (Fig. 3). Finally, few DEGs encoded proteins related to chaperone, dioxygenase, isomerase, ligase, and translation activities (“Others” in Table 2).

Overview of differential expressed genes (DEGs) from H. seropedicae HRC54 in the presence of sugarcane apoplastic fluid, highlighting predominant functions with up and down-regulation such as transport, oxidoreductase activity and signal transduction; and unique regulations as motility, secretion system and hemagglutinin. The blue arrows represent upregulation and red arrows represent down-regulation
Expression of H. seropedicae HRC54 genes involved in specific metabolic subsystems was altered in response to sugarcane apoplastic fluid
Oxidoreductase activity
Six downregulated and nine upregulated genes were involved in oxidoreductase activity at the early stages of plant–bacteria interactions, which is associated with response to oxidative stress (Knief et al. 2011; Imlay 2014). Among the six downregulated genes, four were involved in the cytochrome C enzymatic chain (ccoP, coxB, coxA, and cox11), which are likely required for bacterial adaptation to different oxygen concentrations (Kulajta et al. 2006). Similarly, in H. seropedicae SmR1 inoculated on wheat plants, genes from the cytochrome C enzymatic chain were suppressed after 3 days (Pankievicz et al. 2016). The remaining two downregulated genes encoded glutamate synthase subunits alpha and beta (gltB and gltD). Six of the nine upregulated genes were related to NADP or NADPH redox activity (nfnB, yahK, Hs2057, Hs2266, Hs3119, and Hs3748), and the remaining three encoded peroxiredoxin (PRDX2I), an amino acid dehydrogenase (glud1_2), and an aldo/keto reductase (Hs2082). nfnB encodes NAD(P+)H nitroreductase, which is involved in the metabolism of nitrogen-containing compounds. yahK encodes NAD(P+)-dependent alcohol dehydrogenase required for glycolysis. In addition, Hs3748 encodes NADH dehydrogenase, which is involved in metabolic processes that generally require oxidoreductase activity. The Hs3119 open reading frame (ORF) was annotated as a transhydrogenase and the Hs2057 and Hs2266 ORFs as NADP-dependent oxidoreductases, but no additional functional information was found. Previous experiments with Paraburkholderia tropica PPe8T revealed the augmentation of genes involved in the oxidoreductase pathway and suppression of genes in the cytochrome C enzymatic chain in response to sugarcane apoplastic fluid (Silva et al. 2018). The differential modulation of these genes could be related to the initial bacterial attempt of adaptation to the presence of sugarcane apoplastic fluid.
Transferases and hydrolases
In H. seropedicae HRC54, the expression patterns of nine genes related to transferase activity were altered in response to sugarcane apoplastic fluid (Fig. 3). This modulation of transferase/hydrolase activity may be required for the metabolic adaption and survival of bacteria under stress and may involve compounds necessary for beneficial plant–microbe interactions (Orellana et al. 2017). The gst, puuE, tyrB, ndk, mdoH, and Hs799 ORFs were upregulated. tyrB encodes an aromatic amino acid transaminase, and puuE encodes 4-aminobutyrate aminotransferase, both related to amino acid metabolism and nitrogen group transfer. mdoH encodes a membrane glycosyltransferase involved in the transfer and metabolism of carbohydrates. ndK encodes a nucleoside-diphosphate kinase, a transferase related to ATP binding, and gst encodes a protein similar to glutathione S-transferase, a transferase involved in the transfer of aryl and acyl groups in glutathione metabolism. Glutathione S-transferase overexpression has been observed during Enterobacter lignolyticus growth in the presence of lignin (Orellana et al. 2017). The Hs799 ORF encoded a protein similar to glutamine amidotransferase, but no additional functional information was found. The downregulated genes thiD, selU, fadA, and tktA were annotated as hydroxymethylpyrimidine/phosphomethylpyrimidine, tRNA 2-selenouridine synthase, acetyl-CoA C-acyltransferase, and transketolase, respectively. Acetyl-CoA C-acyltransferase suppression was observed in Burkholderia kururiensis exposed to rice plant extract (Coutinho et al. 2015). thiD is involved in thiamine metabolism, and fadA and tktA are involved in carbohydrate, specifically fatty acid and pentose phosphate, metabolism. selU is involved in transfers during RNA biogenesis. Three genes related to hydrolase activity were differentially expressed, and only cbiG, which encodes a cobalamin biosynthesis protein, was downregulated. cbiG acts on the carbon–carbon bonds and is involved in porphyrin and chlorophyll metabolism. The ostB and Hs4740 ORFs were upregulated. ostB encodes trehalose 6-phosphate phosphatase, which is involved in starch and sucrose metabolism. The Hs4740 ORF encoded a protein similar to glycoside hydrolase, which is also involved in carbohydrate metabolism, but no additional functional information was found. H. seropedicae SmR1 associated with maize has been reported to exhibit differential expression patterns of hydrolases (Balsanelli et al. 2016). Overall, the overexpression of genes related to transferase and hydrolase activities suggests that the bacteria altered their gene expression pattern to adapt to and metabolize nutrients in sugarcane apoplastic fluid.
Signal transduction
Nine genes involved in signal transduction were differentially expressed, of which eight were downregulated and one was upregulated (Fig. 3). During plant–bacteria interactions, chemotaxis and signal transduction systems are key to other processes such as host adhesion and recognition (Falke et al. 1997; Batista et al. 2018). The upregulated gene glrR is a response regulator that enables bacteria to adapt and respond to the environment. The signal transduction genes aer; mcp; and the Hs2330, Hs3454, Hs3539, Hs4087, Hs505, and Hs707 ORFs were downregulated. mcp encodes a methyl-accepting chemotaxis protein, which serves as an aerotaxis receptor. Similar expression patterns have been reported during the growth of H. seropedicae SmR1 associated with maize and in the presence of the flavonoid naringenin (Tadra-Sfeir et al. 2015; Balsanelli et al. 2016) as well as in Nitrospirillum amazonense CBAmC in response to sugarcane apoplastic fluid (Terra et al. 2020). The Hs3454, Hs3539, Hs2330, and Hs505 ORFs were also annotated as chemotaxis proteins, but no additional functional information was found. These DEGs involved in signal transduction may be related to bacterial adaptation, signaling, and recognition. However, the annotation of these genes was automatic and no specific function was found; therefore, additional experiments are warranted to elucidate their biological function and involvement in the interaction process.
Transport system
Several genes encoding sorbitol, mannitol, and branched-chain amino acids of the ABC transport system and saccharide transporters were suppressed. ABC transporters are extremely important for bacterial metabolism because of their function in nutrient uptake and exchange (Fatht and Kolter 1993). Several genes related to ABC transporters (Fig. 3), such as smog and ugpC involved in saccharide transport as well as livH and livM involved in amino acid transport, were suppressed. Similarity analysis did not identify the orthologous of the Hs1900 OFR, but its location in the H. seropedicae HRC54 genome was very close to that of the Hs1901 and Hs1902 ORFs (livH and livM), suggesting that Hs1900 is related to the liv enzymatic chain. However, additional experiments are required to confirm this hypothesis. Similar expression profiles of genes related to transport systems were found in H. seropedicae SmR1 associated with wheat plants (Pankievicz et al. 2016) as well as in P. tropica PPe8T and N. amazonense CBAmC in response to sugarcane apoplastic fluid (Silva et al. 2018; Terra et al. 2020). The Hs80 ORF, annotated as a tripartite tricarboxylate transporter, was downregulated, but its function remains unknown. Only two genes involved in this pathway were upregulated in the presence of sugarcane apoplastic fluid. ctpV (copper transport) and the Hs2747 ORF were annotated as ABC transporter substrate-binding proteins. Copper is involved in homeostatic processes that regulate plant defense mechanisms, allowing infection and colonization during interaction (Thiebaut et al. 2014). Suppression of these genes suggests that the bacteria specifically modulated carbohydrate metabolism, probably in response to sugars present in the apoplastic fluid.
Four genes related to transmembrane transport, a more specific pathway within the transport system, were differentially expressed in H. seropedicae HRC54 in the presence of sugarcane apoplastic fluid. acrA and acrB, which are part of the multi-drug efflux system in Gram-negative bacteria, were upregulated. This family participates in the antimicrobial resistance pathway, specifically the surface adhesion chain. The resistance nodulation and cell division family (RND) proteins are responsible for bacterial surface adhesion. Genes encoding these proteins were are also upregulated in Azospirillum sp. associated with wheat plants (Camilios-neto et al. 2014) and Burkholderia kururiensis exposed to rice plant extract (Coutinho et al. 2015). During competition with other bacteria in the presence of Mimosa plant extract, genes of the RND family were induced in Burkholderia phymatum (Klonowska et al. 2018). The upregulated expression of the RND family genes in different plant–bacteria interaction experiments highlights the importance of this family in the interaction process. The Hs4729 ORF, a putative porin, was also upregulated. This protein is part of small exchange channels in the membrane and is involved in the membrane exchange and metabolism of poly-beta-hydroxybutyrate, an important compound in interaction (Tirapelle et al. 2013). The upregulation of this gene has also been reported in H. seropedicae SmR1 associated with wheat and maize (Balsanelli et al. 2016; Pankievicz et al. 2016). The bile acid gene Hs3295, a sodium transporter, was the only downregulated gene among those belonging to the transmembrane transport system. This gene may be related to photorespiratory metabolism in plants, but its function in bacteria remains unknown. Changes in the expression patterns of these genes highlight the importance of transport systems, which can help the bacteria to metabolize host-derived nutrients. However, further experiments are required to confirm this hypothesis.
Binding and cell division
Four genes related to binding were differentially expressed (Fig. 3). Meanwhile, polB and the Hs2304 and Hs2300 ORFs were downregulated. polB encodes a type II DNA polymerase responsible for DNA repair during DNA synthesis. The Hs2304 and Hs2300 ORFs were annotated as a DNA-binding response regulator and tetratricopeptide repeat protein, respectively. The DNA-binding response regulator is a part of the output domain of response regulators related to cellular processes, while the tetratricopeptide repeat protein is a motif with no specific function reported to date. hfq, an RNA-binding protein (RBP), was upregulated. RBPs are responsible for RNA regulation and metabolism and play crucial roles in cellular function, transport, and localization.
Regarding cell division, only one gene was upregulated and the others were downregulated. pal, a peptidoglycan-associated lipoprotein involved in bacterial survival under endophytic conditions (Godlewska et al. 2009), was upregulated. Although its role in virulence or interaction remains unclear, pal upregulation indicates the attempt of H. seropedicae HRC54 to identify sugarcane apoplastic fluid as a possible host plant signal. Cell division proteins are important during the initial stages of plant–bacteria interactions (dos Santos et al. 2010; Knief et al. 2011). ftsZ, required for the formation of a contractile ring structure (Z ring) at the future cell division and bacterial multiplication site, was downregulated. The ring assembly regulates the timing and site of cell division. In contrast, upregulation of this protein was reported in Gluconactobacter diazotrophicus associated with sugarcane (Lery et al. 2011) and Rhizobium tropici in response to heat stress (Gomes et al. 2012). Additional experiments with prolonged exposure to sugarcane apoplastic fluid may provide more details of genes related to cell division and their roles in plant–bacteria interaction.
Motility and secretion system
The flagelin and flagellar transcription genes fliC and flhC, which are important primary genes in bacterial flagellar assembly, were downregulated. This suppression of the motility system can be related to the bacterial switch from free swimming to motile habit for attaching to the host surface. Regarding the secretion system, three genes were upregulated at only 2 h after the addition of apoplastic fluid. Lip, dotU, and vgrG are members of the type VI secretion system (T6SS), involved in bacterial symbiosis and other processes of the plant–bacteria interaction. T6SS comprises nine genes, of which one-third were upregulated in H. seropedicae HRC54 in response to sugarcane apoplastic fluid. The motility system is important for bacterial interaction and is typically related to chemotaxis (Pedrosa et al. 2011; Monteiro et al. 2012), which is an initial step during interaction (Pankievicz et al. 2016).
Suppression of the motility system-related genes has also been reported in H. seropedicae SmR1 associated with maize and wheat plants as well as in response to naringenin and sugarcane extract (Cordeiro et al. 2013; Tadra-Sfeir et al. 2015; Balsanelli et al. 2016; Pankievicz et al. 2016). Similar suppression of these genes has also been reported in other species such as P. tropica PPe8T and N. amazonense CBAmC in response to sugarcane apoplastic fluid (Silva et al. 2018; Terra et al. 2020). In the present experiment, H. seropedicae HRC54 genes related to flagelin and flagellar transcription were suppressed in response to sugarcane apoplastic fluid, suggesting that the bacteria recognized the fluid as part of the host plant and initiated changes in the motility system essential to the endophytic life.
T6SS is believed to be exclusive to pathogenic bacteria (Jani and Cotter 2010). Nevertheless, recent studies have shown the importance of this secretion system in competition with other bacteria and further successful interaction with plants (Filloux 2009; Jani and Cotter 2010). Interestingly, three members of the T6SS (Lip, dotU, and vgrG) were overexpressed in H. seropedicae HRC54 in response to sugarcane apoplastic fluid. T6SS was also upregulated in B. kururiensis in the presence of rice plant extract as well as in P. tropica in response to sugarcane apoplastic fluid (Coutinho et al. 2015; Silva et al. 2018). These results highlight the importance of T6SS at the initial stages of beneficial plant–microbe interactions and the need for the comprehensive investigation of this system.
Hemagglutinins and chaperones
fhaB and the Hs2067 ORF, which encode filamentous hemagglutinin N-terminal domain proteins, were downregulated. Hemagglutinins may be related to bacterial virulence, but more recent data suggest that in some Gram-negative bacteria, these proteins are involved in adhesion and surface attachment as well as biofilm formation—aspects that are often related to bacterial invasion; endophytic bacteria may use similar mechanisms for interaction with plants (Bernal et al. 2018). Genes related to the chaperone activity, including trxA (thioredoxin) and hsIU (ATP-dependent protease), were upregulated in H. seropedicae HRC54 in response to sugarcane apoplastic fluid (Fig. 3). ATP-dependent proteases are required for the degradation of specific intracellular molecules. Thioredoxin is part of the cellular antioxidant system, which is important for the protection of bacteria from oxidative damage caused by reactive oxygen species (ROS). With the addition of sugarcane apoplastic fluid, the bacteria were exposed to a “new” environment, with a high probability of the presence of ROS. However, identification of only one gene is not sufficient to support this hypothesis.
Genes encoding hemagglutinin are relevant to plant–microbe interactions. Such genes have been identified in the genome of H. seropedicae SmR1 (Pedrosa et al. 2011). In the present experiment, two genes encoding hemagglutinin (fhaB and Hs2067) were suppressed in H. seropedicae HRC54 in response to apoplastic fluid. Although hemagglutinin has generally been believed to be related to the virulence of pathogenic bacteria, more recent data have shown its importance in surface attachment and biofilm formation (Ariyakumar and Nishiguchi 2009; Gottig et al. 2009; Bernal et al. 2018; Taulé et al. 2021). Interestingly, in the present experiment, genes related to another system believed to be exclusive to pathogenic bacteria (T6SS) were also differently expressed in HRC54 in response to sugarcane apoplastic fluid. Perhaps, these systems are related to plant–bacteria interactions and warrant further exploration.
Several ORFs annotated with general functions such as lyase activity, transcription, and protein activation in bacterial metabolism, including Hs245, Hs947, and Hs430, were downregulated. trpS, bpeT, pmm-pgm, RP-L31, and the Hs4167 ORF were upregulated in response to sugarcane apoplastic fluid. trpS and RP-L31 are related to the translation and metabolism of tryptophan, which is an important amino acid for bacterial growth (Glick 2015). pmm-pgm is a phosphomannomutase/phosphoglucomutase, which is related to glycolysis and involved in virulence factor synthesis in Pseudomonas aeruginosa (Regni et al. 2006). The Hs4167 ORF was annotated as a Tim44 domain-containing protein, which is responsible for the translocation of nuclear-encoded proteins across the mitochondrial inner membrane. These processes could be relevant to plant–bacteria interaction process and warrant further research.
Unknown and other functions
Several metabolic functions were affected by at least one of the altered genes (Fig. 3). Hs38, a putative membrane protein; Hs245, a glyoxalase/bleomycin resistance/extradiol dioxygenase; Hs430, a class I SAM-dependent methyltransferase; and Hs947, related to the TetR family transcriptional regulators, were downregulated. Meanwhile, trpS, bpeT, pmm-pgm, RP-L31, and the Hs4167 ORF were upregulated. TrpS is a tryptophan–tRNA ligase and RP-L31 is a type B 50S ribosomal protein, both related to translation. They are also members of the LysR transcriptional regulator family, a diverse family of genes related to virulence, motility, and quorum sensing. Similarity analysis showed that some DEGs were annotated as unknown functions in the COG and KEGG databases (Table S1). However, based on the position of their ORFs in the H. seropedicae HRC54 genome, it is reasonable to assume that they serve some specific functions hitherto unknown. For instance, the Hs3922 and Hs3926 ORFs were identified as proteins with domains of unknown function (DUFFs), whereas Hs3924 and Hs3925 are related to cytochrome C oxidase. Nevertheless, more detailed studies are warranted to confirm this hypothesis. Such domains or putative proteins in H. seropedicae strains have not been previously reported; therefore, future comprehensive studies of some of these differentially expressed proteins are essential to identify their functions at the initial stages of plant–bacteria interactions.
Expression patterns of 12 genes were validated with RT-qPCR
To confirm the expression patterns of H. seropedicae HRC54 genes in response to sugarcane apoplastic fluid, 12 genes were randomly selected for validation using RT-qPCR, of which 6 each were upregulated and downregulated (Table S3). The upregulated genes showed higher relative expression levels and the downregulated genes showed lower relative expression levels in samples exposed to sugarcane apoplastic fluid. The expression patterns of 9 of the 12 genes were confirmed by the assay (Fig. 4). The Hs4308 ORF showed a lower relative expression level in samples exposed to sugarcane apoplastic fluid, which was contradictory to the results of RNA-Seq analysis, in which this gene was upregulated. Similarly, smoG and ugpC showed higher relative expression levels in samples exposed to sugarcane apoplastic fluid, which was contradictory to the results of transcriptomic analysis (Fig. 4). This small variation in the results can be attributed to the fact that RT-qPCR is a more sensitive method. However, given that the expression patterns of majority of the genes were confirmed by the RT-qPCR assay, our transcriptomic data derived from in silico analyses can be considered reliable.

Relative expression of genes from H. seropedicae HRC54 in the presence of sugarcane apoplastic fluid using RT-qPCR. Validation was performed using up (A) and downregulated genes (B). The expression level was calculated by qBASE software using Delta CT method (2−ΔΔCT). The symbols represents a significant difference in t test (p < 0.05), and NS represents a non-significant difference in t test (p > 0.05)
Conclusions
DEGs explored in the present study constitute the early response of H. seropedicae HRC54 to compounds present in the apoplastic fluid of the sugarcane variety RB867515. The transcriptomic profile of H. seropedicae HRC54 during 2 h in the presence of sugarcane apoplast fluid showed the modulation of genes related to carbohydrate and amino acid metabolism, motility, secretion, oxidoreductase activity, and signal transduction. This expression pattern is consistent with bacterial response to host plant signals and adaptation to and survival under endophytic life conditions, suggesting that the bacteria recognized sugarcane apoplast fluid as the host plant environment. Additionally, in H. seropedicae HRC54, metabolism was altered to use compounds in the apoplastic fluid as a nutrient source and genes related to motility, secretion, transferase activity, and signal transduction necessary to establish interaction with the host plant were modulated. Furthermore, many putative proteins or proteins with DUFFs were differentially expressed in the transcriptomic data. Therefore, additional experiments are warranted to determine the importance of such proteins in the sugarcane–H. seropedicae interaction.
Accession numbers
The raw nucleotide sequences data for the six libraries were deposited in the National Center for Biotechnology Information (NCBI), in Sequence Read Archive (Leinonen et al. 2011) and are available under the BioProject accession number PRJNA694965 and BioSamples accession numbers SAMN17587759, SAMN17587760, SAMN17587761, SAMN17587762, SAMN17587763, SAMN17587764.
References
Alves GC, Videira SS, Urquiaga S, Reis VM (2014) Differential plant growth promotion and nitrogen fixation in two genotypes of maize by several Herbaspirillum inoculants. Plant Soil 387:307–321. https://doi.org/10.1007/s11104-014-2295-2
Amaral FP, Bueno JCF, Hermes VS, Arisi ACM (2014) Gene expression analysis of maize seedlings (DKB240 variety) inoculated with plant growth promoting bacterium Herbaspirillum seropedicae. Symbiosis 62:41–50. https://doi.org/10.1007/s13199-014-0270-6
Andrews S (2010) FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.bbsrc.ac.uk/projects/fas
Ariyakumar DS, Nishiguchi MK (2009) Characterization of two host specific genes, mannose sensitive hemagglutinin (mshA) and uridyl phosphate dehydrogenase (UDPDH) that are involved in the Vibrio fischeri-Euprymna tasmanica mutualism. FEMS Microbiol Lett 299:65–73. https://doi.org/10.1021/nl061786n.Core-Shell
Asis CA, Shimizu T, Khan Akao MKS (2003) Organic acid and sugar contents in sugarcane stem apoplast solution and their role as carbon source for endophytic diazotrophs. Soil Sci Plant Nutr 49:915–920. https://doi.org/10.1080/00380768.2003.10410356
Baldani JI, Baldani VLD (2005) History on the biological nitrogen fixation research in graminaceous plants: Special emphasis on the Brazilian experience. An Acad Bras Cienc 77:549–579. https://doi.org/10.1590/S0001-37652005000300014
Baldani JI, Baldani VLD, Seldin L, Dobereiner J (1986) Characterization of Herbaspirillum seropedicae gen. nov., sp. now., a root- associated nitrogen-fixing bacterium. Int J Syst Bacteriol 36:86–93. https://doi.org/10.1099/00207713-36-1-86
Baldani JI, Pot B, Kirchhof G et al (1996) Emended description of Herbaspirillum; inclusion of [Pseudomonas] rubrisubalbicans, a milk plant pathogen, as Herbaspirillum rubrisubalbicans comb. nov.; and classification of a group of clinical isolates (EF group 1) as Herbaspirillum species 3. Int J Syst Bacteriol 46:802–810. https://doi.org/10.1099/00207713-46-3-802
Baldani JI, Reis VM, Videira SS et al (2014) The art of isolating nitrogen-fixing bacteria from non-leguminous plants using N-free semi-solid media: a practical guide for microbiologists. Plant Soil. https://doi.org/10.1007/s11104-014-2186-6
Balsanelli E, Tadra-Sfeir MZ, Faoro H et al (2016) Molecular adaptations of Herbaspirillum seropedicae during colonization of the maize rhizosphere. Environ Microbiol 18:2343–2356. https://doi.org/10.1111/1462-2920.12887
Batista MB, Teixeira CS, Sfeir MZT et al (2018) PHB biosynthesis counteracts redox stress in Herbaspirillum seropedicae. Front Microbiol 9:1–12. https://doi.org/10.3389/fmicb.2018.00472
Bernal P, Llamas MA, Filloux A (2018) Type VI secretion systems in plant-associated bacteria. Environ Microbiol 20:1–15. https://doi.org/10.1111/1462-2920.13956
Brader G, Compant S, Mitter B et al (2014) Metabolic potential of endophytic bacteria. Curr Opin Biotechnol 27:30–37. https://doi.org/10.1016/j.copbio.2013.09.012
Brusamarello-Santos LCC, Pacheco F, Aljanabi SMM et al (2012) Differential gene expression of rice roots inoculated with the diazotroph Herbaspirillum seropedicae. Plant Soil 356:113–125. https://doi.org/10.1007/s11104-011-1044-z
Camilios-neto D, Bonato P, Wassem R et al (2014) Dual RNA-seq transcriptional analysis of wheat roots colonized by Azospirillum brasilense reveals up-regulation of nutrient acquisition and cell cycle genes. BMC Genomics 15:1471–2164
Chandra A, Roopendra K, Verma I (2019) Transcriptome analysis of the effect of GA3 in sugarcane culm. 3 Biotech 9:1–12. https://doi.org/10.1007/s13205-019-1908-0
CONAB de CNA (2020) Acompanhamento da Safra Brasileira-Cana de açúcar-V.7-SAFRA 2020/21-N.2-Segundo levantamento-Agosto 2020. Acompan da Safra Bras Grãos 7:64
Conesa A, Gotz S, Garcia-Gomez JM et al (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3676. https://doi.org/10.1093/bioinformatics/bti610
Cordeiro FA, Tadra-Sfeir MZ, Huergo LF et al (2013) Proteomic analysis of Herbaspirillum seropedicae cultivated in the presence of sugar cane extract. J Proteome Res 12:1142–1150. https://doi.org/10.1021/pr300746j
Coutinho BG, Licastro D, Mendonça-previato L et al (2015) Plant-influenced gene expression in the Rice Endophyte Burkholderia kururiensis M130. Mol Plant–microbe Interact 28:10–21. https://doi.org/10.1094/MPMI-07-14-0225-R
da Breda FAF, da Silva TFR, dos Santos SG et al (2018) Modulation of nitrogen metabolism of maize plants inoculated with Azospirillum brasilense and Herbaspirillum seropedicae. Arch Microbiol. https://doi.org/10.1007/s00203-018-1594-z
da Fonseca Breda FA, da Silva TFR, dos Santos SG et al (2019) Modulation of nitrogen metabolism of maize plants inoculated with Azospirillum brasilense and Herbaspirillum seropedicae. Arch Microbiol 201:547–558. https://doi.org/10.1007/s00203-018-1594-z
da Silva PRA, Vidal MS, Soares CDP et al (2018) Sugarcane apoplast fluid modulates the global transcriptional profile of the diazotrophic bacteria Paraburkholderia tropica. PLoS ONE 13:1–19. https://doi.org/10.1371/journal.pone.0207863
de Bordonal RO, Carvalho JLN, Lal R et al (2018) Sustainability of sugarcane production in Brazil. A review. Agron Sustain Dev. https://doi.org/10.1007/s13593-018-0490-x
de Oliveira ALM, De Canuto EL, Urquiaga S et al (2006) Yield of micropropagated sugarcane varieties in different soil types following inoculation with diazotrophic bacteria. Plant Soil 284:23–32. https://doi.org/10.1007/s11104-006-0025-0
de Souza R, Ambrosini A, Passaglia LMP (2015) Plant growth-promoting bacteria as inoculants in agricultural soils. Genet Mol Biol 38:401–419. https://doi.org/10.1590/S1415-475738420150053
Dong Z, Canny MJ, McCully ME et al (1994) A nitrogen-fixing endophyte of sugarcane stems (a new role for the apoplast). Plant Physiol 105:1139–1147. https://doi.org/10.1104/pp.105.4.1139
dos Santos MF, Muniz de Pádua VL, de Matos NE et al (2010) Proteome of Gluconacetobacter diazotrophicus co-cultivated with sugarcane plantlets. J Proteomics 73:917–931. https://doi.org/10.1016/j.jprot.2009.12.005
dos Santos SG, da Silva RF, Alves GC et al (2020) Inoculation with five diazotrophs alters nitrogen metabolism during the initial growth of sugarcane varieties with contrasting responses to added nitrogen. Plant Soil 451:25–44. https://doi.org/10.1007/s11104-019-04101-1
Falke JJ, Bass RB, Butler SL et al (1997) The two-component signaling pathway of bacterial chemotaxis: a molecular view of signal transduction by receptors, kinases, and adaptation enzymes. Annu Rev Cell Dev Biol 13:457–512. https://doi.org/10.1146/annurev.cellbio.13.1.457
Fatht MJ, Kolter R (1993) ABC transporters : bacterial exporters. Microbiol Rev 57:995–1017
Filloux A (2009) The type VI secretion system: a tubular story. EMBO J 28:309–310. https://doi.org/10.1038/emboj.2008.301
Glick BR (2015) Beneficial plant–bacterial interactions. Springer Int Publ. https://doi.org/10.1007/978-3-319-13921-0
Godlewska R, Wiśniewska K, Pietras Z, Jagusztyn-Krynicka EK (2009) Peptidoglycan-associated lipoprotein (Pal) of Gram-negative bacteria: function, structure, role in pathogenesis and potential application in immunoprophylaxis: Minireview. FEMS Microbiol Lett 298:1–11. https://doi.org/10.1111/j.1574-6968.2009.01659.x
Gomes MAF, Souza MD, Boeira RC, Toledo LG (2008) Nutrientes vegetais no meio ambiente: ciclos bioquímicos, fertilizantes e corretivos. Embrapa Meio Ambient Doc 66:62
Gomes DF, Stefânia J, Schiavon AL, Andrade DS (2012) Proteomic profiling of Rhizobium tropici PRF 81: identification of conserved and specific responses to heat stress. BMC Microbiol 12:1471–2180. https://doi.org/10.1186/1471-2180-12-84
Gottig N, Garavaglia BS, Garofalo CG et al (2009) A filamentous hemagglutinin-like protein of Xanthomonas axonopodis pv. citri, the phytopathogen responsible for citrus canker, is involved in bacterial virulence. PLoS ONE. https://doi.org/10.1371/journal.pone.0004358
Guimarães SL, Baldani JI, Baldani VLD (2003) Efeito da inoculação de bactérias diazotróficas endofíticas em arroz de sequeiro. Rev Agron 37:25–30. http://www.ia.ufrrj.br/revista/Vol.37-2003
Haslam RP, Downie AL, Raveton M et al (2003) The assessment of enriched apoplastic extracts using proteomic approaches. Ann Appl Biol. https://doi.org/10.1111/j.1744-7348.2003.tb00272.x
Hellemans J, Mortier G, De Paepe A et al (2007) qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol 8:R19. https://doi.org/10.1186/gb-2007-8-2-r19
Imlay JA (2014) Transcription factors that defend bacteria against reactive oxygen species. Annu Rev Microbiol 69:150612172656007. https://doi.org/10.1146/annurev-micro-091014-104322
Jani AJ, Cotter PA (2010) Type VI Secretion: not just for pathogenesis anymore. Cell Host Microbe 8:2–6. https://doi.org/10.1016/j.chom.2010.06.012
Junior RBM, Canellas LP, da Silva LG, Olivares FL (2008) Promoção de enraizamento de microtoletes de cana-de-açúcar pelo uso conjunto de substâncias húmicas e bactérias diazotróficas endofíticas. Rev Bras Cienc Do Solo 32:1121–1128
Kandel S, Joubert P, Doty S (2017) Bacterial endophyte colonization and distribution within plants. Microorganisms 5:77. https://doi.org/10.3390/microorganisms5040077
Kanehisa M, Goto S (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28:27–30. https://doi.org/10.1093/nar/27.1.29
Klonowska A, Melkonian R, Miché L et al (2018) Transcriptomic profiling of Burkholderia phymatum STM815, Cupriavidus taiwanensis LMG19424 and Rhizobium mesoamericanum STM3625 in response to Mimosa pudica root exudates illuminates the molecular basis of their nodulation competitiveness and symbiotic ev. BMC Genomics 19:1–22. https://doi.org/10.1186/s12864-018-4487-2
Knief C, Delmotte N, Vorholt JA (2011) Bacterial adaptation to life in association with plants—a proteomic perspective from culture to in situ conditions. Proteomics 11:3086–3105. https://doi.org/10.1002/pmic.201000818
Kulajta C, Thumfart JO, Haid S et al (2006) Multi-step assembly pathway of the cbb3-type cytochrome c oxidase complex. J Mol Biol 355:989–1004. https://doi.org/10.1016/j.jmb.2005.11.039
Leinonen R, Sugawara H, Shumway M (2011) The sequence read archive. Nucleic Acids Res 39:2010–2012. https://doi.org/10.1093/nar/gkq1019
Lery LMS, Hemerly AS, Nogueira EM et al (2011) Quantitative proteomic analysis of the interaction between the endophytic plant-growth-promoting bacterium Gluconacetobacter diazotrophicus and sugarcane. Mol Plant–microbe Interact 24:562–576. https://doi.org/10.1094/MPMI-08-10-0178
Levy A, Salas Gonzalez I, Mittelviefhaus M et al (2018) Genomic features of bacterial adaptation to plants. Nat Genet 50:138–150. https://doi.org/10.1038/s41588-017-0012-9
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25:402–408. https://doi.org/10.1006/meth.2001.1262
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. bioRxiv. https://doi.org/10.1101/002832
Magoc M, Wood D, Salzberg SL (2013) EDGE-pro: estimated degree of gene expression in prokaryotic genomes. Evol Bioinform. https://doi.org/10.4137/EBO.S11250
Martins DS, Reis VM, Schultz N et al (2020) Both the contribution of soil nitrogen and of biological N2 fixation to sugarcane can increase with the inoculation of diazotrophic bacteria. Plant Soil 454:155–169. https://doi.org/10.1007/s11104-020-04621-1
Monteiro RA, Schmidt MA, de Baura VA et al (2008) Early colonization pattern of maize (Zea mays L. Poales, Poaceae) roots by Herbaspirillum seropedicae (Burkholderiales, Oxalobacteraceae). Genet Mol Biol 31:932–937. https://doi.org/10.1590/s1415-47572008005000007
Monteiro RA, Balsanelli E, Wassem R et al (2012) Herbaspirillum-plant interactions: microscopical, histological and molecular aspects. Plant Soil 356:175–196. https://doi.org/10.1007/s11104-012-1125-7
Olivares FL, Baldani VLD, Reis VM et al (1996) Occurrence of the endophytic diazotrophs Herbaspirillum spp. in roots, stems, and leaves, predominantly of Gramineae. Biol Fertil Soils 21:197–200. https://doi.org/10.1007/s003740050049
Oliveira ALM, Urquiaga S, Döbereiner J, Baldani JI (2002) The effect of inoculating endophytic N2-fixing bacteria on micropropagated sugarcane plants. Plant Soil 242:205–215. https://doi.org/10.1023/A:1016249704336
Oliveira StoffelsSchmid ALMMM et al (2009) Colonization of sugarcane plantlets by mixed inoculations with diazotrophic bacteria. Eur J Soil Biol 45:106–113. https://doi.org/10.1016/j.ejsobi.2008.09.004
Orellana R, Chaput G, Markillie LM et al (2017) Multi-time series RNA-seq analysis of Enterobacter lignolyticus SCF1 during growth in lignin-amended medium. PLoS ONE 12:1–21. https://doi.org/10.1371/journal.pone.0186440
Pankievicz VCS, Camilios-Neto D, Bonato P et al (2016) RNA-seq transcriptional profiling of Herbaspirillum seropedicae colonizing wheat (Triticum aestivum) roots. Plant Mol Biol. https://doi.org/10.1007/s11103-016-0430-6
Pechanova O, Hsu C-Y, Adams JP et al (2010) Apoplast proteome reveals that extracellular matrix contributes to multistress response in poplar. BMC Genomics 11:674. https://doi.org/10.1186/1471-2164-11-674
Pedrosa FO, Monteiro RA, Wassem R et al (2011) Genome of Herbaspirillum seropedicae strain SmR1, a specialized diazotrophic endophyte of tropical grasses. PLoS Genet. https://doi.org/10.1371/journal.pgen.1002064
Pereira W, Leite JM, Hipólito GDS et al (2013) Acúmulo de biomassa em variedades de cana-de-açúcar inoculadas com diferentes estirpes de bactérias diazotróficas. Rev Cienc Agron 44:363–370. https://doi.org/10.1590/S1806-66902013000200020
Pessoa DDV, Vidal MS, Baldani JI, Simoes-Araujo JL (2016) Validation of reference genes for RT-qPCR analysis in Herbaspirillum seropedicae. J Microbiol Methods 127:193–196. https://doi.org/10.1016/j.mimet.2016.06.011
Pinski A, Betekhtin A, Hupert-Kocurek K et al (2019) Defining the genetic basis of plant–endophytic bacteria interactions. Int J Mol Sci. https://doi.org/10.3390/ijms20081947
Raju G, Shanmugam K, Kasirajan L (2020) High-throughput sequencing reveals genes associated with high-temperature stress tolerance in sugarcane. 3 Biotech 10:1–13. https://doi.org/10.1007/s13205-020-02170-z
Ramos AC, Melo J, de Souza SB et al (2020) Inoculation with the endophytic bacterium Herbaspirillum seropedicae promotes growth, nutrient uptake and photosynthetic efficiency in rice. Planta 252:1–8. https://doi.org/10.1007/s00425-020-03496-x
Regni C, Shackelford GS, Beamer LJ (2006) Complexes of the enzyme phosphomannomutase/phosphoglucomutase with a slow substrate and an inhibitor. Acta Crystallogr Sect F Struct Biol Cryst Commun 62:722–726. https://doi.org/10.1107/S1744309106025887
Reis VM, Urquiaga S, Pereira W et al (2009) Eficiência agronômica do inoculante de cana-de-açúcar aplicado em três ensaios conduzidos no Estado do Rio de Janeiro durante o primeiro ano de cultivo. Embrapa Agrobiol Pesqui e Desenvolv. https://www.embrapa.br/busca-de-publicacoes/-/publicacao/663945
Renan OP, Nivaldo S, Rafael CM et al (2016) Growth analysis of sugarcane inoculated with diazotrophic bacteria and nitrogen fertilization. African J Agric Res 11:2786–2795. https://doi.org/10.5897/AJAR2016.11141
Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 1:139–140. https://doi.org/10.1093/bioinformatics/btp616
Sattelmacher B, Horst WJ (2007) The apoplast of higher plants: compartment of storage, transport and reactions. Springer, Dordrecht, pp 1–447
Schultz N, Oliveira RP, Chaves VA et al (2014) Inoculation of sugarcane with diazotrophic bacteria. Rev Bras Cienc Do Solo 38:407–414
Schultz N, Reis VM, Urquiaga S (2015) Resposta da cana-de-açúcar à adubação nitrogenada: fontes nitrogenadas, formas de aplicação, épocas de aplicação e efeito varietal. Embrapa Agrobiol Seropédica 298:52
Tadra-Sfeir MZ, Faoro H, Camilios-Neto D et al (2015) Genome wide transcriptional profiling of Herbaspirillum seropedicae SmR1 grown in the presence of naringenin. Front Microbiol 6:1–8. https://doi.org/10.3389/fmicb.2015.00491
Tatusov RL, Galperin MY, Natale DA, Koonin EV (2000) The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res 28:33–36. https://doi.org/10.1093/nar/28.1.33
Taulé C, Vaz-Jauri P, Battistoni F (2021) Insights into the early stages of plant–endophytic bacteria interaction. World J Microbiol Biotechnol 37:1–9. https://doi.org/10.1007/s11274-020-02966-4
Tejera N, Ortega E, Rodes R, Lluch C (2006) Nitrogen compounds in the apoplastic sap of sugarcane stem: some implications in the association with endophytes. J Plant Physiol 163:80–85. https://doi.org/10.1016/j.jplph.2005.03.010
Terra LA, de Soares CP, Meneses CHSG et al (2020) Transcriptome and proteome profiles of the diazotroph Nitrospirillum amazonense strain CBAmC in response to the sugarcane apoplast fluid. Plant Soil 451:145–168. https://doi.org/10.1007/s11104-019-04201-y
Thiebaut F, Rojas CA, Grativol C et al (2014) Genome-wide identification of microRNA and siRNA responsive to endophytic beneficial diazotrophic bacteria in maize. BMC Genomics 15:766. https://doi.org/10.1186/1471-2164-15-766
Tirapelle EF, Müller-Santos M, Tadra-Sfeir MZ et al (2013) Identification of proteins associated with polyhydroxybutyrate granules from Herbaspirillum seropedicae SmR1—old partners,new players. PLoS ONE 8:e75066. https://doi.org/10.1371/journal.pone.0075066
Untergasser A, Nijveen H, Rao X et al (2007) Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res 35:W71–W74. https://doi.org/10.1093/nar/gkm306
Vacheron J, Desbrosses G, Bouffaud M-L et al (2013) Plant growth-promoting rhizobacteria and root system functioning. Front Plant Sci 4:1–19. https://doi.org/10.3389/fpls.2013.00356
Zhao S, Fernald RD (2005) Comprehensive algorithm for quantitative real-time polymerase chain reaction. J Comput Biol 12:1047–1064. https://doi.org/10.1089/cmb.2005.12.1047.Comprehensive
Acknowledgements
The authors thank the National Council of Scientific and Technological Development—CNPq for the fellowships (Grant number 308818/2016-4 and Doctor's scholarship 154782/2016-5); and Embrapa (project MP2 number 02.13.08.006.00.00) and CNPq/INCT-FBN (No. 573828/2008-3) for the financial support.
Author information
Authors and Affiliations
Contributions
DDVP and JLSA were responsible for the bioinformatic analysis; MZTS and EMS for the RNA-Seq experiment and CMS for the confocal microscopy. DDVP drafted the manuscript and MSV, JIB and JLSA corrected it. The authors are aware of the content and results presented in this publication.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest in the publication.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Pessoa, D.D.V., Dos-Santos, C.M., Vidal, M.S. et al. Herbaspirillum seropedicae strain HRC54 expression profile in response to sugarcane apoplastic fluid. 3 Biotech 11, 292 (2021). https://doi.org/10.1007/s13205-021-02848-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13205-021-02848-y