
Abstract
Tamarind seed has been a source of valuable nutrients such as protein (contains high amount of many essential amino acids), essential fatty acids, and minerals which are recognized as additive to develop perfect balanced functional foods. The objective of present work was to optimize the process parameters for extraction and hydrolysis of protein from tamarind seeds. Papain-derived hydrolysates showed a maximum degree of hydrolysis (39.49%) and radical scavenging activity (42.92 ± 2.83%) at optimized conditions such as enzyme-to-substrate ratio (1:5), hydrolysis time (3 h), hydrolysis temperature (65 °C), and pH 6. From this study, papain hydrolysate can be considered as good source of natural antioxidants in developing food formulations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Tamarindus indica L. is grown widely in in Asian countries. Its seeds are an abundant and cheaply available by-product of the tamarind pulp industry. India produces 100,000 ton of tamarind seed from 250,000 tons of tamarind (Singh et al. 2007). It is rich in protein; containing a high amount of essential amino acids, like isoleucine, leucine, lysine, histidine, methionine, phenylalanine, threonine, tryptophan, and valine. In addition to this, it contains non-essential amino acids such as cysteine, tyrosine, arginine, alanine, aspartic, glutamic, glycine, proline, and serine (de Lumen et al. 1986). It is also a good source of essential fatty acids and minerals particularly calcium, phosphorous, and potassium. Furthermore, tamarind kernel powder, tamarind seed polysaccharides, and tamarind gum are very useful in food and pharmaceutical industries. The compounds extracted from tamarind seed have applications such as excipients in drug delivery systems, in curing diseases and disorders and as an immunity booster. Various extracted components from tamarind seeds are useful as additives, emulsifying, and gelling agent in the food industry which is reviewed by Bagul et al. (2015).
The increasing demand for continuous improvement in nutritional and functional properties of protein has contributed to the diversion of the research work from conventional sources to unconventional plant-derived protein (Henry and Kettlewell 1996). Significant pharmacology and medicinal values were found in seeds dietary proteins which are reported as bioactive protein (Duranti et al. 2008). Information available for the nutritional composition and extraction of proteins from seeds such watermelon, wood apple and kiwi seeds (Sonawane et al. 2016; Deng et al. 2014). However, very few reports are available on the production of biofunctional peptides such as glycin rich peptides from guva (Pelegrini et al. 2008) antifungal peptides from kale seeds (Lin and Ng 2008) and antioxidant peptides from ber seeds (Kanbargi et al. 2016).
Hence, efforts were under taken to optimize parameters for tamarind seed protein extraction and enzymatic hydrolysis followed by its characterization for antioxidant capacity.
Materials and methods
Materials
Tamarind seeds [Periyakulam 1 (PKM-1)] were procured from local market of Nashik, Maharashtra, India. Seeds were winnowed to remove dried leaves, fruit shells, and other lighter material from them. Sorting was done to ensure that good quality seeds of tamarind were selected before further processing. Parasitic-worm-infected seeds were removed and damaged ones were separated. Seeds were then thoroughly washed to remove the adhered pulp and to remove partially hollow infected seeds. The seed was sundried until constant weight. They were stored in airtight containers (for 1 year) for further use.
Chemicals
Sodium chloride, sodium hydroxide, urea, Tris–HCl, citric acid, ascorbic acid, sodium phosphate dibasic, sodium phosphate monobasic, boric acid and 85% orthophosphoric acid, hydrochloric acid, sulfuric acid, petroleum ether (bp. 60–80 °C), and ethanol were purchased from SD Fine Chemicals (Mumbai, India). Sodium dodecyl sulfate, mercaptoethanol, cysteine hydrochloride, coomassie brilliant blue-250, polyethylene glycol, and dialysis bag were purchased from Hi-Media (Mumbai, India). All chemicals and reagents used in the present study were of analytical grade. Alcalase, papain, and protease were obtained from Advanced Enzymes Technologies Pvt Ltd. (Mumbai, India).
Preparation of tamarind kernel powder (TKP)
Roasting was used to remove the seed coat. Sand bath was used in the process to achieve an even roasting effect. During roasting, a temperature difference develops between seed coat and kernel and also water from the shell evaporates making it brittle which results in cracking of the coat. These cracked seed coats were easily removed by little pounding. Simple separation method like winnowing was used for separating coats from kernels. Tamarind seeds, after removal of the seed coat, were taken for grinding. The flour mill was cleaned and seeds were ground to obtain a fine powder. The kernel powder was passed through 40 mesh size to ensure the uniform size of the particle. Tamarind kernel powder (50 g) was packed in Wattman number I paper. In a closed jar, 200 ml of pet ether was added and the packed TKP was immersed into the solvent. Assembly was kept for 16–18 h at room temperature. The defatted flour was dried using oven drying (45 °C) before protein extraction (Tsopmo et al. 2010; Sonawane and Arya 2017b).
Centesimal composition of the tamarind seed flours
The powdered samples were subjected to proximate analysis in terms of moisture, fat, protein, ash, and carbohydrate contents which eventually decides the approaches to be followed for the processing of these samples. The analyses were carried out using official methods of analysis (AOAC 1990).
Extraction of protein from tamarind seed by sequential method
100 mg of defatted samples were weighed in a conical flask. 50 ml of the first solvent (distilled water pH 7) was added. The mixture was kept at room temperature for 1 h with continuous stirring. After, centrifugation was done at 10,000 rpm for 10 min. The supernatant was collected for the protein analysis. In the residue, the next solvent (5% NaCl solution) was added in the same ratio as previous. Similar procedure was followed for 70% ethanol and 0.25% NaOH. The yield of extracted protein was estimated by Bradford assay (Bradford 1976). Effect of different physical parameters like time, stirring speed, and sample-to-solvent ratio was also studied by keeping one-factor constant at a time.
Enzymatic hydrolysis of tamarind kernel protein
Protein samples were weighted and taken into the conical flask. Distilled water was added and stirred to make a homogenized mixture. pH of the protein solution was adjusted to optimum pH depending on the enzyme that had been used for hydrolysis, using 0.1 N HCl and 0.1 M NaOH. The enzyme was weighed according to the substrate-to-enzyme ratio (5:1 to 100:1), mixed with the protein solution, and stirred vigorously. The flask was then placed in the water bath set at optimum temperature conditions. After desired time (~ 1–4 h), the flask was taken out and the reaction was stopped by heating the mixture at 90 °C for 10 min. The mixture was cooled and analyzed for degree of hydrolysis and antioxidant activity. Optimum physical parameters, i.e., pH, temperature, and the method of inactivation for the enzyme used in the hydrolysis, are listed in Table 3. Optimization was carried out using parameters type of enzyme, enzyme-to-substrate ratio, hydrolysis time, and temperature.
Degree of hydrolysis
The degree of hydrolysis (DH) was determined by the TCA method of Hoyle and Merritt’s (1994) with slight modifications. In the first part, 500 μl of hydrolyzed protein samples were mixed with 500 μl of 20% TCA solution. The mixture was allowed to rest for 30 min. After 30 min., samples were centrifuged at 10,000 rpm. The soluble protein content of the supernatant was determined by the method of Lowry et al. (1951), modified by Hartree (1972). Results were expressed as mg of protein. Finally, degree of hydrolysis was determined using following equation:
Free radical scavenging assay
Antioxidant activity was measured using Hitachi spectrophotometer with the improved ABTS method (Cai et al. 2004; Re et al. 1999; Sonawane and Arya 2013). The ABTS reagent was prepared freshly and used within 3 days. The reagent was made by mixing 7 mM ABTS and 2.45 mM potassium persulfate. This reagent was incubated for 16–17 h at 37 °C. The ABTS cations were diluted with 0.01 M Phosphate buffer saline to set OD at 0.7 (± 0.02) at 734 nm. 3.9 ml (absorbance of 0.700 ± 0.02) of ABTS was added to the 0.1 ml of the hydrolyzed sample and mixed thoroughly and absorbance was measured at 734 nm immediately after 6 min. The standard curve was linear between 0 and 20 μM trolox. Results were expressed in μM Trolox equivalents (TE)/g.
DPPH free radical scavenging assay
The ability to scavenge DPPH free radicals was determined based on the method (Sahreen et al. 2010; Abdille et al. 2005; Sonawane and Arya 2015) with slight modification in the mixture of test sample concentration and DPPH concentration. 0.15 mM of DPPH prepared in methanol was diluted to set the absorbance below 1.2 (± 0.02) at 517 nm and added to the 1 ml of the test sample in a test tube and vigorous shaking. This was then kept for 15 min incubation in dark room and absorbance was measured at 517 nm. The standard curve was linear between 0 and 10 μM/ml of Trolox which expressed in μM trolox equivalents (TE)/g.
Radical formation by ferric-reducing antioxidant power (FRAP)
Ferric-reducing antioxidant power (FRAP) assay was developed by Benzie and Strain (1996) with slight modification (Thaipong et al. 2006) in the mixture of test sample and FRAP reagent concentration. First, FRAP reagent was prepared by mixing the following solutions: tenfold 300 mM acetate buffer + onefold TPTZ (10 mM in 40 mM HCl) + onefold FeCl3 (20 mM). This 1 ml of FRAP reagent was added to the 0.1 ml of sample extract which was then shaken vigorously and absorbance was read at 593 nm after incubation of 15 min at 37 °C. The standard curve was linear between 0 and 100 μM trolox. Results were expressed in μM trolox equivalents (TE)/g.
Determination of molecular weight of peptides
The molecular weight of peptides formed during hydrolysis was determined by the SDS-PAGE. For electrophoresis, 30% acrylamide-bis-acrylamide solution mixture was used for the preparation of 4% stacking gel and 12% resolving gel. 10–20 μl of sample mixed with the sample buffer was loaded into the well. Gel was run at 70 V for approximately 90–120 min for the visualization of bands, silver staining was done (Sonawane and Arya 2017a, b).
Statistical Analysis
Statistical analysis of the data was carried out using Tukey’s multiple comparison tests (p < 0.05) using the SPSS software package version 16 of IBM.
Results and discussion
Centesimal composition of tamarind seed flour
The result of the proximate analysis is compiled in Table 1. Proximate analysis was done for both whole tamarind seed (with seed coat) and tamarind kernel (without seed coat). These values were compared with reported values from the literature (Morad et al. 1978; Ishola et al. 1990). The moisture content of the whole seed was 23.60%, while reported moisture content was in the range of 9.4–11.3% (Morad et al. 1978; Ishola et al. 1990). The protein content of whole seed was about 16.63%, while reported value was in the range of 13.3–26.9% (Morad et al. 1978; Ishola et al. 1990). The fat content of the whole seed observed was 6.13%, whereas reported values were in the range of 4.5–16.2% (Morad et al. 1978; Ishola et al. 1990). The carbohydrate content was found to be around 51.27%, while its reported value was in the range of 50.0–57.0% (Morad et al. 1978; Ishola et al. 1990). The ash content was 2.37%, while reported value was 2.4–4.2% (Morad et al. 1978; Ishola et al. 1990). For the seed kernel, moisture content was 14.77% and its reported value was in the range of 11.4–22.7%. The protein content of the kernel was 20.34% and reported was 15.0–20.9%. The fat content of the seed kernel was 3.5%, while reported was in the range of 3.9–16.2% and the carbohydrate content was found to be around 58.51%, while reported carbohydrate was in the range of 65.1–72.2%. Seed kernel showed a lower level of a fat percentage than the whole seed. This can be attributed to the pre-treatments that were given to the whole seed. When seeds were roasted to remove seed coat, a small amount of oil leached out during the process. This might be the reason for the low fat percentage observed in seed kernel. Seed kernel also showed more protein percentage than that of the whole seed (Table 1).
Protein extraction by sequential extraction method and optimization of processes parameters
According to the solubility-fractionation scheme of Osborne (Ewart 1968; Page 1985), the proteins were fractionated into four groups (albumin, globulin, prolamin, and glutelin) according to their solubility in distilled water (pH 7.0), 5% NaC1 solution (pH 7.0), 70% ethanol, and 0.25% NaOH solution (pH 10.0), respectively. Same scheme was applied for extraction, where the solutions mentioned above were used as a protein extraction solvent. Results of sequential protein extraction are mentioned in Table 2. In stage I, 15.31 μg/mg of albumin protein was obtained. In the second stage of extraction, an yield of 6.68 μg/mg of globulin was obtained. In the third and fourth stages, 18.82 μg/mg of prolamin and 87.65 μg/mg of glutelin were extracted, respectively. The yield of extracted protein combined to obtain overall yield for sequential protein extraction, which was 128.26 μg/mg (Table 2).
Physical parameters like time, stirring speed, and sample-to-solvent ratio was optimized during extraction.
Optimization of time for protein extraction
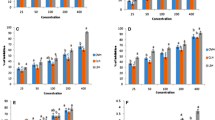
Time was optimized by varying protein extraction time at all the different stages. Yield at each stage was determined after every 1, 2, and 3 h (Fig. 1). Maximum protein yield (26.82 μg/mg) was observed during stage I (2 h) followed by at 3 h (26.11 μg/mg), with no significant differences. For stage II, yield was maximum, i.e., 19.68 and 22.70 μg/mg at both 2 and 3 h with no significant differences. For both stages III and IV, maximum yield observed was 18.38 and 71.48 μg/mg after an 1 h of extraction.

Effect of different extraction times on sequential protein extraction
Optimization of stirring speed
To determine the effect of stirring on the extraction, 300 and 800 rpm stirring speed was chosen, where 300 rpm was speed used in all earlier extraction, while 800 rpm was the maximum speed of an instrument. When yield was checked after the respective time of extraction (Fig. 2), no significant differences at first three stages (I, II, and III) were observed except for stage IV, wherein an increase in yield was observed at 800 rpm.

Effect of different stirring speeds on sequential protein extraction
Optimization of sample-to-solvent ratio
The sample-to-solvent ratio was optimized by taking three ratios into consideration viz., 1:50, 1:100, and 1:150. The highest yield was obtained for 1:100 ratios at stage I, stage II, and stage IV of extraction (Fig. 3), which was 27.14, 25.46, and 156.37 μg/mg, respectively. However, for extraction of prolamin, i.e., stage III, a ratio which gave a maximum yield of 18.38 μg/mg was 1:50. The total protein content observed for tamarind seed kernel powder during sequential extraction was 227.35 as compared to 128.26 without optimization (Table 2). Hence, the yield obtained with this method was considerably good. As the protein yield was high for albumin and glutelin which were subjected to enzymatic hydrolysis.

Effect of different sample-to-solvent ratios on sequential protein extractions
Protein hydrolysis by enzymatic method
Tamarind kernel protein, i.e., albumin and glutelin, was hydrolyzed with the help of proteolytic enzymes. Hydrolysis was done at optimum pH and temperature level of the particular enzyme, as given in Table 3. For initial stage, sample-to-enzyme ratio was taken as 1:10. Simultaneously, the antioxidant activity of the hydrolyzed peptide was determined by DPPH assay. It was observed that the antioxidant activity was dependent on the amino acid composition of peptides and degree of hydrolysis and enzyme used for hydrolysis (Pihlanto 2006).
Screening of type of enzyme for hydrolysis
Table 3 shows the degree of hydrolysis (DH) and % radical scavenging activity for albumin and glutelin protein, respectively. At the given conditions, i.e., at 1:10 enzyme-to-substrate ratio, 2 h of hydrolysis time and the respective optimum temperature and pH conditions are shown in Table 3, and trypsin showed maximum DH of 49.73% with 43.99% radical scavenging activity. Papain showed 27.73% of DH and 45.09% of radical scavenging activity, while chymotrypsin showed 44.26% DH and the highest radical scavenging activity of 47.99% (Table 4). However, statistically, there was no significant difference observed for the % radical scavenging activities of these three enzymes. In case of glutelin, the highest DH values were shown by trypsin (33.50%), chymotrypsin (32.07%) and protease (31.23%) with no significant differences (Table 4). Papain showed DH of 21.07 which was much lower than the other enzymes. However, the % radical scavenging activity shown by all the six enzymes.
When the same experiments were performed for the mixture of albumin and glutelin, the highest DH value was obtained for trypsin (39.76%) and chymotrypsin (34.51%) and the DH for papain (25.34) was a bit lower than both trypsin and chymotrypsin. However, the % radical scavenging activity was 38.97, 39.42, and 37.81 for papain, trypsin, and chymotrypsin, respectively, which shows no significant difference (Table 4).
Ideally, the enzyme is selected based on its highest performance for degree of hydrolysis (formation of large number of small peptides which aids good protein digestibility) and % radical scavenging activity. However, the cost of the enzyme plays a significant role in designing the cost-effective nutraceutical product. In all the three cases, papain showed equivalent % radical scavenging activity to trypsin and chymotrypsin and cost of the papain was lower than that of the trypsin and chymotrypsin. Hence, papain was selected for hydrolysis of tamarind kernel protein (50:50 mixtures of albumin and glutelin).
Optimization of enzyme-to-substrate ratio
The enzyme-to-substrate ratio was optimized for papain by trying hydrolysis for different ratios such as 1:5, 1:10, 1:50, and 1:100 (Table 5). The enzyme was inactivated after 2 h and DH and radical scavenging activity was analyzed with given assays. The degree of hydrolysis was higher at the lower ratio of enzyme to substrate such as 29.53% found at 1:10 and 33.49% at 1:5. These values were statistically similar. At enzyme-to-substrate ratio of 1:5, radical scavenging activity obtained was equal to 42.92%, which was significantly higher than that of the activity obtained at 1:10 enzyme-to-substrate ratio, i.e., 37.57%. Therefore, an enzyme-to-substrate ratio of 1:5 was optimized.
Optimization of hydrolysis time
Hydrolysis time was optimized by checking DH value and % radical scavenging activity at 1, 2, 3, and 4 h by keeping 1:5 enzyme-to-substrate ratio and its optimum temperature and pH constant. Maximum DH value of 32.82 and 33.03% was obtained at 3 and 4 h, respectively. Statistically, there was no difference between the two was observed. Similarly, % radical scavenging activity was highest at 3 (41.35) and 4 h (40.83) with no statistical difference (Table 5). Hence, an hydrolysis time of 3 h was considered optimal for the process.
Optimization of temperature for hydrolysis
For optimization of process temperature, different temperatures between 45 and 85 °C were taken and the maximum DH and activity was checked by keeping enzyme-to-substrate ratio (1:5), hydrolysis time (3 h), and pH (6) constant. The degree of hydrolysis and radical scavenging activity was maximum 38.38 and 40.74%, respectively, for 65 °C. Activity, as well as DH, decreases with increase in temperature, as shown in Table 5. The main reason might be the loss of activity of enzyme due to high temperature (Chaplin and Bucke 1990). Hence, at 75 and 85 °C, DH and the radical scavenging activity were very low.
Optimization of pH for hydrolysis
Different pH over the range of 4–8 were checked for the highest DH and radical scavenging activity by keeping enzyme-to-substrate ratio (1:5), hydrolysis time (3 h), and hydrolysis temperature (65 °C) constant. Maximum degree of hydrolysis (39.49%) and radical scavenging activity (42.92 ± 2.83%) was observed at pH 6 (Table 5). This could be due to the fact that at different pH (except optimum); the charge on structure varies with consequent structural alterations. This results in changing the binding of substrate, the catalytic efficiency, and the amount of active enzyme (Chaplin and Bucke 1990).
Antioxidant activity of protein hydrolysates
Antioxidant activity of the protein hydrolysates was checked by three assays DPPH radical scavenging assay, ABTS assay, and FRAP assay. The antioxidant activity was measured in terms of Trolox Equivalent per mg of protein. Papain-derived hydrolysates showed an activity of 178.83 μM TE/mg of protein by DPPH assay, 66.94 μM TE/mg of protein by ABTS assay, and 94.71 μM TE/mg of protein by FRAP assay (Table 6). The protein hydrolysate from ber seed using papain shows 9.14 μM TE/mg of protein and 17.54% activity by DPPH. Similarly, 15.71% of DPPH activity at concentration of 5 mg/ml shown by C. lanatus seed protein hydrolysate (Sonawane and Arya 2017a) and 15.02% DPPH activity observed at 40 µg/ml from L. acidissima seed protein hydrolysate (Sonawane and Arya 2017a). The difference in the activity of protein hydrolysate is due to the different seeds and different methods of extractions used (Kanbargi et al. 2016). The sequence of amino acids or composition of amino acids, hydrophobicity of protein molecules, and size are responsible to affect on antioxidant activity (Kong and Xiong 2006; Sarmadi and Ismail 2010). There are reports which show that few amino acids like Arg, Lys, Gly, His, Leu, and Tyr show antioxidant activity (Xie et al. 2008).
SDS-PAGE for molecular weight determination of peptides formed
SDS-PAGE was performed to determine the molecular weights of different peptides formed during the hydrolysis. It was seen that broad range of peptides was formed during the hydrolysis process (Fig. 4). There were around 14–15 bands of protein fractions and peptides were seen with a molecular weight ranging from 70–74 kDa to even lower than 14 kDa.

SDS-PAGE for papain hydrolysates
Glutelins are storage proteins. Native glutelins weighted approximately 300 kDa. The peptide subunits of gluteins are of two types, high molecular weight subunit (80–120 kDa) and low molecular weight subunit (40–55 kDa). It was seen (Table 4) that the DH value for the glutelin protein was low. Therefore, there were a large amount of protein which was not hydrolyzed and appeared in the SDS-PAGE at high molecular weight region.
Conclusions
Proteins were efficiently extracted using sequential extraction method which were further subjected to hydrolysis through screening of different enzymes for obtaining good antioxidant activity polypeptide. From above study, it can be concluded that bioactivity of the peptide depends on the type of enzyme used for the hydrolysis, degree of hydrolysis and the type of peptide formed. Optimum temperature, pH condition during the hydrolysis also plays very important role to retain the enzyme in its active form. Therefore, according to this study, papain hydrolyzed tamarind kernel protein (albumin and glutelin) at optimized conditions; showed good antioxidant activity. This may foreknow their future uses into either functional foods or dietary supplements.
References
Abdille MH, Singh RP, Jayaprakash GK, Jena BS (2005) Antioxidant activity of the extracts from Dillenia indica fruits. Food Chem 90:891–896
AOAC (1990) Official methods of analysis, 15th edn. Association of Official Analytical Chemists, Washington, DC
Bagul MB, Sonawane SK, Arya SS (2015) Tamarind seeds: chemistry, technology, applications and health benefits: a review. Indian Food Ind 34(3):28–35
Benzie IFF, Strain JJ (1996) The ferric reducing ability of plasma as a measure of “antioxidant power”: the FRAP assay. Anal Biochem 239:70–76
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Cai YZ, Luo Q, Sun M, Corke H (2004) Antioxidant activity and phenolic compounds of 112 Chinese medicinal plants associated with anticancer. Life Sci 74:2157–2184
Chaplin MF, Bucke C (1990) Enzyme technology. CUP Arch 1990:20
De Lumen BO, Becker R, Reyes PS (1986) The nutritive value of tamarind seed for broiler chicks. Anim Feed Sci Technol 22(4):285–293
Deng J, Sun T, Cao W, Fan D, Cheng N, Wang B, Gao H, Yang H (2014) Extraction optimization and functional properties of proteins from kiwi fruit (Actinidia chinensis Planch.) seeds. Int J Food Prop 17(7):1612–1625
Duranti M, Consonni A, Magni C, Sessa F, Scarafoni A (2008) The major proteins of lupin seed: characterisation and molecular properties for use as functional and nutraceutical ingredients. Trends Food Sci Technol 19:624–633
Ewart JAD (1968) Fractional extraction of cereal flour protein. J Sci Food Agric 19:241
Hartree EF (1972) Determination of protein: a modification of the lowry method that gives a linear photometric response. Anal Biochem 48(2):422–427
Henry J, Kettlewell P (1996) Cereal grain quality. Chapman and Hall, London
Hoyle NT, Merritt JOHN (1994) Quality of fish protein hydrolysates from herring (Clupea harengus). J Food Sci 59(1):76–79
Ishola MM, Agbaji EB, Agbaji AS (1990) A chemical study of Tamarindus indica (Tsamiya) fruits grown in Nigeria. J Sci Food Agric 51:141–143
Kanbargi KD, Sonawane SK, Arya SS (2016) Functional and antioxidant activity of Ziziphus jujube seed protein hydrolysates. Food Meas 10:226–235
Kong BH, Xiong YL (2006) Antioxidant activity of zein hydrolysate in liposome system and the possible mode of action. J Agric Food Chem 54:6059–6068
Lin P, Ng TB (2008) A novel and exploitable antifungal peptide from kale (Brassica alboglabra) seeds. Peptides 29:1664–1671
Lowry OH, Rosebrough NJ, Farr AL, Randal RJ (1951) Protein analysis with Folin-phenol reagent. J Biol Chem 193:265–275
Morad MM, El Magoli SB, Sedky KA (1978) Physico-chemical properties of Egyptian tamarind seed oil. Fette Seifen Anstrichmittel 80:357–359
Page S (1985) Characterization of Oat (Avena sativa L.) residual proteins. Cereal Chem 62(4):276–279
Pelegrini PB, Murad AM, Silva LP, dos Santos RCP, Costa FT, Tagliari PD, Bloch C Jr, Noronha EF, Miller RNG, Franco OL (2008) Identification of a novel storage glycine-rich peptide from guava (Psidium guajava) seeds with activity against Gram-negative bacteria. Peptides 29:1271–1279
Pihlanto A (2006) Antioxidative peptides derived from milk proteins. Int Dairy J 16(11):1306–1314
Re R, Pellegrini N, Proteggente A, Pannala A, Yang M, Rice-Evans C (1999) Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic Biol Med 26:1231–1237
Sahreen S, Khan M, Khan R (2010) Evaluation of antioxidant activities of various solvent extracts of Carissa opaca fruits. Food Chem 122:1205–1211
Sarmadi BH, Ismail A (2010) Antioxidative peptides from food proteins. Peptides 31:1949–1956
Singh D, Wangchu L, Moond SK (2007) Processed products of Tamarind. Nat Prod Rad 6(4):315–321
Sonawane S, Arya SS (2013) Antioxidant activity of jambhul, wood apple, ambadi and ambat chukka: an indigenous lesser known fruits and vegetables of India. Adv Food Sci Technol 5(03):270–275
Sonawane SK, Arya SS (2015) Effect of drying and storage on bioactive components of jambhul and wood apple. J Food Sci Technol 52:2833–2841
Sonawane SK, Arya SS (2017a) Citrullus lanatus protein hydrolysate optimization for antioxidant potential. Food Meas 11(4):1834–1843
Sonawane SK, Arya SS (2017b) Bioactive L acidissima protein hydrolysates using Box-Behnken design. 3 Biotech 7:218
Sonawane SK, Bagul MB, LeBlanc JG, Arya SS (2016) Nutritional, functional, thermal and structural characteristics of Citrullus lanatus and Limonia acidissima seed flours. Food Meas 10(1):72–79
Thaipong K, Boonprakob U, Crosby K, Cisneros-Zevallos L, Byrne HD (2006) Comparison of ABTS, DPPH, FRAP, and ORAC assays for estimating antioxidant activity from guava fruit extracts. J Food Compos Anal 19:669–675
Tsopmo A, Cooper A, Jodayree S (2010) Enzymatic hydrolysis of oat flour protein isolates to enhance antioxidative properties. Adv J Food Sci Technol 2(4):206–212
Xie Z, Huang J, Xu X, Jin Z (2008) Antioxidant activity of peptides isolated from alfalfa leaf protein hydrolysate. Food Chem 111:370–376
Acknowledgements
The authors gratefully acknowledge Advance Enzyme, Mumbai, India for providing free samples of enzyme used in the study. The authors also thank the Department of Biotechnology (DBT), Government of India for financial support to carry out this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Authors do not have any conflict of interest.
Rights and permissions
About this article

Cite this article
Bagul, M.B., Sonawane, S.K. & Arya, S.S. Bioactive characteristics and optimization of tamarind seed protein hydrolysate for antioxidant-rich food formulations. 3 Biotech 8, 218 (2018). https://doi.org/10.1007/s13205-018-1240-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13205-018-1240-0