
Abstract
Adulteration of milk is a common practice that concerns regulatory agencies, industry, and the population. Despite the growing need for checking adulteration, the current methods employed generally have low performance and are highly dependent on manual labor. This study aims to calibrate and validate a compact equipment (MilkoScan FT1) that adopts a Fourier transform infrared spectroscopy methodology to monitor adulteration in raw milk. Almost 2500 milk samples were used for reference spectrum construction and 1650 samples were used to validate the identification of the following five most commonly used adulterants (at three different concentrations each): (1) cornstarch, (2) sodium bicarbonate, (3) sodium citrate, (4) formaldehyde, and (5) saccharose, plus the additions of two levels of water or whey. To define the calibration with the best performance in milk adulteration identification, 12 calibrations involving 8, 10, 12, 14, 16, or 18 factors, with one or two outlier eliminations, were developed. The results of sensitivity and specificity analyses, as well as Kruskal–Wallis and Dunn multiple comparison tests, revealed that the calibration that best identified the adulterants was the one involving 14 factors, with a single elimination of outliers, exhibiting for all adulterants simultaneously, 84% sensitivity and 100% specificity. The calibration showed excellent sensitivity to cornstarch (>98%), sodium bicarbonate (100%), sodium citrate (99%), and formaldehyde (>84%), indicating that this calibration has good capacity for adulteration detection. Thus, this methodology is a viable option for the dairy industry to identify adulteration of raw milk.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Food adulteration is a very old practice that has become more sophisticated over time (Kamal and Karoui 2015). Foods with high nutritional value, such as milk, are the most vulnerable to adulteration; as a result, the demand for products with guaranteed authenticity is growing (Karoui and De Baerdemaeker 2007).
In recent years, several adulteration practices have been observed in milk, such as the addition of melamine in China (Lim et al. 2016) and the additions of various other compounds, such as sodium citrate, sodium hydroxide, sodium chloride, saccharose, phosphates, carbonates, bicarbonates, hydrogen peroxide, urea, and formaldehyde (Botelho et al. 2015), which have been reported in Brazil and other countries such as India. Such adulteration creates great concern for the entire production chain.
Owing to the sophisticated adulteration practices for milk that have been adopted by the industry, producers, and transporters, it is necessary to develop technologies for identifying such fraud. However, the current methods employed for this purpose generally have low analytical performance, are highly dependent on manual labor, and require the use of reagents (Harding 1995). These characteristics hinder the implementation of large-scale monitoring programs, which hampers the analysis of all the adulterants in milk routinely (Cassoli 2010). Another barrier is the identification of adulteration as soon as it is developed because, historically, adulteration must occur so that methodologies can be developed to identify it (Cassoli 2010).
Because adulteration practices have become increasingly refined, analytical methods must also become increasingly sophisticated. Some examples of such methods include the use of Fourier transform infrared spectroscopy (FTIR) (Nicolaou et al. 2010; Cassoli et al. 2011; Jawaid et al. 2013) and other spectroscopic and chromatographic techniques (Bogialli et al. 2004; Sassine et al. 2004; Andersen et al. 2005; Chen et al. 2005; Garcia et al. 2012), as well as milk scanning techniques to identify adulteration (Santos et al. 2012).
The FTIR technique has been gaining interest for raw milk quality control, especially because of its high level of analytical capacity, low sample manipulation and use of fewer reagents, resulting in less time, lower costs, and a higher number of samples that can be analyzed in the same time interval (Rodriguez-Saona and Allendorf 2011).
The basic assumption behind the application of spectroscopic techniques is based on the generation of a food “fingerprint” (Karoui and De Baerdemaeker 2007). A dairy product with a certain chemical composition that is exposed to a light source has a characteristic spectrum that results from the absorption of various chemical components (Karoui and De Baerdemaeker 2007). Because the exact composition of any natural material changes depending on the variety, season, location, and other characteristics, it is necessary to have a set of representative spectra or “standards” that the test material can be compared with to establish its quality or authenticity (Karoui and De Baerdemaeker 2007).
Different FTIR instruments are available on the market currently that range from compact (used for quality control during the reception of milk by the industry) to high performance (used by central laboratories with greater analytical capacities of up to 600 samples/h). Compact equipment provides the industry with the possibility of including FTIR in its raw material quality control programs, or even replacing traditional (chemical) methods that are recommended by inspectors.
Thus, in an attempt to fulfill this need to develop rapid, effective, and low-cost analytical methods for identifying adulterated milk, this work aims to calibrate and validate a compact FTIR instrument (MilkoScan FT1, Foss Analytical) for monitoring raw milk adulteration. The hypothesis is that the FTIR equipment, which is being increasingly used by industries to evaluate the composition of milk, can identify the main adulterants in raw milk.
The results of this study can be applied directly to the detection of adulterants and will thus help in protecting the public health by improving the quality of marketed milk.
Materials and methods
Local development
The analyzed raw milk samples were obtained from a dairy located in the middle region of Minas Gerais, Brazil, that processes approximately 800,000 L of milk per day. The industry uses the MilkoScan FT1 equipment for quality control of the milk to analyze its fat, protein, casein, total solids, urea nitrogen, freezing point, pH, etc.
Reference spectrum construction
For the reference spectrum construction, 2497 samples of raw milk were collected over 6 months (from the beginning of August 2014 to the end of January 2015, to represent variations in milk composition at different periods of the year) from carrier trucks that arrived daily at the industry. Milk samples were collected from each carrier truck over the tank lid in 50 mL sterile vials using appropriate tools. All the samples were readily analyzed. These samples represent the milk of every truck that arrived at the industry during that 6-month period.
The analyses were performed with the FTIR MilkoScan FT1 equipment. The equipment has the ability to scan the entire middle infrared region with wavelengths between 2.0 and 10.8 μm (5012–926 cm−1), and the spectra were exported and stored in electronic files (Foss Analytical 2011b). The equalization of the equipment was performed monthly as recommended by the manufacturer (Foss Analytical 2011a) to ensure spectral acquisition standardization during the experimental period (Hansen 1998).
Prior calibration of the device, for milk composition characterization (levels of fat, protein, casein, total solids, urea nitrogen, freezing point, pH, etc.) was evaluated using standard samples (VALACTA®) with the FOSS Integrator software, which automatically calculates all of the calibration parameters [standard error (SE), standard error calibration (SEC), and determination coefficient (R2)] and includes the calibration feature “slope intercept” to perform all the necessary calculations to optimize the equipment performance.
Calibration development
The spectral database was created using principal component analysis (or factor analysis) with the Abnormal Screening Module (ASM) software (Foss Analytical 2011b). One of the purposes of principal component analysis (PCA) is to reduce the amount of data, i.e., to adjust the information from the original data set and replace it with a smaller set of latent variables (Dunn et al. 1989). In this type of analysis, the first principal components (PCs) contain almost all the useful information for the dataset, whereas the remaining PCs mainly contain noise (Dunn et al. 1989). Therefore, a large number of components should be chosen to better separate the useful information from the noise (Dunn et al. 1989).
Hence, 12 calibrations were developed for the reference spectrum and validation samples. Different numbers of factors (or principal components) in the statistical model were considered: 8, 10, 12, 14, 16, or 18 factors with one or two outlier eliminations. During the first elimination of outliers, sample spectra with large differences compared with the average spectral values were discarded. Next, the calibration curve was adjusted and used for the calculation of the “scores.” During the second elimination of outliers, after first excluding the outliers, the calibration curve was adjusted, new differing spectra were then excluded, and the calibration curve was again adjusted and used to calculate newer “scores.” All calibrations were carried out using the middle infrared region, with wavelengths between 2.0 and 10.8 μm (5012–926 cm−1).
The “score” calculation was based on the “spectral distance” (Euclidean distance) between the test and the reference samples. This distance is large or small for samples that resemble or differ from the reference (Foss Analytical 2011b).
The follow references were adopted to facilitate the understanding of the results: (1) calibration 1: 8 factors with a single elimination of outliers; (2) calibration 2: 8 factors with two eliminations of outliers; (3) calibration 3: 10 factors with a single elimination of outliers; (4) calibration 4: 10 factors with two eliminations of outliers; (5) calibration 5: 12 factors with a single elimination of outliers; (6) calibration 6: 12 factors with two eliminations of outliers; (7) calibration 7: 14 factors with a single elimination of outliers; (8) calibration 8: 14 factors with two eliminations of outliers; (9) calibration 9: 16 factors with a single elimination of outliers; (10) calibration 10: 16 factors with two eliminations of outliers; (11) calibration 11: 18 factors with a single elimination of outliers; and (12) calibration 12: 18 factors with two eliminations of outliers.
Sample preparation for validation
The milk used in this step was collected randomly from 25 carrier trucks and stored under refrigeration (<10 °C) until use. The milk collected from each truck (65 L per collection day) was subsequently divided into 1-L samples to receive adulterants at different concentrations.
Three or six percent water was added to each 1 L milk sample, and the following compounds were added: (1) cornstarch (at 500, 750, and 1000 mg L−1); (2) sodium bicarbonate (at 600, 1200, and 1800 mg L−1); (3) sodium citrate (at 500, 750, and 1000 mg L−1); (4) formaldehyde (at 150, 300, and 450 mg L−1); and (5) saccharose (at 250, 500, and 750 mg L−1). In total, there were 750 samples of adulterated milk containing water, and these samples were analyzed 25 times.
To evaluate adulteration with whey, 3 or 6% whey was added to each 1 L sample of milk, and the same compounds at the concentrations listed above were also added. In total, there were, therefore, another 750 samples of adulterated milk with whey, and these samples were also analyzed 25 times.
The control samples were raw milk, raw milk with water (3 or 6%), and raw milk with whey (3 or 6%), and these were also analyzed 25 times each. Therefore, 1625 samples were analyzed, including the following: 1500 samples of raw milk adulterated with water or whey (3 or 6%) with the addition of five adulterants (at three concentrations each), 25 control samples of raw milk, and 25 samples adulterated with only water or whey (3 or 6%).
Milk characterization and validation
The analyses were performed according to the procedures recommended by the MilkoScan FT1 manufacturer (Foss Analytical 2011a). The levels of fat, protein, casein, non-fat solids, acidity, and density were measured to characterize the milk composition. The “scores” were calculated according to the data obtained with the 12 calibrations developed in the previous step (“Calibration development” section), using wavelengths between 2.0 and 10.8 μm (5012–926 cm−1). Samples with scores greater than three were considered adulterated.
To evaluate the accuracy of each calibration for the detection of adulterated milk, the sensitivity (ability to correctly identify adulterated samples) and specificity (ability to correctly identify unadulterated samples) were calculated (Erdreich and Lee 1981).
The Kruskal–Wallis test was used for the detection of each adulterant using the sensitivity from each calibration (which involved 8, 10, 12, 14, 16, or 18 factors with one or two eliminations of outliers). This test evaluates the position of the averages with a ranking organization and provides a classification order from the best to the worst average (lower average values are considered better). After determining the ranking, the Dunn test was used to make multiple comparisons between the results.
To define the calibration with the best performance, the calibrations providing results with the highest specificity were identified. Then, those providing results with the best sensitivity were selected from this group.
All the estimates were calculated using R software (version 3.2.4; R Foundation, Vienna, Austria) and a statistical significance level of 0.05.
Results and discussion
Reference spectrum construction
Milk composition is affected by a variety of factors including season, lactation stage, type of feed given to the animals, physiological state, milking interval, and genetic factors, among other aspects (Heck et al. 2009). Thus, milk samples from different regions and feed systems are also subject to different seasonal effects that influence the main components of raw milk (Chen et al. 2014). Consequently, samples must represent this variability in the construction of the reference spectrum (Soyeurt et al. 2009).
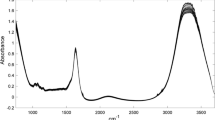
Therefore, the reference spectrum (Fig. 1) was constructed from the analysis of milk samples collected over 6 months (from August to January, a period which includes the end of the dry season, the transitional period, and the beginning of the rainy season in Brazil) to incorporate seasonal variation. The averages (after the removal of outliers) of the levels of fat, protein, casein, non-fat solids, and acidity (measured as grams of lactic acid per deciliter of milk) and density during these 6 months of data collection are illustrated in Table 1.

Representative FTIR spectrum for the raw milk samples used in the reference spectrum construction
Calibration development
After developing the 12 calibrations for the reference spectrum and validation samples, which considered 8, 10, 12, 14, 16, or 18 factors with one or two outlier eliminations in the statistical model, the results obtained for the calibration using 10 factors (principal components) could explain more than 90% of the spectral variation of the samples used in the reference spectrum construction. According to Heuer et al. (2001) and Karoui and De Baerdemaeker (2007), it is necessary to choose factors that represent approximately 90% of the spectral sample variability to construct a calibration model.
Some studies have obtained similar numbers of factors in their calibrations. Cassoli et al. (2011) found that nine factors were sufficient to explain 90% of the spectral variation of the reference sample. Santos et al. (2013a) developed models using six and seven factors that explained more than 97% of the variance in multispectral data, whereas Etzion et al. (2004) obtained better results in determining the concentrations of proteins in raw milk using nine factors in a partial least squares (PLS) model.
Validation
Because milk adulteration usually occurs due to the addition of substances at low concentrations (Cassoli et al. 2011), the validation-step calibrations were developed using a number of factors that varied from 8 to 18 as described above.
The factors used in this study were calculated using calibration models that were automatically selected using the “auto-select optimal” tool of the ASM software (Foss Analytical 2011b). The selection of factors was based on the predicted residual error of sum of squares (PRESS) value, which indicates how well the model fits the calibration data (Jawaid et al. 2013).
The results described in Table 2 reveal that among the 12 compared calibrations, calibration 12 (which included 18 factors with two eliminations of outliers) presented the best classification based on the Kruskal–Wallis test.
Calibration 12 had the lowest value in the ranking but did not differ (p > 0.05) from calibrations 10 and 8 when compared, using the Dunn test (Table 3).
The use of the calibration that includes the maximum number of factors is not recommended when there are other calibrations that are not significantly different. In such cases, a calibration with a smaller number of factors should be chosen (Tobias 1995). As the number of factors used increases, the proportion of components that explain little sample variation increases, more noise is introduced into the calibration, and the small components cause collinearity problems (Geladi and Kowalski 1986). According to these criteria, calibrations 8 and 10 could be chosen because they best identified adulterations in milk, were not significantly different from calibration 12, and involved fewer numbers of factors.
To better evaluate the calibrations obtained, the sensitivity and specificity of each calibration have been plotted (Fig. 2). However, examination of these data clearly reveal that calibrations 8 and 10 do not exhibit 100% specificity; in other words, based on the control samples (those without any type of adulteration), these calibrations resulted in some false positives.

Sensitivities and specificities of the tested calibrations. Sensitivity, which was measured for the adulterated samples, and specificity, which was measured for the controls (unadulterated samples), are presented as % values for the 12 evaluated calibrations. Calibration 7 has been chosen as optimal for identifying adulterations in milk
Thus, if the choice of the calibrations that better identify adulterations in milk is based on those that exhibit the maximum specificities, in other words, no false positive values (calibrations 3, 5, 6, 7, 9, or 11), then calibration 7 (14 factors and one removal of outliers), although it has not been classified by the Kruskal–Wallis test as one of the best calibrations, should be chosen because, as shown in Table 3, this calibration does not statistically differ (p > 0.05) from calibrations 8 and 10 (previously ranked better at identifying adulterants in raw milk) and includes fewer factors.
The numbers of factors used to identify adulterants and contaminants in milk by infrared spectroscopy vary widely according to the compounds studied. Kasemsumran et al. (2007) used near-infrared spectroscopy and determined that the best calibration model for milk adulteration with whey used four factors in a PLS model. Sivakesava and Irudayaraj (2002) defined 15 as the optimal number of factors using the PLS method to identify tetracycline in milk. Heuer et al. (2001) obtained a predictive model that consisted of 19 principal components (factors) for the determination of acetone in cow milk. These results highlight the need for proper equipment calibration, especially when it is used to detect compounds that do not naturally belong in milk.
Regarding the sensitivity analyses for calibration 7 for all the adulterants added at different water or whey concentrations (Table 4), the data reveal that calibration 7 can define all the adulterated samples with greater sensitivity, regardless of the concentration and type of adulterant. For example, the sensitivities were 100% after the addition of sodium bicarbonate at all the tested concentrations of this adulterant and after the addition of water or whey.
Using calibration 7, the addition of cornstarch and sodium citrate resulted in sensitivities lower than 100% at the lowest concentrations of these adulterants (500 mg L−1). In contrast, the addition of formaldehyde resulted in lower sensitivity when it was added at 150 or 300 mg L−1 together with whey (3 and 6%). This characteristic demonstrates the capacity of whey to mask the addition of formaldehyde. The sensitivity was close to 100% at the other adulterant concentrations.
Botelho et al. (2015) found sensitivities of 83.3, 100, and 93.8% to the addition of 0.5–10% w/v (5–10 g L−1) of cornstarch, sodium citrate, and formaldehyde, respectively, based on a screening method that used attenuated total reflectance (ATR) spectroscopy in the mid-infrared range and multivariate classifications. Cassoli et al. (2011) found maximum sensitivity (98%) in the detection of 0.075% (750 mg L−1) sodium citrate in raw milk. Compared to the results reported by Cassoli et al. (2011), the present work produced better sensitivity values for this and other sodium citrate concentrations.
At one of the concentrations used in this study, saccharose (up to 750 mg L−1) resulted in the lowest detection rate, particularly when whey was added to the samples. However, these concentrations were very small, which may explain the low identification rate of this adulterant. He et al. (2010) were able to identify glucose adulterations in raw milk at 1000 mg dL−1 using infrared spectroscopy combined with two-dimensional correlation analysis. This concentration is much higher than that used in the present study. Liu et al. (2015) were able to identify 240 mg of saccharose adulterant per liter of raw milk using infrared spectroscopy combined with multivariate chemometric techniques. However, these studies did not investigate any other adulterants, such as water or whey, capable of masking the addition of saccharose.
To measure the effect of added water or whey per se, the sensitivities of all 12 calibrations were calculated following the addition of water or whey. When water was added at 6%, the sensitivity was 100% for all the tested calibrations, except for calibration 11, for which the sensitivity was 95%. This result implies that regardless of the adulterants used, the developed calibration could detect water at the tested concentration. However, the sensitivities of the calibration for the diluents (3% water or whey) were low, particularly when whey was added. However, even with these low sensitivities, the calibrations were able to detect the adulterants and not just the diluents.
The sensitivities of the different methods used to identify water or whey in raw milk are highly variable and depend on the procedure used. Santos et al. (2012) were able to identify 7% water adulteration in raw milk using the image scanning method. Liu et al. (2015) detected the addition of 1% water and 0.5% of whey with infrared spectroscopy combined with multivariate chemometric techniques. Santos et al. (2013b) and Santos et al. (2013a) identified whey adulterations in milk at concentrations greater than 7.5 g L−1 using soft independent modeling by class analogy (SIMCA) classification models and mid-infrared spectroscopy. Cassoli et al. (2011) observed low sensitivity even when high concentrations (20%) of whey were added to raw milk using an FTIR method. Das et al. (2011) identified adulterations in milk with 5% whey and 10% water with a novel method using an impedance sensor. In contrast, Motta et al. (2014) obtained 1 µg mL−1 as the detection limit for whey in raw milk via proteomic analysis and liquid chromatography; however, this technique requires more extensive sample preparation and a longer analysis time compared with infrared spectroscopy. These results suggest that milk adulteration with whey is difficult to identify probably owing to the chemical similarity of whey and milk and because whey is a by-product obtained during the cheese manufacturing process.
In this study, 25 samples of raw milk combined with various contaminants were examined. The contaminants included water or whey plus cornstarch, sodium bicarbonate, sodium citrate, saccharose, and formaldehyde (at three different concentrations) in addition to the control samples. Approximately 2500 samples were used to construct the reference spectrum, which served as a standard for the comparison of the test sample spectra (adulterated). Using sensitivity and specificity analyses based on the evaluation of the milk with the developed calibration as well as with the ranking from the Kruskal–Wallis test and a Dunn multiple comparison test, the results revealed that the developed calibration expressed a good overall capacity for the detection of adulterations of raw milk. The calibration that best identifies the adulterants (calibration 7) exhibits 84% sensitivity (or 16% false negative rate) and 100% specificity (0% false positive rate), and does not differ statistically from calibration 10, which exhibits 89% sensitivity (Fig. 2).
Conclusion
Based on the results obtained in this study, it can be concluded that the equipment that employs the FTIR methodology, after calibrations, displayed excellent sensitivity and specificity. Thus, this approach is a viable option for the identification of adulterants of raw milk.
As a rapid methodology that does not require reagents or sample preparation, FTIR is an optimal alternative for detecting adulterations, particularly because the equipment is already in use in the most industry to determine milk composition. Thus, this methodology has the potential to contribute to protect public health and improve the quality of marketed milk.
References
Andersen WC, Roybal JE, Gonzales SA et al (2005) Determination of tetracycline residues in shrimp and whole milk using liquid chromatography with ultraviolet detection and residue confirmation by mass spectrometry. Anal Chim Acta 529:145–150. doi:10.1016/j.aca.2004.08.012
Bogialli S, Capitolino V, Curini R et al (2004) Simple and rapid liquid chromatography-tandem mass spectrometry confirmatory assay for determining amoxicillin and ampicillin in bovine tissues and milk. J Agric Food Chem 52:3286–3291. doi:10.1021/jf0499572
Botelho BG, Reis N, Oliveira LS, Sena MM (2015) Development and analytical validation of a screening method for simultaneous detection of five adulterants in raw milk using mid-infrared spectroscopy and PLS-DA. Food Chem 181:31–37. doi:10.1016/j.foodchem.2015.02.077
Cassoli LD (2010) Validação da metodologia de infravermelho com transformada de Fourier para identificação de adulteração em leite cru. Ph.D. Thesis, Escola Superior de Agricultura “Luiz de Queiroz”. University of São Paulo, Piracicaba. https://goo.gl/l8aZmo
Cassoli LD, Sartori B, Zampar A, Machado PF (2011) An assessment of Fourier transform infrared spectroscopy to identify adulterated raw milk in Brazil. Int J Dairy Technol 64:480–485. doi:10.1111/j.1471-0307.2011.00711.x
Chen C-Y, Li W-J, Peng K-Y (2005) Determination of Aflatoxin M 1 in milk and milk powder using high-flow solid-phase extraction and liquid chromatography: tandem mass spectrometry. J Agric Food Chem 53:8474–8480. doi:10.1021/jf052142o
Chen B, Lewis MJ, Grandison AS (2014) Effect of seasonal variation on the composition and properties of raw milk destined for processing in the UK. Food Chem 158:216–223. doi:10.1016/j.foodchem.2014.02.118
Das S, Sivaramakrishna M, Biswas K, Goswami B (2011) Performance study of a “constant phase angle based” impedance sensor to detect milk adulteration. Sens Actuators A Phys 167:273–278. doi:10.1016/j.sna.2011.02.041
Dunn WJ, Scott DR, Glen WG (1989) Principal components analysis and partial least squares regression. Tetrahedron Comput Methodol 2:349–376. doi:10.1016/0898-5529(89)90004-3
Erdreich LS, Lee ET (1981) Use of relative operating characteristic analysis in epidemiology. A method for dealing with subjective judgement. Am J Epidemiol 114:649–662
Etzion Y, Linker R, Cogan U, Shmulevich I (2004) Determination of protein concentration in raw milk by mid-infrared Fourier transform infrared/attenuated total reflectance spectroscopy. J Dairy Sci 87:2779–2788. doi:10.3168/jds.S0022-0302(04)73405-0
Foss Analytical (2011a) MilkoScan FT1: user manual (Rev.1). FOSS Analytical A/S, Hillerod, Denmark, pp 1–86
Foss Analytical (2011b) MilkoScan FT1 software manual (Rev.3). FOSS Analytical A/S, Hillerod, Denmark, pp 1–154
Garcia JS, Sanvido GB, Saraiva SA et al (2012) Bovine milk powder adulteration with vegetable oils or fats revealed by MALDI-QTOF MS. Food Chem 131:722–726. doi:10.1016/j.foodchem.2011.09.062
Geladi P, Kowalski BR (1986) Partial least-squares regression: a tutorial. Anal Chim Acta 185:1–17. doi:10.1016/0003-2670(86)80028-9
Hansen PW (1998) Urea determination in milk using Fourier transform infrared spectroscopy and multivariate calibration. Milchwissenschaft 53:251–255
Harding F (ed) (1995) Milk quality (Chapter 6). Blackie Academic & Professional, London
He B, Liu R, Yang R, Xu K (2010) Adulteration detection in milk using infrared spectroscopy combined with two-dimensional correlation analysis. Library (Lond). doi:10.1117/12.841580
Heck JML, van Valenberg HJF, Dijkstra J, van Hooijdonk ACM (2009) Seasonal variation in the Dutch bovine raw milk composition. J Dairy Sci 92:4745–4755. doi:10.3168/jds.2009-2146
Heuer C, Luinge HJ, Lutz ET et al (2001) Determination of acetone in cow milk by Fourier transform infrared spectroscopy for the detection of subclinical ketosis. J Dairy Sci 84:575–582. doi:10.3168/jds.S0022-0302(01)74510-9
Jawaid S, Talpur FN, Sherazi STH et al (2013) Rapid detection of melamine adulteration in dairy milk by SB-ATR-Fourier transform infrared spectroscopy. Food Chem 141:3066–3071. doi:10.1016/j.foodchem.2013.05.106
Kamal M, Karoui R (2015) Analytical methods coupled with chemometric tools for determining the authenticity and detecting the adulteration of dairy products: a review. Trends Food Sci Technol 46:1–22. doi:10.1016/j.tifs.2015.07.007
Karoui R, De Baerdemaeker J (2007) A review of the analytical methods coupled with chemometric tools for the determination of the quality and identity of dairy products. Food Chem 102:621–640. doi:10.1016/j.foodchem.2006.05.042
Kasemsumran S, Thanapase W, Kiatsoonthon A (2007) Feasibility of near-infrared spectroscopy to detect and to quantify adulterants in cow milk. Anal Sci 23:907–910. doi:10.2116/analsci.23.907
Lim J, Kim G, Mo C et al (2016) Detection of melamine in milk powders using near-infrared hyperspectral imaging combined with regression coefficient of partial least square regression model. Talanta 151:183–191. doi:10.1016/j.talanta.2016.01.035
Liu J, Ren J, Liu ZM, Guo BH (2015) A new comprehensive index for discriminating adulteration in bovine raw milk. Food Chem 172:251–256. doi:10.1016/j.foodchem.2014.09.063
Motta TMC, Hoff RB, Barreto F et al (2014) Detection and confirmation of milk adulteration with cheese whey using proteomic-like sample preparation and liquid chromatography-electrospray-tandem mass spectrometry analysis. Talanta 120:498–505. doi:10.1016/j.talanta.2013.11.093
Nicolaou N, Xu Y, Goodacre R (2010) Fourier transform infrared spectroscopy and multivariate analysis for the detection and quantification of different milk species. J Dairy Sci 93:5651–5660. doi:10.3168/jds.2010-3619
Rodriguez-Saona LE, Allendorf ME (2011) Use of FTIR for rapid authentication and detection of adulteration of food. Annu Rev Food Sci Technol 2:467–483. doi:10.1146/annurev-food-022510-133750
Santos PM, Wentzell PD, Pereira-Filho ER (2012) Scanner digital images combined with color parameters: a case study to detect adulterations in liquid cow’s milk. Food Anal Methods 5:89–95. doi:10.1007/s12161-011-9216-2
Santos PM, Pereira-Filho ER, Rodriguez-Saona LE (2013a) Rapid detection and quantification of milk adulteration using infrared microspectroscopy and chemometrics analysis. Food Chem 138:19–24. doi:10.1016/j.foodchem.2012.10.024
Santos PM, Pereira-Filho ER, Rodriguez-Saona LE (2013b) Application of hand-held and portable infrared spectrometers in bovine milk analysis. J Agric Food Chem 61:1205–1211. doi:10.1021/jf303814g
Sassine A, Moura S, Léo VM, Bustillos OV (2004) Cypermethrin residues determination in the milk of a lactating dairy cow by gas chromatography-ion trap mass spectrometry. J Anal Toxicol 28:238–241
Sivakesava S, Irudayaraj J (2002) Rapid determination of tetracycline in milk by FT-MIR and FT-NIR spectroscopy. J Dairy Sci 85:487–493. doi:10.3168/jds.S0022-0302(02)74099-X
Soyeurt H, Bruwier D, Romnee J-M et al (2009) Potential estimation of major mineral contents in cow milk using mid-infrared spectrometry. J Dairy Sci 92:2444–2454. doi:10.3168/jds.2008-1734
Tobias RD (1995) An introduction to partial least squares regression. Proceedings of the annual SAS users group international conference, 20th, Orlando, FL 2–5. http://support.sas.com/techsup/technote/ts509.pdf
Acknowledgments
The authors would like to thank the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) funding agencies.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing financial interests.
Rights and permissions
About this article

Cite this article
Coitinho, T.B., Cassoli, L.D., Cerqueira, P.H.R. et al. Adulteration identification in raw milk using Fourier transform infrared spectroscopy. J Food Sci Technol 54, 2394–2402 (2017). https://doi.org/10.1007/s13197-017-2680-y
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13197-017-2680-y