
Abstract
In the present study, near-infrared spectroscopy (NIRS) was explored as a fast and reliable screening method for the detection of adulteration of skim milk powder (SMP). Sixty genuine SMP were adulterated with acid whey (1–25 % w/w), starch (2 and 5 %) and maltodextrin (2 and 5 %) for a total of 348 adulterated samples. Two chemometric approaches were employed. In the first approach, an untargeted one class model for genuine skim milk powder was developed by Soft Independent Modelling of Class Analogy. In the second approach, adulterant-specific regression models were developed to assess the amount of each adulterant by partial least square regression and principal component regression. The class modelling approach had the advantage that several adulterants could be detected with the same chemometric model, including situations where multiple adulterants are present in the test sample or where yet unknown adulterants are present. Regression models showed a better sensitivity with genuine SMP samples completely discriminated from samples adulterated with 5 % acid whey and 2 % of starch or maltodextrin. NIRS proved to be a useful tool for the rapid and cost-efficient untargeted and/or targeted detection of adulterations in SMP.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Milk powder is a dairy product which is manufactured by evaporating milk to dryness (residual moisture content ≈5 %). Milk powder retains most of the desired nutritional and organoleptic properties (colour, flavour, solubility) of fluid milk. Drying milk has the obvious advantages of reducing transport and storage costs and to extend milk shelf-life. Milk powders can be used for direct human consumption or as ingredients in a wide range of food products including baked goods, confectionery, dairy desserts, ice cream and infant formula.
Recently, the phenomenon of adulteration of milk powder has increased worldwide, favoured by the lack of strong deterrence in national and international regulations and the difficulties of carrying out multiple control tests to unveil several potential adulterations. Milk powder adulteration can compromise its nutritional quality, its safety (because of the potential adulteration with toxic compounds, e.g. melamine) and results in unfair competition towards legitimate businesses. The most frequent adulteration consists in the addition of whey which is a very cheap by-product of cheese manufacturing. Liquid whey can be of two different types: (i) sweet whey (also known as cheese whey) i.e. the by-product from the manufacturing of rennet types of hard cheese and (ii) acid whey (also known as sour whey) i.e. the by-product from the manufacturing of acid types of cheese. Sweet and acid whey has very similar composition, acid whey being slightly richer in proteins and more acidic than sweet whey. Other common contaminants in milk and milk powders are starch, sucrose, urea and other nitrogen replacers including melamine and, less frequently, maltodextrin (MD) and sodium hydroxide.
The detection of fraudulent addition of sweet whey to milk or milk powder is easily carried out by checking the presence or absence of glycomacropeptide also known as caseinomacropeptide which is a bioactive 64-amino acid residue glycopeptide released enzymatically in whey from k-casein by chymosin during cheese making. It is thus present in the sweet whey but not in milk (Chavez et al. 2008). Unfortunately, it is absent in acid whey either. For the detection of acid whey adulteration in dairy products, methods based on the whey protein/total protein ratio are preferentially used. The whey protein/total protein ratio can be measured colorimetrically (De Koning and Van Rooijen 1971), by polarimetry (Lechner and Klostermeyer 1981), by SDS-PAGE (Basch et al. 1985), by capillary electrophoresis (Kanning et al. 1993) or by spectroscopy. These last methods are based on the zero- or first-order derivative (De Block et al. 1997; Cartuyvels et al. 1999) or on fourth-order derivative (Meisel 1995; Miralles et al. 2000) and rely on the different derivative spectroscopy responses of the amino acids tryptophan and tyrosine and on the fact that the tryptophan/tyrosine ratio is about 0.59 for whey proteins and 0.19 for caseins. However, the natural variability in whey/total protein ratio in milk and corresponding powder results in a substantially high limit of detection for whey in skim milk powder (SMP). Lüthi-Peng and Puhan (1999) reported a range for the whey/total protein ratio of 12.7–21.0 % for raw cow milk. Miralles et al. (2000) reported that the addition of acid and rennet whey to UHT milk results in a significant difference in whey/total protein ratio only upon addition between 2.5 and 5 %.
On the other hand, for the detection of starch in milk and dairy products, the iodine test (Banks et al. 1971) is the method of choice because of its incomparable sensitivity (0.01 % w/w) and the simplicity of the analysis. In this test, iodine dissolved in an aqueous solution of potassium iodide quantitatively reacts with the amylose-coiled chain of starch and the amount of starch is quantified by potentiometric or amperometric titration or by visible spectroscopy (amylose-iodine complex gives a purple black colour).
Detection of MD with the iodine test may give false-negative results if the glucose chain is too short to accommodate the I3− ions. Detection of MD in milk-based powder therefore requires time-consuming chromatographic technique such as HPAEC–PAD (Rocklin and Pohl 1983) even though a relatively fast screening method for the detection of MD in milk powder and infant formula based on electrospray ionization–mass spectrometry analysis and selective enzymatic hydrolysis has been reported (Sanvido et al. 2010).
Near-infrared spectroscopy (NIRS) is a fast and high-throughput analytical methodology which has had countless applications for food authentication and detection of adulterations in recent years as reviewed, for instance, by Rodriguez-Saona and Allendorf (2011) and Cozzolino (2012). NIRS radiation (800–2500 nm) triggers transitions over two (first overtone), three (second overtone) or higher vibrational energy levels of the sample molecules (Osborne 2000; Barton 2002). Each functional group of a molecule shows characteristic NIRS absorption at specific wavelengths. NIRS spectra thus represent a molecular fingerprint of the test material that can be used to verify the nature of the sample either quantitatively, i.e. to detect and quantify the amount of a target adulterant, or qualitatively, i.e. by discriminating and predicting the real nature of an unknown sample (e.g. organic food). Remarkable advantages of NIRS over the traditional wet analytical techniques are that NIRS is incomparably faster and more high-throughput. This is even more so if one considers that several parameters (not only chemical composition but also physical properties and quality attributes) can be estimated simultaneously from the same NIRS spectra and the information contained in an NIRS spectrum can be stored for future use. Another remarkable advantage of NIRS is that it is a non-destructive analysis, which makes it suited for the in-line quality control. NIRS is also environmentally friendly because it does not require the use of solvents and chemicals and does not produce any waste. Finally, the routine use of NIRS instruments from the sample preparation for analysis (where needed) to the spectrum acquisition is extremely simple and requires only minimally trained personnel.
NIRS applications for the authentication of milk powders have been reported for the detection of sweet whey, starch and sucrose (Borin et al. 2006), foreign proteins (Maraboli et al. 2002), melamine (Balabin and Smirnov 2011; Lu et al. 2009) and whey concentrate powder (Mendenhall and Brown 1991).
The aim of this study was to evaluate the performance of NIRS for the detection of acid whey, starch and maltodextrin adulteration in SMP.
Materials and Methods
Skim Milk Powders and Adulterants
Sixty SMP were obtained in the framework of regular control schemes carried out by the Dutch Food Safety Authority (NVWA, The Netherlands). The two samples of acid whey powders (AWP) were obtained from Denkavit Nederland BV (Voorthuizen, The Netherlands) and Arion Dairy Products BV (Groesbeek, The Netherlands). Corn starch (Chemical Abstracts Service (CAS) number 9005-25-8) and maltodextrin (MD, dextrose equivalent 16.5–19.5, CAS number 9050-36-6) were purchased from Sigma-Aldrich (St. Louis, MO).
Design of the Adulteration
The whole lot of 60 SMP was divided in two sub-lots. For the adulteration with acid whey, a full factorial design was applied on a sub-lot of 27 SMP which were adulterated with four levels of each of the two AWP for a total of 243 samples (27 genuine and 216 adulterated SMP). The adulteration levels that were selected were the following: 1, 5, 15 and 25 % (w/w). For the adulteration with starch and MD, a full factorial design was applied on the 33 SMP of the second sub-lot at two different adulteration levels: 2 and 5 % (w/w) for a total number of 66 samples adulterated with starch and 66 samples adulterated with MD. AWP, starch and MD powders were thoroughly mixed with the SMP for at least 1 h using a rotator (Reax 2, Heidolph, Germany). Since the amount of each SMP sample collected was insufficient for an independent spiking of each adulterant concentration, the adulterated samples were prepared with serial spiking: the lowest level of adulteration was prepared and measured by NIRS. After each NIRS measurement, the proper amount of the adulterant was added to meet the second lowest concentration and so forth.
NIRS Measurements
NIRS spectra were recorded on genuine SMP and the adulterated SMP by means of a FOSS NIRSystems 6500 SY-I system (Foss Electric, Hillerød, Denmark) equipped with a spinning module over the VIS/NIRS range (400∼2498 nm) with a 2-nm sampling interval under the reflectance mode. Measurements were taken on 1.0−1.5-cm-thick (about 5 g) portion, evenly distributed into standard ring cups (diameter of 3.75 cm). All the measurements were performed in duplicate on two independent samples of approximately 5 g for each SMP or mixture, and the final spectra that were used for chemometric modelling were the average of those two replicates.
Chemometric Modelling
Principal component analysis (PCA) was performed to check any natural clustering in the genuine SMP and detect the presence of outliers. Soft Independent Modelling of Class Analogy (SIMCA) was used to develop a one class model for genuine SMP. Partial least square regression (PLSR) and principal components regression (PCR) were performed to construct regression models for the prediction of the concentration of each adulterant in SMP.
For each regression model, a theoretical critical signal height (CSH) for type I errors and for single future predictions was calculated based on the approach described in Vogelgesang and Hadrich (1998). This CSH will correspond to the upper limit of the prediction interval (at a fixed level of confidence, α) at zero concentration (genuine SMP). In practice, that means that there is less than 100× α % chance that each single future prediction on genuine SMP will be greater than the calculated CSH. This CSH can be calculated according to the following formula:
where AVG is the average of predicted % adulterant in genuine SMP (corresponding to the intercept of the calibration equation), t (1 − α;n − 1) is the t-value for a one-sided (1 − α)×100 % confidence interval and n − 2 degrees of freedom, s X is the residual standard deviation, \( \overline{x} \) is the mean of all the adulteration levels of all the calibration analyses and n the number of samples. The normality of the residual distribution for the genuine and adulterated SMP was checked by means of a Shapiro–Wilk test for normality (p = 0.623, n = 179). In practice, if the level of adulteration predicted by the NIRS model in a new test sample is greater than the value calculated by Eq. 1 for a certain level of confidence 1 − α, there is less than α% to be wrong in classifying the test sample as adulterated.
The limit of detection (minimum amount of adulterant that can be predicted with a fixed level of certainty) is calculated from the regression equation developed on the calibration set for y = CSH. Once the CSH is calculated for a desired level of significance for type I error, the identification limit (ID, the minimum concentration of adulterant for which the level of significance for a type II error is less than of a fixed value) can be derived as the % adulteration which lower limit of the prediction interval corresponds to CSH. Such ID limit can be calculated at the desired level of significance for type II error from the following equation:
and solving for ID, where t (1 − β; n − 2) is the t-value for a one-sided (1 − β)×100 % confidence interval, n − 2 are the degrees of freedom, s X is the residual standard deviation and n the number of samples.
For the development of SIMCA, PLSR and PCR models, six transforms or combinations of transforms were applied: (i) standard normal variate (SNV), (ii) first derivative, (iii) second derivative, (iv) SNV + first derivative, (v) SNV + second derivative, (vi) multiplicative scatter correction (MSC), (vii) MSC + first derivative and (viii) MSC + second derivative. The first and the second derivatives were calculated using the Savitzky–Golay algorithm (Savitzky and Golay 1964) modified as suggested by Gorry (1990) with 21 points of windows and a second-order polynomial function. SNV is an approach to compensate for scattering by solids in NIR spectrometry. It consists in a row autoscaling: for each sample, the sample mean is subtracted from each variable value and the obtained values are dived by the sample standard deviation. MSC is another standard approach to compensate for scattering by solids in NIR spectrometry. Each sample spectrum is regressed linearly against an ideal spectrum to yield a slope and intercept which are then used to “correct” the sample spectrum at each wavelength. The transformed data were then mean-centred, and the algorithms were run. PCA, SIMCA, PLSR and PCR models were performed in Pirouette 4.5 (Infometrix, Seattle, USA). Only the data in the range 1100–2500 was used for the chemometric modelling.
Results and Discussion
Development of the Predictive Models
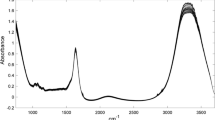
In Fig. 1, the normalized NIRS spectra for one SMP sample and the two AWP as well as starch and MD used in the present study are shown. The acid whey spectra resemble the spectrum for lactose reported for instance by Ni et al., (2010) but do not exhibit clear absorption peaks at 2050 and 2176 nm which are found in the spectrum of whey concentrate (Baer et al. 1983). This was expected, since AWP average composition is as follows: lactose (65–70 %), whey proteins (11–13 %) and ash (10 %). Slightly different NIRS spectra were recorded for the two AWP samples. These differences are especially apparent in the regions 1450–1600, 1950–2200 and 2280–2400 nm and reflect compositional differences among different AWP and/or different spray-drying conditions applied. For obvious reasons, the starch and the MD NIRS spectra also resemble that of lactose and are different from that of a standard SMP sample with more intense peaks at 1450 and 1940 nm and a much lower signal in the region ranging from 2120 to 2400 nm.

Near-infrared spectra (after SNV transform) of a genuine milk powder, the two acid whey, starch and maltodextrin samples used in the study
To evaluate the potential of NIRS to detect adulteration of SMP with AWP, starch and MD, two approaches were tested: (i) an untargeted class modelling approach for genuine SMP and (ii) a targeted regression approach to predict the adulterants level in SMP. Before the chemometric modelling of the NIRS data, a PCA analysis was performed on the 60 genuine SMP only (Fig. 2a). Data were transformed (SNV + second derivative) and then mean-centred. Four principal components were included in the PCA model which explained ≈95 % of the total variance. From the PCA score plot of the first two PCs (data not shown), four milk powders appeared quite different from the bulk of the samples. However, only three samples showed a Mahalanobis distance (the distance of the sample from the multivariate mean) exceeding the 95 % probability threshold computed for the four principal components included in the model (Fig. 2b). Since three SMP samples exceeding the 95 % probability threshold are expected based on statistical considerations and the excess was <60 % of the computed threshold, it was decided to retain all the SMP for the subsequent adulteration step and multivariate analysis.

a First two components of the PCA score plot (SNV + second derivative + mean-centreing) for the genuine skim milk powders. The amount of variance explained by each factor is reported in brackets. b Mahalanobis distance for the genuine skim milk powders in the PCA model. The horizontal line represents the threshold value calculated for four factors selected in the PCA model
The idea behind approach (i) was to build up a class model for genuine SMP based on a library of NIRS spectra and that non-authentic samples would classify as outliers with respect to that class which would allow for non-specific anomaly testing in SMP. To that purpose, SIMCA was used as class modelling technique. For discrimination purposes, SIMCA develops principal component models for each training set class. When the set of measurements of a new sample is projected into the PC space of each training class, the new sample is assigned to the class(es) it best fits. In the present study, we developed one SIMCA model for the genuine SMP based on 44 SMP samples. The SIMCA model was based on NIRS spectra after SNV and second derivative transformation of the data and included eight factors which explained 99 % of the total variance. The remaining 364 samples (16 genuine SMP and 348 adulterated with acid whey, starch and MD) were then projected in the PC space of the genuine SMP class, and for each of them, the multivariate distance from the SMP class was calculated and compared with a critical threshold value. If the sample multivariate distance was lower than the critical value, the sample was considered as belonging to the SMP class. The sensitivity and the specificity of the model are reported in Table 1, and the results are visually represented in Fig. 3. Four SMP samples exhibited a multivariate distance from the SMP class above the critical value and were then misclassified as adulterated samples, whereas in total 60 adulterated samples were misclassified as genuine SMP. The misclassified samples were most of the SMP adulterated with 1 % acid whey, but also, some SMP adulterated with 5 % acid whey as well as some adulterated with 2 % of MD were misclassified. It was noted that the inclusion of <8 factors in the SIMCA model resulted in substantial lower type I errors (genuine SMP predicted as adulterated) but a higher type II error (adulterated samples predicted as genuine), whereas the opposite was observed upon inclusion of >8 factors. A closer look at the SIMCA model scores may provide a qualitative indication of the type of adulterant present in the test sample. The samples adulterated with starch and MD exhibited high positive score along PC1, whereas sample adulterated with acid whey showed high negative scores along PC2 (Fig. 4).

Distance of the samples of the validation set from the genuine milk powder class modelled by SIMCA (SNV + second derivative + mean-centreing). Horizontal line corresponds to the critical distance to the genuine milk powder class. New samples falling beyond the line are classified as adulterated milk powders

SIMCA (SNV + second derivative + mean-centreing) score plot of the first two principal components for the samples of the validation set
For approach (ii), two different algorithms, namely PLSR and PCR, and several data transforms were tested in this study. On average, PLS regression models performed better (lowest root mean square error of prediction (RMSEP, expressed as % added adulterant)) than PCR models. Light scattering correction was crucial to improve the predictive capability of the models and second derivation performed on average better than first derivation. For the prediction of %AWP adulteration, the regression models were developed using a training set of 180 samples (20 genuine SMP + 160 samples from adulteration of each SMP with acid whey at all the adulteration levels). The optimal number of factors for each regression model was chosen based on the lowest RMSEP on the 96 samples of the validation set (40 genuine SMP and 56 adulterated). The lowest RMSEP was obtained by PLS after application of the SNV transform + second derivative. This model included 11 factors which explained approximately 99 % of the total X variance. The model provided a root mean square error of calibration (RMSEC) of 0.59 % (% of added adulterant) and resulted in a RMSEP of 1.50 %. The amount of Y variance explained by the model (r 2) in external validation was 0.982. The fit between the predicted % of adulteration and the actual level of adulteration for the selected model in calibration and in validation is shown in Fig. 5a, b. Since SMP is comparatively higher in proteins (with a whey protein/casein ratio of approximately 15–20 %) and lower in lactose, the adulteration of SMP with AWP results in a decrease of the total protein content of the mixture compared to the original SMP, as well as in the increase in the whey protein/casein ratio and the increase in lactose content. In Fig. 6, the regression vector for the selected model is shown. The regression vector can be thought of as a weighted sum of the loadings included in the model. Variables with high coefficients contribute significantly to a prediction. From Fig. 5, it appears that the region between 2250 and 2315 nm is especially important for the correct prediction of the % of added AWP as well as the region between 1650 and 1720 nm. These regions are associated to the combination of C–H stretching and bending modes and the first overtone for the C–H vibrational modes, respectively (Miller 1989). Finally, in the regression vector, peaks at 1950, 1980 and 2040 nm are also apparent. These bands might be related to the different protein contents/compositions in AWP-adulterated samples compared to genuine SMP because the first is associated to the vibration modes of CONH2 amide groups and the latter to vibration modes of CONHR amide groups (Miller 1989).

Plot of the % of added adulterant as predicted by the best regression model for each adulterant towards the actual concentrations. a, b Results on calibration and validation set for acid whey (PLS, SNV + second derivative + mean-centreing). c, d Results on calibration and validation set for starch (PCR, MSC + second derivative + mean-centreing). e, f Results on calibration and validation set for maltodextrin (PLS, SNV + second derivative + mean-centreing). Diagonal line is the line of perfect fit

Regression vectors for the PLS models developed for quantification of acid whey (PLS, SNV + second derivative + mean-centreing), starch (PCR, MSC + second derivative + mean-centreing) and maltodextrin adulteration (PLS, SNV + second derivative + mean-centreing)
For the regression analysis of the starch adulteration, 24 SMP were used for the model calibration with a total of 72 samples (24 genuine SMP and 48 from adulteration of each SMP with 2 and 5 % of starch). The remaining 27 samples (nine genuine SMP samples + 18 adulterated SMP) + the genuine SMP used for the development of the regression model for acid whey were used for the validation (in total 54 samples). The best performing model (lowest RMSEP) was a PCR model with MSC + second derivative transformation of NIRS spectral data. This model included nine factors which explained approximately 99 % of the total X variance. The model provided a RMSEC of 0.40 % and resulted in a RMSEP of 0.36 %. The amount of Y variance explained by the model (r 2) in external validation was 0.980. The fit between the predicted % of adulteration and the actual level of adulteration for the selected model in calibration and in validation is shown in Fig. 5c, d. The RMSEP obtained for the best model for starch showed that a lower amount of starch can be detected by NIRS in SMP compared to acid whey. The regression vector for the model shows six important peaks (Fig. 6): at 1410, 1910, 1950, 2180, 2260 and 2280 nm. The peak at 1410 would be associated with the first overtone of vibrational modes of –OH functional groups. The peaks at 1910, 1950 and 2180 nm might be associated with the CHO functional groups of starch and those at 2260 and 2280 would correspond to combination of C–H stretching and bending modes (Miller 1989).
The regression analysis on samples adulterated with MD was performed as described for starch. The best model was a PLS model based on SNV + second derivative of the NIRS spectral data which gave a RMSEP of 0.41 % and a RMSEC of 0.64 %. Six factors were selected which explained 98 % of the total X variance, whereas the amount of Y variance explained was 0.903. The fit is provided in Fig. 5e, f. Surprisingly, the regression vector for MD (Fig. 6) is somewhat different from that for starch even though the functional group composition of SMP adulterated with starch and MD is supposed to be very similar. Beyond the peaks at 1410 and 1915 nm which are related to –OH and CHO functional groups, additional peaks at around 2040 and 2420 nm seem to be very important for the prediction. This last peak would be associated to combinations of vibrational modes of C–C and C–H bonds (Miller 1989).
Based on the calibration equations for the three regression models, CSH were calculated as 0.93 and 1.30 % for a confidence level of 95 % (CSH95) and 99 % (CSH99), respectively, for acid whey adulteration and as 0.62 and 0.73 % for α = 0.05 and as 0.88 and 1.01 % for α = 0.01 for starch and MD adulteration, respectively. The ID limits calculated for the three regression models are summarized in Table 2.
This means, for instance, that there is less than 1 % chance of wrongly classifying a genuine sample as adulterated with acid whey if the predicted values is greater than the CSH99, i.e. 1.33 %, and there is <1 % chance of wrongly classifying a sample adulterated with at least 2.71 % acid whey as genuine if the predicted value is lower than the CSH99.
The calculated CSHs can be compared with the results of the prediction on the samples of the validation sets. For acid whey, only two genuine samples of the validation set had predicted % acid whey greater than the calculated CSH95 which was expected on 40 samples (i.e. based on normal distribution, it is expected that 5 % of the sample have predicted values greater than CSH95 which means two samples out of 40 samples). However, one of the samples adulterated with 5 % acid whey had a predicted level less than both the CSH values which suggests that perhaps the actual limits for type II errors are higher than the calculated theoretical limits reported in Table 2. For starch and MD, the calculated CSHs match with the predicted values on the validation set. In each validation set, three (out of 36) samples were above the calculated CSH95 and one was above the CSH99. Moreover, all the samples adulterated with 2 % starch or MD were well above the calculated CSHs.
The comparison between Fig. 3 and Table 2 shows that the development of adulterant-specific regression models allows a more sensitive detection of all the tested adulterants in SMP. The results based on the analysis of whey/total protein ratio of the sample set used in the present study suggest a significantly higher ID (>10 %, data not shown). The results reported in this study would therefore suggest that the NIRS method would have at least the same sensitivity as compared to methods based on whey/total protein ratio for acid whey detection. As for starch adulteration, NIRS would not represent an improvement in terms of sensitivity compared to the iodine method but it would be applicable also for the detection of pure amylopectin starch (waxy starches) which has the same chemical structure of amylose but does not give a positive reaction to the iodine test because of the absence of amylose linear chain.
However, the prediction may be significantly biased if the test sample is not genuine but already adulterated with substance different from those for which the regression models have been developed. Indeed, each regression model was also tested on SMP adulterated with each of the other two adulterants. When the regression model developed for AWP was used to predict on SMP adulterated with starch (but not with acid whey), very negative values were obtained for most of the samples (data not shown). Analogous results were obtained on SMP adulterated with MD. Analogously, the regression models developed for starch and MD predicted negative levels (down to −4 %) for almost all the SMP adulterated with acid whey (but not with starch or MD). Finally, when the regression model developed for starch is used to predict on samples adulterated with MD, the prediction is biased but high concentration of MD can still be detected. The same holds when the model for MD is used on samples adulterated with starch.
The class modelling approach, on the other hand, proved slightly less sensitive but allows general anomaly testing, i.e. the detection of multiple adulteration at once, including situations where more than one adulterant is used in the same test sample without the need of developing adulterant-specific models. In principle, such class modelling approach can be extended to the detection of whatever adulterant in SMP and, remarkably, it can be used to detect the presence of yet unknown adulterants. However, developing robust methods for the non-targeted detection of adulteration in SMP based on NIRS is complicated by the expected physical-chemical variability in SMP composition which reduces the ability of the method to detect low amount of any adulterant. Such variability depends more on minor constituent composition and drying conditions than on SMP proximate composition (Botros et al. 2013). Therefore, a considerable database needs to be built for use in practice.
As for MD detection, the NIRS method developed in this study would represent a novel tool for the rapid screening of MD in SMP. However, one of the complicating factors in MD detection by NIRS is that MD with different dextrin equivalents (DE, % of reducing sugars relative to glucose in a sugar product) is in principle available for adulteration and those can show differences in the NIRS spectra (Storz and Steffens 2004). Those differences are large enough to be used for the prediction of the DE of the MD test sample. However, our results suggest that the models would be applicable, at least to a certain extent, also to MD with other DE. It is worth mentioning, however, that addition of MD alone to genuine milk or milk powders is not a frequent practice and that MD are mainly added to milk or milk powders adulterated with, for instance, whey protein and fat, to adjust the density and cryoscopy of the resulting liquid milk (Ferrão et al. 2007).
Conclusions
In the present study, the potential of NIR spectroscopy for the detection of adulteration of SMP with AWP, starch and MD was tested with two different chemometric approaches. The class modelling untargeted approach has the advantage that several adulterants can be detected with one single model, including yet unknown adulterants. The targeted regression approach has the advantage of a better sensitivity but independent models have to be developed for each adulterant and the prediction may be significantly biased if two or more adulterants are simultaneously present. NIR spectroscopy analysis of SMP proved thus suitable for a rapid and cost-efficient broad anomaly testing as well as the targeted detection of the tested adulterants. Regardless of the chemometric approach used, NIRS allows the adulteration of SMP with acid whey, starch and MD to be detected at once with acceptable sensitivity with just one spectrum acquisition.
It is necessary to stress that the results of the present analysis have been derived from dry blended mixture of milk powder with acid whey/starch/MD powder and might have been different if the adulteration of fluid milk before spray drying is considered (wet blending). Some extra validation analyses need to be carried out to test the applicability of the chemometric models to such situation.
References
Baer RJ, Frank JF, Loewenstein M (1983) Compositional analysis of nonfat dry milk by using near infrared diffuse reflectance spectroscopy. JAOAC 66:858–863
Balabin RM, Smirnov SV (2011) Melamine detection by mid- and near-infrared (MIR/NIR) spectroscopy: a quick and sensitive method for dairy products analysis including liquid milk, infant formula, and milk powder. Talanta 85:562–568
Barton FE II (2002) Theory and principles of near infrared spectroscopy. Spectrosc Eur 14(1):2–18
Banks W, Greenwoo CT, Muir DD (1971) The characterization of starch and its components. Part 3. The technique of semi-micro, differential, potentiometric, iodine titration, and the factors affecting it. Starch/Starke 23:118–124
Basch JD, Procino FW, Holsinger LG, Farrell HM Jr (1985) Quantitation of caseins and whey proteins of processed milks and whey protein concentrates, application of gel electrophoresis, and comparison with Harland-Ashworth procedure. J Dairy Sci 68:23–31
Borin A, Ferrão MF, Mello C, Maretto DA, Poppi RJ (2006) Least-squares support vector machines and near infrared spectroscopy for quantification of common adulterants in powdered milk. Anal Chim Acta 579:25–32
Botros LL, Jablonski J, Chang C, Bergana MM, Wehling P, Harnly JM, Downey G, Harrington P, Potts AR, Moore JC (2013) Exploring authentic skim and nonfat dry milk powder variance for the development of nontargeted adulterant detection methods using near-infrared spectroscopy and chemometrics. J Agric Food Chem 61:9810–9818
Cartuyvels D, Merchiers M, Van Renterghem R, De Block J (1999) A fast and simple method to determine the whey powder to milk powder ratio using spectroscopy in alkali. Milchwissenschaft 54:268–272
Chavez NA, Salinas E, Jauregui J, Palomares LA, Macias K (2008) Detection of bovine milk adulterated with cheese whey by Western blot immunoassay. Food Agric immunol 19:265–272
Cozzolino D (2012) Recent trends on the use of infrared spectroscopy to trace and authenticate natural and agricultural food products. Appl Spectrosc Rev 47(7):518–530
De Block J, Merchiers M, Van Renterghem R (1997) Quantification of the ratio of whey powder to milk powder using spectroscopy in alkali. Milchwissenschaft 52:213–216
De Koning PJ, Van Rooijen PJ (1971) Estimation of whey proteins in casein co-precipitate or in mixtures with milk powder by the use of a modified ninhydrin reaction. Milchwissenschaft 26:1–6
Ferrão MF, Mello C, Borin A, Maretto DA, Poppi RJ (2007) LS-SVM: a new chemometric tool for multivariate regression. Comparison of LS-SVM and PLS regression for determination of common adulterants in powdered milk by NIR spectroscopy. Quim Nova 30:852–859
Gorry PA (1990) General least-squares smoothing and differentiation by the convolution (Savitzky-Golay) method. Anal Chem 62:570–573
Kanning M, Casella M, Olieman C (1993) Milk and soy proteins analysis using capillary zone electrophoresis. LCGC Int 6:701–706
Lechner E, Klostermeyer H (1981) Nachweis einer VerfaKlschung von magermilchpulver mit molkenpulver (polarographische methode). Milchwissenschaft 36:267–270
Lu C, Xiang B, Hao G, Xu J, Wang Z, Chen C (2009) Rapid detection of melamine in milk powder by near infrared spectroscopy. J Near Infrared Spectrosc 17:59–67
Lüthi-Peng QQ, Puhan Z (1999) The 4th derivative spectroscopic method for the rapid determination of protein and casein in milk. Milchwissenschaft 54:74–77
Maraboli A, Cattaneo T, Giangiacomo R (2002) Detection of vegetable proteins from soy, pea and wheat isolates in milk powder by near infrared spectroscopy. J Near Infrared Spectrosc 10:63–69
Mendenhall IV, Brown RJ (1991) Fourier transform infrared determination of whey powder in nonfat dry milk. J Dairy Sci 74:2896–2900
Meisel H (1995) Application of fourth derivative spectroscopy to quantitation of whey protein and casein in total milk protein. Milchwissenschaft 50:247–251
Miller CE (1989) Chemical principles of near-infrared technology. In: Williams P, Norris K (eds) Near infrared technology in the agricultural and food industries. American Association of Cereal Chemists, St.Paul (MN)
Miralles B, Bartolomé B, Ramos M, Amigo L (2000) Determination of whey protein to total protein ratio in UHT milk using fourth derivative spectroscopy. Int Dairy J 10:191–197
Ni Z, Feng YC, Hu CQ (2010) Analyzing the methods to remove artifacts encountered in the development of a NIR quantitative model for powder medicines. J Anal Bioanalytical Techniques 1:114–120
Osborne BG (2000) Near-infrared spectroscopy in food analysis. Wiley, Chichester, UK
Rocklin RD, Pohl CA (1983) Determination of carbohydrates by anion exchange chromatography with pulsed amperometric detection. J Liq Chromatogr 6:1577–1590
Rodriguez-Saona LE, Allendorf ME (2011) Use of FTIR for rapid authentication and detection of adulteration of food. Annu Rev Food Sci Technol 2:467–483
Sanvido G, Garcia JG, Corilo YE, Vaz BG, Zacca JJ, Cosso RG, Eberlin MN, Peter MG (2010) Fast screening and secure confirmation of milk powder adulteration with MD via electrospray ionization-mass spectrometry [ESI(þ)-MS] and selective enzymatic hydrolysis. J Agric Food Chem 58:9407–9412
Savitzky A, Golay MJE (1964) Smoothing and differentiation of data by simplified least squares procedures. Anal Chem 36:1627–1639
Storz E, Steffens K-J (2004) Feasibility study for determination of the dextrose equivalent (DE) of starch hydrolysis products with near-infrared spectroscopy (NIRS). Starch/Stärke 56:58–62
Vogelgesang J, Hädrich J (1998) Limits of detection, identification and quantification: a statistical approach for practitioners. Accredit Qual Assur 3:242–255
Acknowledgments
The authors gratefully acknowledge the Dutch Ministry of Economic Affairs and Innovation for financial support.
Conflict of Interest
Edoardo Capuano declares that he has no conflict of interest. Rita Boerrigter-Eenling declares that she has no conflict of interest. Alex Koot declares that he has no conflict of interest. Saskia M van Ruth declares that she has no conflict of interest. This article does not contain any studies with human or animal subjects.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Capuano, E., Boerrigter-Eenling, R., Koot, A. et al. Targeted and Untargeted Detection of Skim Milk Powder Adulteration by Near-Infrared Spectroscopy. Food Anal. Methods 8, 2125–2134 (2015). https://doi.org/10.1007/s12161-015-0100-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-015-0100-3