
Abstract
Known for its remarkable biodiversity and high levels of endemism, the Brazilian Atlantic Rainforest has been characterized as one of the most threatened biomes on the planet. Despite strong interest in recent years, we still lack a comprehensive scenario to explain the origin and maintenance of diversity in this region, partially given the relatively low power of analyses involving few independent genetic loci. In this study, we examine a phylogenomic dataset of five ant species to investigate phylogeographical patterns across the Brazilian Atlantic Forest. We sequenced ultraconserved elements to generate hundreds of loci using a bait set developed specifically for hymenopterans. We analyzed the data using Bayesian and maximum likelihood approaches of phylogenetic inference. Results were then integrated with environmental niche modeling of current and past climates, including the Last Glacial Maximum and the last interglacial period. The studied species showed differentiation patterns that were consistent with the north/south division of the Atlantic Rainforest indicated in previous studies for other taxa. However, there were differences among species, both in the location of phylogeographic breaks and in the pattern of genetic variation within these areas. Samples from southern localities tended to show recent genetic structure, although a site in Tapiraí (state of São Paulo) repeatedly showed an intriguing older history of differentiation. All species experienced shifts in areas of suitability through the time. Our study suggests that distinct groups may have responded idiosyncratically to the climatic changes that took place in the Brazilian Atlantic Forest. The amount of intraspecific genetic structure was related to the inferred geographical distribution of habitat suitability according to current and past times. Also, a parallel between the amount of Quaternary climatic suitability and the level of interspecific differentiation was detected for four species. Finally, despite strong contractions at the northeastern region of the forest, the remaining areas appear to have been able to act as refugia.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The remarkable plant and animal diversity in the Brazilian Atlantic Forest had a profound impact on the young Charles Darwin in 1832, leading him to note in his diary from the voyage on the HMS Beagle that “no art could depict so stupendous a scene” (Darwin and Keynes 2004). Indeed, the Brazilian Atlantic Forest has been estimated to harbor the equivalent of 50–60% of the species richness of the entire Amazon Forest, despite only encompassing less than one fourth of its geographical extent (Silva and Casteleti, 2003; Silva et al. 2005; Ribeiro et al. 2009). Such elevated diversity and endemism have led the Brazilian Atlantic Forest to be recognized among the world’s biodiversity hotspots (Myers et al. 2000), yet the evolutionary processes that led to such high diversity remain unclear. For instance, diversity hotspots have been identified across the Brazilian Atlantic Forest for a variety of taxa, including birds (Silva et al. 2004; Giraudo et al. 2008), mammals (Costa et al. 2000), and plants (Murray-Smith et al. 2009; Leão et al. 2014), but the apparent lack of congruence in the location of these hotspots calls into question whether diversification has been mediated through vicariance, or perhaps instead through processes more idiosyncratic to species, such as dispersal ability (Smith et al. 2014).
The lack of clear evolutionary drivers in the Brazilian Atlantic Forest is obvious when compared to its better-known neighbor, the Amazon basin (Haffer 1997; Hayes and Sewlal 2004; Bonaccorso et al. 2006; Oliveira et al. 2017). In fact, one of the earliest diversification hypotheses for the Brazilian Atlantic Forest involves the Pleistocene Refugia Model, made famous for its application to the question of diversification in the Amazon basin (Haffer 1969; Vanzolini and Williams 1981; Vanzolini 1992), which posits that Pleistocene climatic cycles led to vicariance and speciation. However, this model does not seem applicable to the Brazilian Atlantic Forest because extant species often considerably predate the Pleistocene and go back in many cases to the Tertiary (e.g., Simpson 1979; Zamudio and Greene 1997; Lara and Patton 2000; Pellegrino et al. 2005; Álvarez-Presas et al. 2014; Thomé et al. 2014; see also Costa 2003). On the other hand, there is evidence that climatic changes during the Quaternary influenced levels of intraspecific genetic variability in the Brazilian Atlantic Forest, with northern populations often being characterized by higher genetic variability, presumably caused by relative habitat stability, and southern populations showing lower genetic diversity associated with recolonization (e.g., Pellegrino et al. 2005; Grazziotin et al. 2006; Tchaicka et al. 2006; Martins et al. 2009; Resende et al. 2010; Ribeiro et al. 2010; Amaro et al. 2012; but see Batalha-Filho et al. 2012).
In addition to latitudinal effects, climatic fluctuations also affected species differently depending on their altitudinal distribution. For instance, the anuran Hypsiboas faber is distributed from the lowlands up to 1000 m and is likely to have persisted in the southern Brazilian Atlantic Forest during the Last Glacial Maximum, while lowland species such as Hypsiboas albomarginatus and Hypsiboas demilineatus did not (Carnaval et al. 2009). A similar pattern was detected for another anuran, Proceratophrys boiei, found across a broad altitudinal distribution (up to 1200 m), which showed no evidence of population expansion (Amaro et al. 2012). These results suggest that montane habitats might have facilitated the maintenance of genetic variability, given that altitudinal migration could protect local populations from extinction. Indeed, it has been found that the mountainous region of the southern Brazilian Atlantic Forest, the Serra do Mar, might harbor substantial phylogeographic endemism (Carnaval et al. 2014). These idiosyncratic climatic response patterns are now becoming clearer to Brazilian Atlantic Forest (Prates et al. 2016), even though the underlying mechanisms for this inconsistency need to be further investigated. For instance, one possibility is that the niche of each species affects the way they are influenced by the climate and vegetation changes of the Brazilian Atlantic Forest.
Most phylogeographic studies on the Brazilian Atlantic Forest have focused on vertebrates. A more comprehensive view of this region’s diversification should include a variety of taxonomic groups, particularly invertebrates that potentially disperse more slowly than vertebrates and can show fine-scale phylogeographical patterns that could help elucidate a region’s history (Garrick et al. 2004). Ants (Formicidae) are a model system already used to investigate phylogeographical patterns in a wide range of terrestrial ecosystems (e.g., Quek et al. 2007; Solomon et al. 2008; Leppänen et al. 2011). With nearly 13,500 described species (Bolton 2019), ants are eusocial organisms that display a variety of life histories and have a wide range of ecological interactions with other organisms (Hölldobler and Wilson 1990). The feature of having fine-scale and regional biodiversity also makes this group suitable for phylogeographic studies (e.g., Seal et al. 2015).
The goal of this study is to carry out a comparative phylogeography with five ant species from the Brazilian Atlantic Forest. A few studies already used ants to address phylogeographic questions in this environment (e.g., Cardoso et al. 2015; Peres et al. 2015), but here, we use ultraconserved elements (UCEs) to generate data from hundreds of predominantly unlinked loci using a developed probe set for Hymenoptera (Faircloth et al. 2015). Compared to mitochondrial DNA, data from hundreds of unlinked loci allow us to assess the divergence history across the genome, instead of a single locus. While the benefits of multilocus phylogeography have been well described, these methods have not been applied to the Brazilian Atlantic Forest, where most studies tend to rely on mitochondrial DNA or a few loci (e.g., Bragagnolo et al. 2015; Batalha-Filho and Miyaki 2016; Cabanne et al. 2016).
Material and methods
Sampling and DNA extraction
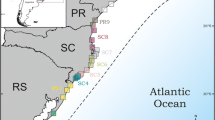
Genomic DNA was extracted from individuals of the following ant species (see details at Table 1): Heteroponera dentinodis (Mayr, 1887) (Heteroponerinae), Hylomyrma reitteri (Mayr, 1887) (Myrmicinae), Octostruma rugifera (Mayr, 1887) (Myrmicinae), Octostruma stenognatha Brown & Kempf, 1960 (Myrmicinae), Odontomachus meinerti Forel, 1905 (Ponerinae), Strumigenys crassicornis Mayr, 1887 (Myrmicinae), and Strumigenys denticulata Mayr, 1887 (Myrmicinae) from 23 localities covering most of the Brazilian coast and the original distribution of the Brazilian Atlantic Forest, although not all species were obtained from each location (Fig. 1). We focused on a multispecies approach given that it is more efficient in providing general inferences than single-species studies (Bernatchez and Wilson 1998). The sequences from Heteroponera dentinodis and Hylomyrma reitteri were used to root intraspecific gene trees for their phylogenetically closest species (see below).

Location of the sampling sites from which the phylogenetically studied specimens were obtained. The colored grid shows which species were sampled in each sampling site. Sampling sites are represented by the columns of the colored grid and are indicated by different letters. A white square means the species was not sampled at that site
Voucher specimens of ants from the same nest series of all species were deposited in the myrmecological collection of the Museu de Zoologia da Universidade de São Paulo, Brazil, and were obtained through of a larger effort to survey the ant fauna of the Brazilian Atlantic Forest between 1999 and 2003 as part of a BIOTA-FAPESP initiative. DNA extraction was carried out using PureLink™ Genomic DNA kits (Invitrogen, USA), and double-stranded DNA concentration was measured on a Qubit 2.0 Fluorometer (Life Technologies, Inc.) using the dsDNA High Sensitivity Assay Kit. The initial DNA concentration for these samples was highly variable, ranging from < 0.2 ng total DNA to 14.2 ng/μl. DNA quality was checked using agarose gel electrophoresis or using the Bioanalyzer (Agilent Technologies) depending on the amount of the DNA. Samples with high molecular weight DNA were sheared by sonication with a Q800R Sonicator (Qsonica Inc.) using 15 s on–15 s off cycles for 50 s with 20% amplitude. Highly fragmented samples were not sheared prior to library preparation.
Sequence capture of UCEs
We prepared libraries with the KAPA HyperPrep Kit (Kapa Biosystems) adjuvant with a bead technique (Fisher et al. 2011), which was used instead of solid-phase reversible immobilization (SPRI) beads (Rohland and Reich 2012). When possible, we used 30 ng of total starting DNA. Between 20 and 30 ng, we used all the available DNA, whereas samples below 20 ng were discarded. Each sample was individually labeled using the iTru dual-indexing adapter system (Glenn et al. 2016), which is similar to the TruSeq layout barcodes (Faircloth and Glenn 2012). We obtained adapter-ligated DNA for all samples with 12–16 PCR cycles (more cycles were performed with low-concentration samples). If necessary, a dual-SPRI cleanup of the samples was performed to remove fragments that were either too long or too short for sequencing.
We then carried out target enrichment (Gnirke et al. 2009) to isolate UCEs that occur in Hymenoptera (Faircloth et al., 2015) using a protocol from Faircloth and Glenn (2012). We created pools of eight samples, and each pool was concentrated to 147 ng/μl in a vacuum centrifuge and enriched for a set of 1510 UCEs using RNA probes (myBaits®, Arbor Biosciences). We assessed the size of the captured UCE products using a Bioanalyzer (Agilent Technologies), followed by quantification of library concentration with real-time PCR with a Kapa Library Quantification Kit (Kapa Biosystems, Inc.). The resulting pools were combined at equimolar ratios and sequenced on an Illumina HiSeq2000 sequencer, using 100-bp paired-end reads at the, Genome Technology Center, University of California Santa Cruz. Data available from the Dryad Digital Repository: https://doi.org/10.5061/dryad.k200rt4.
Bioinformatic processing of UCE data
After sequencing, we converted the BCL files into FASTQ and demultiplexed samples using Illumina bcl2fastq Conversion software (v1.8.4). FastQC 0.11.3 (Andrews 2010) was used to assess the sequencing quality. The Trimmomatic tool (Lohse et al. 2012) implemented in Illumiprocessor (Faircloth 2013) was applied to remove low-quality regions, barcodes, and adapters. The assembly was carried out with Trinity (Grabherr et al. 2011) as implemented in the PHYLUCE pipeline (Faircloth 2015) and using the pre-set parameters. The resulting contigs were also processed with PHYLUCE (Faircloth 2015) to recover UCE loci, which were aligned using MAFFT (Katoh and Standley, 2013), and irregular edges were automatically cleaned. All the post-assembly processing was done together for all species and following the default settings of PHYLUCE (Faircloth 2015).
We pruned the resulting data to generate complete concatenated matrices (without any missing locus for any specimen) and incomplete concatenated matrices (75% completeness needed to retain a locus). Trees from the obtained sequences were inferred using maximum likelihood with RAxML (version 8.0.0) (Stamatakis 2014) with an unpartitioned, GTRGAMMA nucleotide substitution model. To root the trees, we used the phylogenetically closest species for which the largest number of loci was available because not all loci were amplified for each species. A locus that was successfully amplified for one species might not be available for the phylogenetically closest species. We built a concatenated alignment using only the shared loci by selecting the longest sequence in the corresponding outgroup species and locus and analyzed the resulting alignment using RAxML. The obtained placement of the outgroup was then used to root the original trees. We also obtained chronograms using a relaxed lognormal clock model in BEAST (version 1.8.4) (Drummond et al. 2012), with MCMC runs of 100 million generations, trees sampled every 1000 generations with GTR + Γ nucleotide substitution model, and a Yule tree prior. Convergence was checked on Tracer v1.6 (e.g., ESSs > 200), and a burn-in of 10% was applied.
To help explain how climate has influenced species, we applied a niche modeling approach that has been successfully used in this and other biomes (e.g., Morales et al. 2015; Nicolas et al. 2016) and that has been confirmed as accurate even with small numbers of samples (Pearson et al. 2006). Ecological niche models (ENMs) were used to infer how the geographical distribution of the ants might have been influenced by the climatic changes during their recent evolutionary history. We estimated ENMs for Octostruma rugifera, Octostruma stenognatha, Odontomachus meinerti, Strumigenys crassicornis, and Strumigenys denticulata. Records of occurrences were compiled using the Global Biodiversity Information Facility database. To reduce spatial sampling bias, we used a randomization approach implemented in the R package spThin 0.1.0 (Aiello-Lammens et al. 2015) to exclude occurrence records separated by less than 15 km apart while keeping the maximum number of records. We then used the occurrence records to select the extent of the study region used to model the niches. Our aim was to delimit an area that was accessible to species via dispersal, a procedure known for improving both model performance and transferability across time (Barve et al. 2011). Specifically, we generated minimum convex polygons using the occurrence records of each species. We then created 300-km buffers around each polygon. The resulting areas were selected as study regions for each species.
We selected a preliminary set of 19 bioclimatic variables at the spatial resolution of 5 arc minutes from the WorldClim 2 database (Fick and Hijmans 2017). We cropped the variables according to the extent of the selected study regions and performed a pairwise Pearson correlation test. We retained only variables with correlation values < 0.80. ENMs were generated using Maxent 3.4.1 (Phillips et al. 2017). Maxent is one of the best performing algorithms for modeling species distribution from presence-only records (Elith et al. 2006). We used raw output format (Merow et al. 2013) and assessed model performance using 10-fold cross-validation (Wenger and Olden 2012). To distinguish present-day suitable and unsuitable habitats, we applied a minimum training presence threshold, while excluding the 5% of the localities with the lowest predicted values (i.e., admitting a 5% omission error; Peterson et al. 2011; Zwiener et al. 2017). Thresholds were calculated using the R package ENMGadgets 0.0.12 (Barve and Barve 2016).
To estimate the potential past distribution of the species, we projected present-day ENMs onto two-time slices: the Last Glacial Maximum (LGM; ~ 22,000 years ago) and the last interglacial (LIG; ~ 120,000–140,000 years ago). Past climate data used were downloaded from the WorldClim 1.4 database (Hijmans et al. 2005). LGM climate data are available from three global circulation models: CCSM4, MIROC-ESM, and MPS-ESM-P. In this case, we projected present-day ENMs into each global circulation model and then created a composite model by averaging the predictions of all models. To distinguish past suitable and unsuitable habitats, we applied a minimum training presence threshold, excluding the 20% of the localities with the lowest predicted values (i.e., admitting a 20% omission error). We chose a relatively high threshold to avoid overly broad predictions and to emphasize regions with higher habitat suitability.
Results
Descriptive statistics of UCE loci recovered for each species are shown in Table 1. The success of the obtained UCE data is notable given the highly degraded state of some DNA extracts (observed by gel electrophoresis) and the fact that the UCE baits were not developed for ants specifically, but for groups of Hymenoptera. Interestingly, we did not find a significant relationship between the initial DNA concentration and the number of obtained loci (R = 0.016, P = 0.33), suggesting that the level of DNA fragmentation might be more important than the concentration itself in the success of UCE capture.
The trees from concatenated UCE loci for each species are shown in Fig. 2(a–e). Given that the topologies from ML (RAxML) and BI (BEAST) are congruent, BI + ML trees will be discussed further as equivalent, given that the observed differences between them would not alter our conclusions. There were substantial differences in relative timing and geographic structure among species. The shallowest divergences within Octostruma rugifera correspond to sites in the southern Brazilian Atlantic Forest (Morretes, state of Paraná, and Palhoça, state of Santa Catarina; Fig. 2(a)). The oldest divergences in Octostruma stenognatha were found in samples from the northeastern Brazilian Atlantic Forest (state of Bahia; Fig. 2(b)). A similar pattern was found on Odontomachus meinerti (Fig. 2(c)), although with a shallower divergence and a less prominent geographical structure. Also, Odontomachus meinerti shows an intriguing result of an old divergence further south (Tapiraí, state of São Paulo), an outcome that is shared with other species (see next). Strumigenys crassicornis and Strumigenys denticulata presented similar results with a strong north/south differentiation and recent divergences in the south. Also, the deep divergence of the Tapiraí is notable in Strumigenys denticulata (Fig. 2(e)).

Intraspecific relationships within five ant species in the Brazilian Atlantic Forest based on phylogenomic data from UCEs. Chronograms also indicating relative timing of divergence of sequences from the studied species. Pink lines indicate 95% credibility intervals. Letters after location names are the same letters presented in Fig. 1
We estimated relative divergence times between samples, based on the assumption that UCE loci share similar mutation rates. Interestingly, there was a correspondence between the level of intraspecific divergence and the inferred geographical distribution of habitat suitability according to current and past ENMs (overall, ENMs showed reliable performance with AUC > 0.79 for all species). For instance, Octostruma rugifera and Odontomachus meinerti were associated with the shallowest divergences, followed by Strumigenys crassicornis, Octostruma stenognatha, and Strumigenys denticulata, respectively (Fig. 2(a–e)), which tended to correspond to the level of stability in their corresponding areas of suitability throughout the LIG/LGM/present (Fig. 3). This combination of results suggests a scenario of population expansion and/or high dispersal capacity, which could erase any geographical structure in the observed genetic variability of these species.

Environmental niche modeling of climatic suitability in the present time, Last Glacial Maximum (LGM), and last interglacial (LIG) period for the studied species. Occurrence data for each species are shown as white diamonds
Discussion
Our results provide valuable insight into the history of the Brazilian Atlantic Forest by exploring genetic structure and environmental stability in five co-distributed ant species. In general, even though our results supported the traditional north/south division in the Brazilian Atlantic Forest, we also found substantial differences among species in the location of genetic divisions and in the patterns of genetic variation within areas. These differences suggest that species responded idiosyncratically to the climatic changes that took place in the Brazilian Atlantic Forest, and that a single vicariance scenario might not be sufficient to describe the dynamics underlying the diversification of this biome (Batalha-Filho et al. 2012; Raposo do Amaral et al. 2016). This conclusion also supports a recent study in the Amazon, in which species showed idiosyncratic responses to vicariance that related more to their ecology and dispersal abilities than to the timing of vicariant events (Smith et al. 2014).
Regarding the ENMs, four of five ant species experienced large shifts in areas of suitability between the present, the Last Glacial Maximum, and the last interglacial period. A substantial fraction of suitable habitat was located where the Atlantic Ocean is today, similar to what was hypothesized by Cabanne et al. (2016). In the case of Octostruma rugifera, no suitable habitat was recovered for the southern Brazilian Atlantic Forest, which might explain the genetic result of shallowest divergences in this area. In contrast, the congener Octostruma stenognatha also showed shallow divergence, but niche results supported a stable suitability area through the LGM. The highly divergent lineages found in Tapiraí, which was independently uncovered for two species in different genera, suggest unique conditions in this area that deserves to be investigated in more detail in future studies.
Our ENMs and genetic results agree with a previously proposed northern refugium (Carnaval et al. 2009; Thomé et al. 2010) and recognize patterns found in another ant study that shows evidence for very small remnants of forest acting as small putative refugia for the maintenance of diversity (Resende et al. 2010). The finding of a northern distinct clade in our comparative phylogeographic analyses shows that the Bahia and Pernambuco areas are indeed consistent candidates to refugia, as previously proposed (Carnaval and Moritz 2008; Carnaval et al. 2009; Carnaval et al. 2014). It is significant to point out that we can see a parallel between the Quaternary climatic suitability intensity and the level of interspecific differentiation, and how even strong contractions in the Northeastern Brazilian Atlantic Forest were still able to act as refugia. Some authors proposed that glaciation/refugia had minor effects in Brazilian Atlantic Forest, including the proposition of a new “Atlantis Forest hypothesis,” meaning a suitable habitat on the emerged continental shelf (Leite et al. 2016a), and that the fragmentation had small influence on fluctuations in the size of forested areas (Thomé et al. 2014). Although the solely “Atlantis” proposition was heavily criticized (Raposo do Amaral et al. 2016; see also Leite et al. 2016b), both articles agree on the need for widely sampled genomes of a variety of taxa to build a solid understanding about the history of the Brazilian Atlantic Forest.
We did not find a strong genetic structure in nuclear DNA from samples in the southern Brazilian Atlantic Forest region, a pattern also reported by Batalha-Filho and Miyaki (2016) in birds and with a subtropical spider (Peres et al. 2015). Although these results could be explained by a delay in the coalescence of nuclear DNA in relation to mitochondrial genes (Hare 2001) or by a recent divergence leading to incomplete lineage sorting (Maddison et al. 2006), these scenarios are starting to become less possible as a weaker genetic structure in the southern Brazilian Atlantic Forest compared to the northern regions is starting to consistently be found among different taxa. In our case, we can also hypothesize that gene flow was possible because of the ant reproductive biology, given that in general winged males and females fly to a nuptial encounter and, after the mating, the juvenile queen can be carried away by the winds before the landing to establish a new colony (Baer, 2011; Cardoso et al. 2015; Cristiano et al. 2016), but this feature is not exclusive to the southern lineages.
Finally, our results underscore the potential of using UCEs as a valuable source of genetic data for shallower timescales (Smith et al. 2013), especially in ants where they have previously been used mostly for deeper timescales (Blaimer et al. 2015). There is increasing evidence that more genetic markers lead to more accurate inferences about phylogenetic and demographic history, but for ants, there have been a limited number of available loci for Sanger sequencing (but see Ströher et al. 2013). For instance, a recent phylogeographic study failed to find nuclear divergences among populations of the sand dune ant Mycetophylax simplex, and nuclear loci could not be used in the analyses (Cardoso et al. 2015). UCEs, meanwhile, allow for a large and orthologous set of nuclear loci to be captured across diverse species within families, which should provide opportunities for more comparative phylogenetic studies (Smith et al. 2014).
Conclusion
As we reach a decade since the first large-scale phylogeography studies for the Brazilian Atlantic Forest were published, it is becoming clear that the natural history of this biome is complex. Vicariance events have been given prominence in prior hypotheses about diversification but, as our results suggest, appear to be only part of the story. Niche modeling combined with genetic data across five ant species suggests that recolonization does not always explain shallow southern divergences due to low habitat suitability during glacial periods, as some species are predicted to have high suitability in the south at the LGM. Our phylogeographic study also highlights how idiosyncratic patterns are common and not an exception in the explanation to the history of diversification at the Brazilian Atlantic Forest. Finally, our results also underscore the utility of UCEs for carrying out comparative phylogeography across evolutionarily divergent taxa.
References
Aiello-Lammens, M., Boria, R., Radosavljevic, A., Vilela, B., & Anderson, R. (2015). spThin: an R package for spatial thinning of species occurrence records for use in ecological niche models. Ecography, 38, 541–545. https://doi.org/10.1111/ecog.01132.
Álvarez-Presas, M., Sánchez-Gracia, A., Carbayo, F., Rozas, J., & Riutort, M. (2014). Insights into the origin and distribution of biodiversity in the Brazilian Atlantic Forest hot spot: a statistical phylogeographic study using a low-dispersal organism. Heredity, 112, 656–665. https://doi.org/10.1038/hdy.2014.3.
Amaro, R., Rodrigues, M., Yonenaga-Yassuda, Y., & Carnaval, A. (2012). Demographic processes in the montane Atlantic Rainforest: molecular and cytogenetic evidence from the endemic frog Proceratophrys boiei. Molecular Phylogenetics and Evolution, 62, 880–888. https://doi.org/10.1016/j.ympev.2011.11.004.
Andrews, S. (2010). FastQC—a quality control tool for high throughput sequence data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc
Baer, B. (2011). The copulation biology of ants (Hymenoptera: Formicidae). Myrmecological News, 14, 55–58.
Barve, N., & Barve, V. (2016). ENMGadgets: tools for pre and post processing in ENM workflow. R package version 0.0.12. https://github.com/narayanibarve/ENMGadgets. (accessed August 15, 2017).
Barve, N., Barve, V., Jiménez-Valverde, A., Lira-Noriega, A., Maher, S., Peterson, A., Soberón, J., & Villalobos, F. (2011). The crucial role of the accessible area in ecological niche modeling and species distribution modeling. Ecological Modelling, 222, 1810–1819. https://doi.org/10.1016/j.ecolmodel.2011.02.011.
Batalha-Filho, H., & Miyaki, C. (2016). Late Pleistocene divergence and postglacial expansion in the Brazilian Atlantic Forest: multilocus phylogeography of Rhopias gularis (Aves: Passeriformes). Journal of Zoological Systematics and Evolutionary Research, 54, 137–147. https://doi.org/10.1111/jzs.12118.
Batalha-Filho, H., Cabanne, G., & Miyaki, C. (2012). Phylogeography of an Atlantic Forest passerine reveals demographic stability through the Last Glacial Maximum. Molecular Phylogenetics and Evolution, 65, 892–902. https://doi.org/10.1016/j.ympev.2012.08.010.
Bernatchez, L., & Wilson, C. (1998). Comparative phylogeography of Nearctic and Palearctic fishes. Molecular Ecology, 7, 431–452. https://doi.org/10.1046/j.1365-294x.1998.00319.x.
Blaimer, B., Brady, S., Schultz, T., Lloyd, M., Fisher, B., & Ward, P. (2015). Phylogenomic methods outperform traditional multi-locus approaches in resolving deep evolutionary history: a case study of formicine ants. BMC Evolutionary Biology, 15, 271. https://doi.org/10.1186/s12862-015-0552-5.
Bolton, B. 2019. An online catalog of the ants of the world. Available from http://antcat.org. (accessed March 14, 2019).
Bonaccorso, E., Koch, I., & Townsend, P. A. (2006). Pleistocene fragmentation of Amazon species’ ranges. Diversity and Distributions, 12, 157–164. https://doi.org/10.1111/j.1366-9516.2005.00212.x.
Bragagnolo, C., Pinto-da-Rocha, R., Antunes, M., & Clouse, R. (2015). Phylogenetics and phylogeography of a long-legged harvestman (Arachnida: Opiliones) in the Brazilian Atlantic Rain Forest reveals poor dispersal, low diversity and extensive mitochondrial introgression. Invertebrate Systematics, 29, 386. https://doi.org/10.1071/IS15009.
Brown, W. L., Jr.; Kempf, W. W. (1960). A world revision of the ant tribe Basicerotini. Studia Entomologica (n.s.)3:161–250.
Cabanne, G., Calderón, L., Trujillo, A. N., Flores, P., Pessoa, R., d’Horta, F., & Miyaki, C. (2016). Effects of Pleistocene climate changes on species ranges and evolutionary processes in the Neotropical Atlantic Forest. Biological Journal of the Linnean Society, 119, 856–872. https://doi.org/10.1111/bij.12844.
Cardoso, D., Cristiano, M., Tavares, M., Schubart, C., & Heinze, J. (2015). Phylogeography of the sand dune ant Mycetophylax simplex along the Brazilian Atlantic Forest coast: remarkably low mtDNA diversity and shallow population structure. BMC Evolutionary Biology, 15, 106. https://doi.org/10.1186/s12862-015-0383-4.
Carnaval, A., & Moritz, C. (2008). Historical climate modelling predicts patterns of current biodiversity in the Brazilian Atlantic Forest. Journal of Biogeography, 35, 1187–1201. https://doi.org/10.1111/j.1365-2699.2007.01870.x.
Carnaval, A., Hickerson, M., Haddad, C., Rodrigues, M., & Moritz, C. (2009). Stability predicts genetic diversity in the Brazilian Atlantic Forest hotspot. Science, 323, 785–789. https://doi.org/10.1126/science.1166955.
Carnaval, A., Waltari, E., Rodrigues, M., Rosauer, D., VanDerWal, J., Damasceno, R., et al. (2014). Prediction of phylogeographic endemism in an environmentally complex biome. Proceedings of the Royal Society B: Biological Sciences, 281, 20141461–20141461. https://doi.org/10.1098/rspb.2014.1461.
Costa, L. (2003). The historical bridge between the Amazon and the Atlantic Forest of Brazil: a study of molecular phylogeography with small mammals. Journal of Biogeography, 30, 71–86. https://doi.org/10.1046/j.1365-2699.2003.00792.x.
Costa, L., Leite, Y., da Fonseca, G., & da Fonseca, M. (2000). Biogeography of South American forest mammals: endemism and diversity in the Atlantic Forest. Biotropica, 32, 872–881. https://doi.org/10.1111/j.1744-7429.2000.tb00625.x.
Cristiano, M. P., Cardoso, C. D., Fernandes-Salomão, T. M., & Heinze, J. (2016). Integrating paleodistribution models and phylogeography in the grass-cutting ant Acromyrmex striatus (Hymenoptera: Formicidae) in southern lowlands of South America. PLoS One, 11(1), e0146734. https://doi.org/10.1371/journal.pone.0146734.
Darwin, C., & Keynes, R. (2004). Charles Darwin’s “Beagle” diary. Cambridge: Cambridge University Press.
Drummond, A., Suchard, M., Xie, D., & Rambaut, A. (2012). Bayesian phylogenetics with BEAUti and the BEAST 1.7. Molecular Biology and Evolution, 29, 1969–1973. https://doi.org/10.1093/molbev/mss075.
Elith, J., Graham, H. C., Anderson, P. R., Dudík, M., Ferrier, S., Guisan, A., et al. (2006). Novel methods improve prediction of species’ distributions from occurrence data. Ecography, 29, 129–151. https://doi.org/10.1111/j.2006.0906-7590.04596.x.
Faircloth, B. (2013). Illumiprocessor: a trimmomatic wrapper for parallel adapter and quality trimming. https://doi.org/10.6079/J9ILL; https://illumiprocessor.readthedocs.io/en/latest/citing.html. (accessed April 16, 2015).
Faircloth, B. (2015). PHYLUCE is a software package for the analysis of conserved genomic loci. Bioinformatics, 32, 786–788. https://doi.org/10.1093/bioinformatics/btv646.
Faircloth, B., & Glenn, T. (2012). Not all sequence tags are created equal: designing and validating sequence identification tags robust to indels. PLoS One, 7, e42543. https://doi.org/10.1371/journal.pone.0042543.
Faircloth, B., Branstetter, M., White, N., & Brady, S. (2015). Target enrichment of ultraconserved elements from arthropods provides a genomic perspective on relationships among Hymenoptera. Molecular Ecology Resources, 15, 489–501. https://doi.org/10.1111/1755-0998.12328.
Fick, S., & Hijmans, R. (2017). WorldClim 2: new 1-km spatial resolution climate surfaces for global land areas. International Journal of Climatology, 37, 4302–4315. https://doi.org/10.1002/joc.5086.
Fisher, S., Barry, A., Abreu, J., Minie, B., Nolan, J., Delorey, T., Young, G., et al. (2011). A scalable, fully automated process for construction of sequence-ready human exome targeted capture libraries. Genome Biology, 12, R1. https://doi.org/10.1186/gb-2011-12-1-r1.
Forel, A. (1905). Miscellanea myrmécologiques II (1905). Annales de la Société Entomologique de Belgique 49:155–185.
Garrick, R., Sands, C., Rowell, D., Tait, N., Greenslade, P., & Sunnucks, P. (2004). Phylogeography recapitulates topography: very fine-scale local endemism of a saproxylic ‘giant’ springtail at Tallaganda in the great dividing range of south-east Australia. Molecular Ecology, 13, 3329–3344. https://doi.org/10.1111/j.1365-294X.2004.02340.x.
Giraudo, A., Matteucci, S., Alonso, J., Herrera, J., & Abramson, R. (2008). Comparing bird assemblages in large and small fragments of the Atlantic Forest hotspots. Biodiversity and Conservation, 17, 1251–1265. https://doi.org/10.1007/s10531-007-9309-9.
Glenn, T., Nilsen, R., Kieran, T., Finger, J., Pierson, T., Bentley, K., et al. (2016). Adapterama I: universal stubs and primers for thousands of dual-indexed Illumina libraries (iTru and iNext). bioRxiv. https://doi.org/10.1101/049114.
Gnirke, A., Melnikov, A., Maguire, J., Rogov, P., LeProust, E., Brockman, W., et al. (2009). Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nature Biotechnology, 27, 182–189. https://doi.org/10.1038/nbt.1523.
Grabherr, M., Haas, B., Yassour, M., Levin, J., Thompson, D., Amit, I., & Regev, A. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnology, 29, 644–652. https://doi.org/10.1038/nbt.1883.
Grazziotin, F., Monzel, M., Echeverrigaray, S., & Bonatto, S. (2006). Phylogeography of the Bothrops jararaca complex (Serpentes: Viperidae): past fragmentation and island colonization in the Brazilian Atlantic Forest. Molecular Ecology, 15, 3969–3982. https://doi.org/10.1111/j.1365-294X.2006.03057.x.
Haffer, J. (1969). Speciation in amazonian forest birds. Science, 165, 131–137. https://doi.org/10.1126/science.165.3889.131.
Haffer, J. (1997). Alternative models of vertebrate speciation in Amazonia: an overview. Biodiversity and Conservation, 6, 451–476. https://doi.org/10.1023/A:1018320925954.
Hare, M. (2001). Prospects for nuclear gene phylogeography. Trends in Ecology and Evolution, 16, 700–706. https://doi.org/10.1016/S0169-5347(01)02326-6.
Hayes, F., & Sewlal, J. (2004). The Amazon River as a dispersal barrier to passerine birds: effects of river width, habitat and taxonomy. Journal of Biogeography, 31, 1809–1818. https://doi.org/10.1111/j.1365-2699.2004.01139.x.
Hijmans, R., Cameron, S., Parra, J., Jones, P., & Jarvis, A. (2005). Very high resolution interpolated climate surfaces for global land areas. International Journal of Climatology, 25, 1965–1978.
Hölldobler, B., & Wilson, E. (1990). The ants. Cambridge: Belknap Press of Harvard University Press.
Katoh, K., & Standley, D. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution, 30, 772–780. https://doi.org/10.1093/molbev/mst010.
Lara, M., & Patton, J. (2000). Evolutionary diversification of spiny rats (genus Trinomys, Rodentia: Echimyidae) in the Atlantic Forest of Brazil. Zoological Journal of the Linnean Society, 130, 661–686. https://doi.org/10.1111/j.1096-3642.2000.tb02205.x.
Leão, T., Fonseca, C., Peres, C., & Tabarelli, M. (2014). Predicting extinction risk of Brazilian Atlantic Forest angiosperms. Conservation Biology, 28, 1349–1359. https://doi.org/10.1111/cobi.12286.
Leite, Y., Costa, L., Loss, A., Rocha, R., Batalha-Filho, H., Bastos, A., et al. (2016a). Neotropical forest expansion during the last glacial period challenges refuge hypothesis. Proceedings of the National Academy of Sciences, 113, 1008–1013. https://doi.org/10.1073/pnas.1513062113.
Leite, Y., Costa, L., Loss, A., Rocha, R., Batalha-Filho, H., Bastos, A., et al. (2016b). Reply to Raposo do Amaral et al.: The “Atlantis Forest hypothesis” adds a new dimension to Atlantic Forest biogeography. Proceedings of the National Academy of Sciences, 113, E2099–E2100. https://doi.org/10.1073/pnas.1602391113.
Leppänen, J., Vepsäläinen, K., & Savolainen, R. (2011). Phylogeography of the ant Myrmica rubra and its inquiline social parasite. Ecology and Evolution, 1, 46–62. https://doi.org/10.1002/ece3.6.
Lohse, M., Bolger, A., Nagel, A., Fernie, A., Lunn, J., Stitt, M., & Usadel, B. (2012). RobiNA: a user-friendly, integrated software solution for RNA-Seq-based transcriptomics. Nucleic Acids Research, 40, W622–W627. https://doi.org/10.1093/nar/gks540.
Maddison, W., Knowles, L., & Collins, T. (2006). Inferring phylogeny despite incomplete lineage sorting. Systematic Biology, 55, 21–30. https://doi.org/10.1080/10635150500354928.
Martins, F., Templeton, A., Pavan, A., Kohlbach, B., & Morgante, J. (2009). Phylogeography of the common vampire bat (Desmodus rotundus): arked population structure, neotropical pleistocene vicariance and incongruence between nuclear and mtDNA markers. BMC Evolutionary Biology, 9, 294. https://doi.org/10.1186/1471-2148-9-294.
Mayr, G. 1887. Südamerikanische Formiciden. Verhandlungen der Kaiserlich-Königlichen Zoologisch-Botanischen Gesellschaft in Wien 37:511–632
Merow, C., Smith, M., & Silander, J. (2013). A practical guide to MaxEnt for modeling species’ distributions: what it does, and why inputs and settings matter. Ecography, 36, 1058–1069. https://doi.org/10.1111/j.1600-0587.2013.07872.x.
Morales, N., Fernández, I., Carrasco, B., & Orchard, C. (2015). Combining niche modelling, land-use change, and genetic information to assess the conservation status of Pouteria splendens populations in Central Chile. International Journal of Ecology, 2015, 1–12. https://doi.org/10.1155/2015/612194.
Murray-Smith, C., Brummitt, N., Oliveira-Filho, A., Bachman, S., Moat, J., Lughadha, E., & Lucas, E. (2009). Plant diversity hotspots in the Atlantic coastal forests of Brazil. Conservation Biology, 23, 151–163. https://doi.org/10.1111/j.1523-1739.2008.01075.x.
Myers, N., Mittermeier, R., Mittermeier, C., da Fonseca, G., & Kent, J. (2000). Biodiversity hotspots for conservation priorities. Nature, 403, 853–858. https://doi.org/10.1038/35002501.
Nicolas, V., Martínez-Vargas, J., & Hugot, J. (2016). Molecular data and ecological niche modelling reveal the evolutionary history of the common and Iberian moles (Talpidae) in Europe. Zoologica Scripta, 46, 12–26. https://doi.org/10.1111/zsc.12189.
Oliveira, U., Vasconcelos, M., & Santos, A. (2017). Biogeography of Amazon birds: rivers limit species composition, but not areas of endemism. Scientific Reports, 7, 2992. https://doi.org/10.1038/s41598-017-03098-w.
Pearson, R., Raxworthy, C., Nakamura, M., & Townsend, P. A. (2006). Predicting species distributions from small numbers of occurrence records: a test case using cryptic geckos in Madagascar. Journal of Biogeography, 34, 102–117. https://doi.org/10.1111/j.1365-2699.2006.01594.x.
Pellegrino, K., Rodrigues, M., Waite, A., Morando, M., Yassuda, Y., & Sites, J. (2005). Phylogeography and species limits in the Gymnodactylus darwinii complex (Gekkonidae, Squamata): genetic structure coincides with river systems in the Brazilian Atlantic Forest. Biological Journal of the Linnean Society, 85, 13–26. https://doi.org/10.1111/j.1095-8312.2005.00472.x.
Peres, E. A., Sobral-Souza, T., Perez, M. F., Bonatelli, I. A. S., Silva, D. P., Silva, M. J., & Solferini, V. N. (2015). Pleistocene niche stability and lineage diversification in the subtropical spider Araneus omnicolor (Araneidae). PLoS One, 10(4), e0121543. https://doi.org/10.1371/journal.pone.0121543.
Peterson, A., Soberón, J., Pearson, R., Anderson, R., Nakamura, M., Martinez-Meyer, E., & Araújo, M. (2011). Ecological niches and geographical distributions. Princeton: Princeton University Press.
Phillips, S., Anderson, R., Dudík, M., Schapire, R., & Blair, M. (2017). Opening the black box: an open-source release of Maxent. Ecography, 40, 887–893. https://doi.org/10.1111/ecog.03049.
Prates, I., Xue, A., Brown, J., Alvarado-Serrano, D., Rodrigues, M., Hickerson, M., & Carnaval, A. (2016). Inferring responses to climate dynamics from historical demography in neotropical forest lizards. Proceedings of the National Academy of Sciences, 113, 7978–7985. https://doi.org/10.1073/pnas.1601063113.
Quek, S., Davies, S., Ashton, P., Itino, T., & Pierce, N. (2007). The geography of diversification in mutualistic ants: a gene’s-eye view into the neogene history of Sundaland rain forests. Molecular Ecology, 16, 2045–2062. https://doi.org/10.1111/j.1365-294X.2007.03294.x.
Raposo do Amaral, F., Edwards, S., Pie, M., Jennings, W., Svensson-Coelho, M., d’Horta, F., Schmitt, C., & Maldonado-Coelho, M. (2016). The “Atlantis Forest hypothesis” does not explain Atlantic Forest phylogeography. Proceedings of the National Academy of Sciences, 113, E2097–E2098. https://doi.org/10.1073/pnas.1602213113.
Resende, H., Yotoko, K., Delabie, J., Costa, M., Campiolo, S., Tavares, M., Campos, L., & Fernandes-Salomão, T. (2010). Pliocene and Pleistocene events shaping the genetic diversity within the central corridor of the Brazilian Atlantic Forest. Biological Journal of the Linnean Society, 101, 949–960. https://doi.org/10.1111/j.1095-8312.2010.01534.x.
Ribeiro, M., Metzger, J., Martensen, A., Ponzoni, F., & Hirota, M. (2009). The Brazilian Atlantic Forest: how much is left, and how is the remaining forest distributed? Implications for conservation. Biological Conservation, 142, 1141–1153. https://doi.org/10.1016/j.biocon.2009.02.021.
Ribeiro, R., Lemos-Filho, J., Ramos, A., & Lovato, M. (2010). Phylogeography of the endangered rosewood Dalbergia nigra (Fabaceae): insights into the evolutionary history and conservation of the Brazilian Atlantic Forest. Heredity, 106, 46–57. https://doi.org/10.1038/hdy.2010.64.
Rohland, N., & Reich, D. (2012). Cost-effective, high-throughput DNA sequencing libraries for multiplexed target capture. Genome Research, 22, 939–946. https://doi.org/10.1101/gr.128124.111.
Seal, J., Brown, L., Ontiveros, C., Thiebaud, J., & Mueller, U. (2015). Gone to Texas: phylogeography of two Trachymyrmex (Hymenoptera: Formicidae) species along the southeastern coastal plain of North America. Biological Journal of the Linnean Society, 114, 689–698. https://doi.org/10.1111/bij.12426.
Silva, J., & Casteleti, C. (2003). Status of the biodiversity of the Atlantic Forest of Brazil. The Atlantic Forest of South America: biodiversity status, threats, and outlook. (ed. by C. Galindo-Leal and I. Câmara), pp. 43–59. CABS and Island Press, Washington.
Silva, J., Cardoso de Sousa, M., & Castelletti, C. (2004). Areas of endemism for passerine birds in the Atlantic Forest, South America. Global Ecology and Biogeography, 13, 85–92. https://doi.org/10.1111/j.1466-882X.2004.00077.x.
Silva, J., Rylands, A., & da Fonseca, G. (2005). The fate of the amazonian areas of endemism. Conservation Biology, 19, 689–694. https://doi.org/10.1111/j.1523-1739.2005.00705.x.
Simpson, B. (1979). Quaternary biogeography of the high montane regions of South America. The south American Herpetofauna: its origin, evolution, and dispersal (ed. by W. Duellman), pp. 157–188. Monograph of the Museum of Natural History, University of Kansas.
Smith, B., Harvey, M., Faircloth, B., Glenn, T., & Brumfield, R. (2013). Target capture and massively parallel sequencing of ultraconserved elements for comparative studies at shallow evolutionary time scales. Systematic Biology, 63, 83–95. https://doi.org/10.1093/sysbio/syt061.
Smith, B., McCormack, J., Cuervo, A., Hickerson, M., Aleixo, A., Cadena, C., et al. (2014). The drivers of tropical speciation. Nature, 515, 406–409. https://doi.org/10.1038/nature13687.
Solomon, S., Bacci, M., Martins, J., Vinha, G., & Mueller, U. (2008). Paleodistributions and comparative molecular phylogeography of leafcutter ants (Atta spp.) provide new insight into the origins of amazonian diversity. PLoS One, 3, e2738. https://doi.org/10.1371/journal.pone.0002738.
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics, 30, 1312–1313. https://doi.org/10.1093/bioinformatics/btu033.
Ströher, P. R., Li, C., & Pie, M. (2013). Exon-primed intron-crossing (EPIC) markers as a tool for ant phylogeography. Revista Brasileira de Entomologia, 57, 427–430. https://doi.org/10.1590/S0085-56262013005000039.
Tchaicka, L., Eizirik, E., De Oliveira, T., Cândido, J., & Freitas, T. (2006). Phylogeography and population history of the crab-eating fox (Cerdocyon thous). Molecular Ecology, 16, 819–838. https://doi.org/10.1111/j.1365-294X.2006.03185.x.
Thomé, M., Zamudio, K., Giovanelli, J., Haddad, C., Baldissera, F., & Alexandrino, J. (2010). Phylogeography of endemic toads and post-Pliocene persistence of the Brazilian Atlantic Forest. Molecular Phylogenetics and Evolution, 55, 1018–1031. https://doi.org/10.1016/j.ympev.2010.02.003.
Thomé, M., Zamudio, K., Haddad, C., & Alexandrino, J. (2014). Barriers, rather than refugia, underlie the origin of diversity in toads endemic to the Brazilian Atlantic Forest. Molecular Ecology, 23, 6152–6164. https://doi.org/10.1111/mec.12986.
Vanzolini, P. (1992). Paleoclimas e especiação em animais da América do Sul tropical. Estudos Avançados, 6, 41–65. https://doi.org/10.1590/S0103-40141992000200003.
Vanzolini, P., & Williams, E. (1981). Vanishing refuge: a mechanism for ecogeographic speciation. Papéis Avulsos de Zoologia, 34, 251–255.
Wenger, S., & Olden, J. (2012). Assessing transferability of ecological models: an underappreciated aspect of statistical validation. Methods in Ecology and Evolution, 3, 260–267. https://doi.org/10.1111/j.2041-210X.2011.00170.x.
Zamudio, K., & Greene, H. (1997). Phylogeography of the bushmaster (Lachesis muta: Viperidae): implications for neotropical biogeography, systematics, and conservation. Biological Journal of the Linnean Society, 62, 421–442. https://doi.org/10.1111/j.1095-8312.1997.tb01634.x.
Zwiener, V., Padial, A., Marques, M., Faleiro, F., Loyola, R., & Peterson, A. (2017). Planning for conservation and restoration under climate and land use change in the Brazilian Atlantic Forest. Diversity and Distributions, 23, 955–966. https://doi.org/10.1111/ddi.12588.
Acknowledgements
We thank Rogerio R. Silva for providing the specimens used in this project and Rodrigo M. Feitosa for the taxonomic advice. We are grateful to the two anonymous reviewers for their helpful comments that greatly improved the manuscript. We also would like to thank Eduardo A. B. de Almeida for the international and financial logistics support.
Funding
This study was funded by a grant from the Conselho Nacional de Desenvolvimento Científico e Tecnológico (301636/2016-8) to MRP. This study was also funded by scholarships from the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) to Patrícia Regina Ströher (Processo: PDSE 99999.002880/2014-08) and from the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (140262/2013-0).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 15 kb)
Rights and permissions
About this article

Cite this article
Ströher, P.R., Meyer, A.L.S., Zarza, E. et al. Phylogeography of ants from the Brazilian Atlantic Forest. Org Divers Evol 19, 435–445 (2019). https://doi.org/10.1007/s13127-019-00409-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13127-019-00409-z