
Abstract
Intimate and long-lasting relationships of fungi and algae have been known for centuries by scientists, and these ancient symbioses might have provided excellent opportunities for horizontal gene transfer (HGT) of protein encoding genes between the two organismal partners. In this study, we sequenced and assembled 451 Mbp of novel genomic DNA from Trebouxia decolorans (Trebouxiaceae, Chlorophyta), the green algal photobiont of the lichen Xanthoria parietina (Teloschistaceae, Lecanoromycetes, Ascomycota). This alga also occurs as a free-living terrestrial organism. The aim of our work was to search for candidate genes pointing to HGT between lichenized fungi and lichen algae. We found evidence for three putative HGT events of fungal genes into the Trebouxia genome, but these are likely more ancient (over 600 mya) than the origin of lichenization within the fungal Ascomycetes. The three transferred genes are part of gene groups that in other species encode a tellurite-resistance dicarboxylate transporter (TDT) family protein, a class-1 nitrilase/cyanide hydratase (CH), and an oxidoreductase/retinol dehydrogenase. In each case, our phylogenomic analyses show orthologs from Trebouxia as sister to orthologs from all fungi or basally placed within Ascomycetes, while the orthologs from green algae and land plants form separate, independent evolutionary lineages. Alternative hypothesis tests significantly support these HGT events. The presence of these genes in Trebouxia was validated by PCR amplification of separately isolated Trebouxia DNA. The ancient incorporation of fungal genes in the genomes of these particular green algae are intriguing and could be early evidence for symbiotic and co-evolutionary relationships among the major eukaryotic branches of algae and fungi present in early terrestrial life on Earth. These genes could have played a pre-disposition role for some fungi and algae in the origin of lichen symbiosis, but further studies are needed to evaluate this in detail.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Horizontal gene transfer (HGT) involves the transfer of genetic material from one organism to another, belonging to separate evolutionary lineages (Andersson 2005; Keeling and Palmer 2008). HGT has played an important role in the evolution and diversification of prokaryotes (Doolittle et al. 2003; Gogarten et al. 2002); however, the extent of HGT in the macroevolution of eukaryotes remains unclear (see Andersson 2005; Keeling and Palmer 2008). Recent genomic studies have shown HGT between fungi, bacteria and fungi, and also between fungi and plants (Fitzpatrick 2012; Marcet-Houben and Gabaldon 2010; Richards et al. 2009; del Campo et al. 2009; Slot and Hibbett 2007). HGT has also occurred between (micro-)organisms with close associations such as hosts and their parasites and microbial prey and their predators (Loftus et al. 2005; Richardson and Palmer 2007). Even if the number of known eukaryotic HGT events (including fungi) is small, these events might have had considerable impact on functional innovations and adaptation to specific ecological niches, e.g., the ability to grow under anaerobic conditions or in the rumen of herbivorous mammals or have importance for the use of fungi by humans like in wine fermentation (see Fitzpatrick 2012 and references therein). For example, carotenoid synthesis in pea aphids, green peach aphids, and the two-spotted spider mite is feasible owing to HGT of carotenoid biosynthesis genes from fungi into the arthropod genomes (Altincicek et al. 2011; Moran and Jarvik 2010). HGT also facilitated the evolution of plant parasitic mechanisms in Oomycetes (Richards et al. 2011), and the HGT of catalase genes into the fungal pathogenic species Nosema locustae, Stagonospora nodorum, Mycosphaerella fijiensis and Botrytis cinerea helped to overcome host defense mechanisms (Marcet-Houben and Gabaldon 2010). Similarly, polyketide synthase genes are crucial in the production of a large portion of secondary lichen substances and are a result of HGT between bacteria and lichen fungi (Schmitt and Lumbsch 2009). The possibility of DNA transfer between lichen mycobionts and photobionts has been suggested previously (Ahmadjian et al. 1991), but so far no evidence has been presented for this. Among lichen algal photobionts, HGT of group I introns between species of Trebouxia has been reported (Friedl et al. 2000), and explained by the occurrence of viruses which may play a role in facilitating intron mobility (Friedl and Bhattacharya 2002).
Trebouxia is the most common green algal lichen photobiont (Friedl and Büdel 2008; Tschermak-Woess 1988). They have their largest populations when living in lichen symbiotic systems, but at least some species are known to occur free-living (Beck et al. 1998; Bubrick et al. 1984; Tschermak-Woess 1978), but note the questioning of this fact by Ahmadjian (1988). One of these non-obligate lichen photobionts is Trebouxia decolorans, the symbiont of Xanthoria parietina, and was used in this study. Trebouxia is type genus of the green algal class Trebouxiophyceae, a family forming the core of the Chlorophyta together with the mainly marine Ulvophyceae and fresh-water Chlorophyceae. The chlorophytes are sister to the Streptophyta, which includes the land plants, and together they form the green plant lineage of the eukaryotes (Leliaert et al. 2012).
The stem lineage of the green plants is estimated to have originated around 1–1.5 billion years ago (Yoon et al. 2004), and this is supported by fossil evidence of its sister group, the red algae, from the same time frame (Knoll 2014). Core chlorophytes evolved from marine planktonic ancestors, and estimates show their divergence from other green algae around 700–900 million years ago (mya) (Leliaert et al. 2011, 2012). Therefore, the Trebouxiophyceae, despite highly nested within the green plant clade, is likely a very ancient, pre-Cambrian group. The origin of the fungal stem lineage has a similar age estimate as the green plant lineage (i.e., about 1 billion years ago) and within the fungi, the two phyla Ascomycota and Basidiomycota are estimated to have diverged around 650 mya in the Proterozoic (Lücking et al. 2009). Lichen fungi are mostly members of the Ascomycota, and they do not form a monophyletic clade, i.e., lichen symbiosis has evolved several times independently within the fungi, but large non-symbiotic fungal groups have evolved from lichen-forming ancestors as well (Lutzoni et al. 2001). Dating of the ages of Ascomycota lineages using molecular clock methods has been problematic due to the lack of satisfactory fossils that can be used as calibration points of phylogenetic branches. Still, recent studies suggest an initial diversification of ascomycetes in the Ordovician (around 460 mya), followed by additional divergences later throughout the Phanerozoic (Beimforde et al. 2014; Prieto and Wedin 2013). The divergence of Lecanoromycetes (containing the majority of lichen fungi) from the Eurotiomycetes is estimated to early Carboniferous (about 350 mya; Beimforde et al. 2014). Geologic data indicates very ancient fungal-algal relationships, through fossils from as early as the Proterozoic Ediacaran geologic period from southern China (about 600 mya) and the Lower Devonian Rhynie chert in Scotland (about 400 mya old; Honegger et al. 2013; Taylor et al. 1995, 1997; Yuan et al. 2005). In these fossils, fungal hyphae have invaded colonies of coccoid cyanobacteria or unicellular algae, thereby establishing an interface between fungal and algal cells that provided a possible venue for exchange of genetic material between eukaryotic organisms.
Fungi often form conjoint relationships with algae by developing microbiotic soil crusts (Belnap et al. 2001) or biofilms (Lappin-Scott and Costerton 2003). Some of these close associations have resulted in the formation of symbiotic organismic entities called lichens, a stable cohabitation of at least one fungal (mycobiont) and one algal or cyanobacterial (photobiont) organism and often including additional endolichenic fungi and bacteria, all together forming self-contained miniature ecosystems (Grube et al. 2012; Honegger 2012; Nash 2008). Lichens are extremely successful and represented in almost all terrestrial habitats from tropical to polar regions and from sea shores to high altitudes (Seaward 2008). The basis of the lichen symbiosis is still unclear and viewpoints range from mutualism to parasitism (e.g., Ahmadjian 1993; Grube and Hawksworth 2007; Molina et al. 1993; Peveling 1988). More than a hundred years after the discovery of the symbiotic nature of lichens by Schwendener (1867), many basic questions regarding their biology remain unanswered. One of these is how the sophisticated interplay between myco- and photobiont is achieved in order to produce intricate and morphologically different structures such as the leafy or fruticose lichens. Genetic control (e.g., protein signaling) seems necessary, because extracellular communication without physical cellular contact has been shown to occur between lichen symbionts (Bubrick et al. 1985; Molina et al. 1998; Sing and Walia 2014). Moreover, interaction within a lichen symbiosis system is accompanied by changes in gene regulation and expression in both the photobiont and mycobiont (Joneson et al. 2011; Trembley et al. 2002). Of particular interest are lichenized algae, which provide carbohydrate to the fungus (e.g. Eisenreich et al. 2011, Nash 2008) and therefore could be heavily influenced by fungal-derived genes to facilitate this process and thereby enhance the symbiosis. We, therefore, purposefully selected an alga from a lichen symbiotic system to investigate HGT.
In lichens, the fungal species is always unique to each species, but the algal component can often be found in several lichen species, i.e., the alga does not show strict coevolutionary host-specificity, and as mentioned earlier, can sometimes survive without a fungus (Bubrick et al. 1984; Tschermak-Woess 1978). Our photobiont study organism, T. decolorans Ahmadjian, is a green algae isolated from the lichen X. parietina (L.) Beltr., a member of Teloschistaceae (Lecanoromycetes, Ascomycota). T. decolorans has also been reported to be the photobiont of lichens from several other fungal families, such as Lecidiaceae, Parmeliaceae, and Physciaceae (Beck et al. 1998; Beck and Mayr 2012; Friedl et al. 2000; Helms et al. 2001).
For all genes, three possible scenarios were considered (1) no evidence of HGT, (2) evidence of relatively recent transfer from fungi to algae, or (3) evidence of ancient transfer from fungi to green plants. Given the relatively large extent of HGT also in eukaryotes, we expected to find at least some HGT from lichenized fungi or their immediate ancestors to the photobiont algae (Trebouxia; scenario 2) and that this would be found when genomic data from photobiont algae were sequenced and compared with a wide array of eukaryotic genomes. This would be shown as an evolutionary pattern of a highly nested algal gene inside clades of lichenized fungal genes in a phylogenomic analysis. To test this hypothesis, we partially sequenced the genome of Trebouxia and compared its inferred genes with a large genomic database of many other eukaryotes, bacteria, and Archaea, and the result is presented in this study.
Materials and methods
Materials
Genomic DNA was isolated from T. decolorans, the photobiont of X. parietina (collected by A. Beck at Maising, Bavaria, Germany on 15 October 2005; Herbarium voucher: M-0102151, strain AB05.019B2). The photobiont was isolated using a micro-manipulator as described in Beck and Koop (2001) and was grown axenically in ¼ TOM medium (Ahmadjian 1967). The identity of the isolated photobiont was validated by comparing the complete internal transcribed spacer (ITS1 and ITS2) sequence, including 5.8S nrDNA, of the nuclear ribosomal (nr)DNA, from the isolate with that of homologs identified in a total DNA preparation from the thallus that was used to establish the photobiont culture. The obtained algal ITS sequences were identical and proved to be at least 99 % identical to T. decolorans sequences deposited in GenBank as verified by a megablast search. One gram of algal culture was harvested and the DNA was isolated using a FastDNA kit following the manufacturer’s protocol (MP-Biomedical, Illkirch, France). From the extracted DNA, about 10 μg of total DNA was used to construct a library (sheared DNA fragments were around 500 bp size) for 100 × 100 bp paired-end sequencing using an Illumina GAIIx in the Bhattacharya lab (Rutgers University). Standard Illumina protocols (http://www.illumina.com/) were used to generate the library.
Assembly and phylogenomic analyses
Illumina reads were assembled to contigs using the CLC Genomics Workbench (http://www.clcbio.com/). Genes were predicted on a subset of these contigs (length > = 250 bp or average coverage depth > = 5×) using Augustus (Stanke and Morgenstern 2005) under both the Arabidopsis and Aspergillus models. The data is presented on the Web and can be accessed using the URL: http://dbdata.rutgers.edu/data/Trebouxia/. Corresponding amino acid sequences were then clustered at 90 % similarity using CD-HIT (Li and Godzik 2006) and compared using a BLASTP search (e-value ≤ 10−5) with sequences from a local database comprised of the NCBI Refseq (v51) collection with additional data available from the Joint Genome Institute (http://www.jgi.doe.gov/) and other public databases (e.g., TBestDB, dbEST; for details, see Chan et al. 2011; Price et al. 2012). Phylogenomic analyses were done as described in Moustafa et al. (2009) and Yoon et al. (2011). Phylogenetic trees were calculated using RAxML v7.2.8 (Stamatakis 2006; Stamatakis et al. 2008) under the PROTGAMMAILGF model with nodal support calculated using 100 bootstrap replicates. The resulting phylogenetic trees were sorted using PhyloSort (Moustafa and Bhattacharya 2008) to identify instances of sister group relationships between the sequenced T. decolorans gene and the following groups: Archaea, Bacteria, Fungi, Viridiplantae, Virus, and ‘Other’. Three trees in which the T. decolorans sequence, but not other algal or plant sequences, was nested within clades of fungal sequences and supported by a bootstrap value ≥ 0.7 were selected for further analysis. The nucleotide sequences of these contigs of the HGT candidate genes are accessible from GenBank with the accession numbers KF573967, KF573968, and KF573969. For these three selected HGT candidates, the alignment was manually improved and restricted to all unambiguously aligned regions, hits for homologous genes from the newly available Xanthoria (http://genome.jgi-psf.org/pages/blast.jsf?db=Xanpa1), Cladonia P. Browne (http://genome.jgi.doe.gov/Clagr2/Clagr2.home.html), Asterochloris Tschermak-Woess (http://genome.jgi-psf.org/Astpho1/Astpho1.home.html), and Endocarpon Hedw. (http://www.ncbi.nlm.nih.gov/nuccore/APWS00000000) genomes were added if present and the analyses were reconducted using RAxML and MrBayes. RAxML analyses were performed on the CIPRES web portal with the same model as described above. Support values were assessed using the ‘rapid bootstrapping’ option with 1000 replicates. Bayesian analyses were conducted using MrBayes 3.2.1 (Ronquist and Huelsenbeck 2003), and the mixed prior for the amino acid model was used. The Metropolis-coupled Markov chain Monte Carlo (MC3) consisted of four independent runs of 2 million generations, starting with a random tree and employing eight simultaneous chains each, in which one in every 100 trees was sampled. The outputs of MrBayes were examined with the program Tracer v1.5 (Rambaut and Drummond 2007) to check for convergence of different parameters. Topological convergence in the four independent MCMC runs was checked with the ‘compare’ plots in the program AWTY (Nylander et al. 2008). Posterior probabilities (PPs) of clades were obtained from the 50 % majority rule consensus of sampled trees after excluding the initial 25 % of trees as burn-in.
Null hypothesis testing
In order to test if the monophyly of a clade comprising fungi + Trebouxia is significantly supported against the expected monophyly of a clade of Green plants including Trebouxia, we compared the ML tree constrained to recover Green plants + Trebouxia as monophyletic and the unconstrained ML tree obtained from the analysis mentioned in 2.2. Such a topology might be present in suboptimal trees not sampled or not present in the 50 % majority-rule consensus tree of the MCMC sampling, but may not be significantly worse than the obtained topology. For these tests, we used two different methods for all three protein data matrices (sulfite efflux pump/tellurite-resistance dicarboxylate transporter (TDT) family, Class-1 nitrilase/cyanide hydratase (CH), and oxidoreductase/retinol dehydrogenase). The first method was the Shimodaira–Hasegawa (SH) test (Shimodaira and Hasegawa 1999), and the second was the expected likelihood weight (ELW) test following Strimmer and Rambaut (2002). The SH and ELW tests were performed using Tree-PUZZLE 5.2 (Schmidt et al. 2002) with the three protein data matrices on a sample of 200 unique trees, the best trees agreeing with the null hypotheses, and the unconstrained ML tree. These trees were inferred in Tree-PUZZLE employing the same substitution model as in ML analysis.
Verification of HGT candidate genes
The three candidate genes for HGT from fungi to Trebouxia were screened for synapomorphic amino acid residues and motifs that support this direction of gene transfer. Additionally, primers for the gene were designed from the nucleotide contigs of T. decolorans: sulphite-efflux pump/tellurite-resistance dicarboxylate transporter (TDT): Con699F: CTA TGC GCA CAG CAT TGT CT Con699R: GTA CTG CAT TGA GGG GCA AT; Nitrilase: Con18734F: GCT GAT CTG CTG GTT TTT CC, Con18734R: CAG AGT CCA GGT GTG AAG CA; Oxidoreductase: Contig45971F: ACA CCT CAC AAC CAC GAC AA, Con45971R: ACC CAG CAG TGA TGG AAA AG. The gene fragments were amplified using genomic DNA from T. decolorans as well as from Trebouxia impressa (strain AB96.011X1; photobiont of the lichen Candelariella reflexa) and Dictyochloropsis reticulata (strain AB06.006A2; photobiont of the lichen Lobaria pulmonaria). The PCR reaction was as follows: 95 °C for 2 min, 5 cycles of 95 °C for 45 s, 57 °C for 45 s, 72 °C for 30 s, followed by 33 cycles of 95 °C for 45 s, 55 °C for 45 s, 72 °C for 30 s, and a final extension of 2 min at 72 °C. The PCR products obtained were sequenced and the resulting sequences were compared to the sequences identified using Illumina genome sequencing. These experiments have been conducted in the Beck lab (Botanische Staatssammlung München).
Results
Illumina sequencing from T. decolorans resulted in 4.7 million 100-bp reads and 451 Mbp of trimmed data. De novo assembly produced 200,241 contigs (N50 = 317 bp) that totalled 55.3 Mbp of genome data. A subset of contigs with length >250 bp or average coverage >5× was retained for further analysis. This set consisted of 24,655 sequences (N50 = 590 bp) totaling 13.4 Mbp and encoded 18,178 putative proteins. BLASTp analysis of these sequences returned 4098 proteins with significant (e-value ≤ 10−5) hits to 3609 unique target peptides. Most top hits were to algae and plants (Viridiplantae; 85.6 %), as expected, with significantly fewer hits to bacteria (3.5 %), fungi (2.9 %), viruses (0.3 %), and archaea (0.2 %). The remaining (others; 7.5 %) was comprised of hits to metazoan and eukaryotic groups other than plants and fungi.
The search for trees that support a monophyletic relationship between a Trebouxia-derived protein sequence and fungal sequences supported by a bootstrap value ≥70 % returned 48 phylogenies. Only three of these trees were identified as showing potential instances of HGT, based on the following two criteria: (1) the Trebouxia-derived sequence was included in a well-supported monophyletic clade with the fungi also in an independent analysis using a refined alignment (sometimes as the most basal branch, i.e., sister to the fungi), and (2) a wide variety of eukaryotic and bacterial groups were present in the tree (e.g., including at least two additional sequences from Green plants), ensuring a broad taxonomic sampling in the alignment, because only such trees presented the opportunity for alternative hypothesis testing of Trebouxia + Fungi against Trebouxia + Green Plants. Thirty-seven of the other 45 phylogenies were excluded due to small taxonomic sampling (criterion 2) and eight phylogenies due to lack of support in the independent analysis (criterion 1).
Sulfite efflux pump/tellurite-resistance dicarboxylate transporter (TDT) family
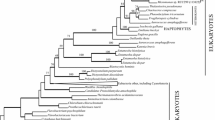
The 360 amino acid alignments of the sulfite efflux pump/tellurite-resistance dicarboxylate transporter (TDT) family consisted of 76 sequences, 27 of eukaryotic and 49 of prokaryotic origin. The alignment was manually refined to a length of 343 amino acids to remove ambiguous regions. The tree was rooted with a monophyletic Archaea at the base, and sequences from Bacteria formed a paraphyletic grade towards sequences from members of Viridiplantae, Rhodophyta, and fungi (Fig. 1). In the resulting tree (Fig. 1, Supplement S1), a clade placing sequences from the two lichen photobionts T. decolorans and Asterochloris sp. were the sister group to twenty Basidiomycetes and Ascomycete fungi. This clade was firmly supported with bootstrap and posterior probability values of 100 % and 1.00, respectively. Four additional green algal members from Chlorophyta were positioned as sister to two Rhodophyta in a separate clade (Fig. 1), but this relationship was well supported (84 %) only in the Bayesian analysis. Alternative hypothesis testing significantly rejected monophyly of the Green plants + Trebouxia + Asterochloris (p ≤ 0.047 in SH and p ≤ 0.049 in ELW tests).

Phylogenetic tree derived from Bayesian analysis of the alignment of the sequences from the sulfite efflux pump/tellurite-resistance dicarboxylate transporter (TDT) family. Fungal sequences (Fungi) are marked in blue, green plants (Viridiplantae) in green, red algae (Rhodophyta) in red, Archaea sequences in purple, and all bacteria in black. The Trebouxia sequence is in bold text, and bolded branches are supported by posterior probability values ≥ 0.90 and bootstrap values ≥ 0.8. Triangles within the complete phylogeny indicate collapsed branches of sequences from the same species. The inset tree (b) shows the overall relationships of larger classification groups based on this tree. (The complete tree with accession and GenBank numbers for the sequences is provided in Supplement S1.)
The most similar hit to this sequence in a Blastp search in GenBank was a sequence from Dothistroma septosporum (Dorog.) M. Morelet strain NZE10 (Mycosphaerellaceae, Capnodiales, Dothideomycetes EME40420.1). PCR analysis confirmed that identical DNA sequences (i.e., to the Trebouxia genome data) encoding the sulfite efflux pump are present in T. decolorans and T. impressa. However, this fragment could not be amplified from genomic DNA of the more distantly related green alga Dictyochloropsis symbiontica (also Trebouxiophyceae).
Class-1 nitrilase/cyanide hydratase (CH)
A 160 amino acid alignment, encoding the N-terminus of the class-1 nitrilase/cyanide hydratase (CH) protein from T. decolorans was aligned to 110 sequences spanning species from bacteria, Archaea, and eukaryotes. The tree was again rooted using Archaea sequences as outgroup. The close relationship of the Trebouxia-derived sequence with 11 fungal sequences was shown as Trebouxia placed as sister to all but one Ascomycota fungi and supported with a bootstrap score of 87 % and posterior probability of 1.00 (Fig. 2, supplement S2). One sequence from Ascomycota (Phaeosphaeria nodorum) was nested alone within proteobacteria. The green alga Coccomyxa and genomes from one bryophyte and three angiosperm plants were placed with high support values in two unrelated parts of the tree near Metazoa and Planctomycetes, not closely related to the Fungi and Trebouxia. Alternative hypothesis testing significantly rejected monophyly of the Green plants + Trebouxia (p ≤ 0.000 in SH and ELW tests).


Phylogenetic tree derived from Bayesian analysis of the alignment of the sequences from the class-1 nitrilase/cyanide hydratase (CH) family. Fungal sequences (Fungi) are marked in blue, green plants (Viridiplantae) in green, red algae (Rhodophyta) in red, animal (Metazoa) sequences in brown, other eukaryotes in pink, Archaea sequences in purple, and all bacteria in black. The Trebouxia sequence is in bold text, and bolded branches are supported by posterior probability values ≥ 0.90 and RAxML bootstrap values ≥ 0.8. Triangles within the complete phylogeny indicate collapsed branches of sequences from the same species. The inset tree (c) shows the overall relationships of larger classification groups based on this tree. (The complete tree with accession and GenBank numbers for the sequences is provided in Supplement S2.)
The most similar hit to the Trebouxia sequence from a Blastp search in GenBank was Chaetomium globosum Kunze strain CBS 148.51 (Chaetomiaceae, Sordariales; XP_001227445.1; a taxon also included in the alignment above). PCR analysis confirmed that an identical DNA sequence (i.e., to the Trebouxia genome data) encoding the class-1 nitrilase is present in T. decolorans. However, successful amplification of this fragment could not be achieved from genomic DNA of the related algae T. impressa and D. symbiontica, despite repeated attempts.
Oxidoreductase/retinol dehydrogenase
The Trebouxia oxidoreductase fragment spanned up to 145 aligned amino acids and including the C-terminus of the protein. The resulting alignment was comprised of 100 taxa from bacteria and eukaryotes (Viridiplantae, Fungi, Amoebozoa, Metazoa, Cryptophyta, and Haptophyta). The tree was rooted using a monophyletic bacterial clade of Bacteriodetes for convenience, since no Archaea was found to be included in this aligned data set. The monophyly of a clade consisting of T. decolorans plus 12 ascomycete fungi was supported by a bootstrap score of 82 % and a posterior probability of 0.99 (Fig. 3, Supplement S3). Trebouxia was basally placed within the fungal clade, with only one sequence derived from Nectria haematococca as more basal within the fungal subclade. The sister clade to the fungal clade (albeit with lower support; 84 % bootstrap and 0.82 posterior probability) consisted of the Viridiplantae sequences from the green algae Coccomyxa and Micromonas as well as 11 angiosperm sequences together with other bacterial and two eukaryotic sequences (Amoebozoa and Hapthophyceae). The other sequences from eukaryotes, a bryophyte (Viridiplantae), and Guillardia, a cryptophyte, were both separately positioned far away from the other eukaryotic sequences and placed close to sequences from Actinobacteria. Alternative hypothesis testing significantly rejected monophyly of the Green plants + Trebouxia (p ≤ 0.003 in SH and p ≤ 0.001 in ELW tests).


Phylogenetic tree derived from Bayesian analysis of the alignment of the sequences from the oxidoreductase/retinol dehydrogenases family. Fungal sequences (Fungi) are marked in blue, green plants (Viridiplantae) in green, animal (Metazoa) sequences in brown, other eukaryotes in pink, and all bacteria in black. The Trebouxia sequence is in bold text, and bolded branches are supported by posterior probability values ≥ 0.90 and RAxML bootstrap values ≥ 0.8. Triangles within the complete phylogeny indicate collapsed branches of sequences from the same species. The inset tree (c) shows the overall relationships of larger classification groups based on this tree. (The complete tree with accession and genbank numbers for the sequences is provided in Supplement S3.)
The most similar hit to this sequence in a Blastp search in GenBank was from Verticillium dahlia Kleb. strain VdLs.17 (mitosporic Plectosphaerellaceae, Glomerellales, Ascomycota; EGY18625.1). PCR analysis confirmed that identical DNA sequences (i.e., to the Trebouxia genome data) encoding oxidoreductase are present in T. decolorans. However, this fragment was not successfully amplified from genomic DNA of the related symbiotic green algae T. impressa and D. symbiontica, despite repeated attempts.
Analysis for recombination between fungal and algal sequences of the candidate genes
In order to test if the HGT event is indeed ancient—as suggested by the position of the Trebouxia sequence below the split of the Basidiomycetes and Ascomycetes—we used the program RAT (Etherington et al. 2005, downloaded on Dec. 9th 2014 from: https://github.com/ethering/RAT) and analysed the amino acid alignments of the gene trees. Settings were as suggested by the programmers, but maximum numbers of contributing sequences set to 25. The last setting had only influence on the detection of possible recombination within bacterial, not within eukaryotic sequences. In no case evidence for recombination was detected. Evidence for possible recombination was detected only between fungal sequences.
Discussion
Horizontal gene transfer between different organismic lineages is usually detected through phylogenetic or phylogenomic analysis of a gene that shows a different relationship to other taxa than what would be expected based on other genes or other paralogs of that gene in those taxa. Such an aberrant pattern that can then only (or most likely) be explained by rare horizontal gene transfer, instead of the usual vertical inheritance seen as a result through eons and/or millennia of evolution and speciation. It should be noted that the obvious and apparent paraphyly of bacteria at the base of the non-monophyletic Eukaryotes in our phylogenomic results (Figs. 1, 2, and 3) are expected due to frequent, historical and on-going HGT, and gene exchange among prokaryotes and also (historically) among the very basal branches in the Eukaryotes (Boto 2014; Pilar 2012). As mentioned earlier, HGT is less common in the more derived and extant branches of the Eukaryotes (Boto 2014). The direction of the transfer is generally inferred from the closest relatives of the gene in question, and if the gene is nested inside a group of organisms in which it is taxonomically not classified (with Trebouxia inside fungi, for example), the surrounding taxa are likely the origin of the gene. The situation becomes more complex if sister group relationships do not fit the overall inferred phylogeny of the eukaryotic Tree of Life, as in the case of two of our genes (Figs. 1 and 2), where Trebouxia orthologs are sister to fungi, not nested inside the fungi (as in Fig. 3). Several scenarios are possible to explain such a pattern, and caution is required as our conclusions are only based on the genome data and (limited) taxon sampling available to us at this time. Subsequent analyses are needed in the future, based on the expanded genomic datasets from Trebouxia as well as much wider taxon sampling including more green algae, basal fungal groups, and lichen-forming fungi. However, our results tell three stories of ancient HGT between fungi and lichen algae, and we are looking forward to see how these scenarios develop over the upcoming years by the ongoing genome sequencing efforts by scientists worldwide.
Our null hypothesis was that HGT events would be between lichenized fungi and lichen algae. If so, a Trebouxia sequence would be highly nested inside a fungal clade and placed close to sequences from lichenized fungi (such as Cladonia grayi, Ascomycota; see Fig. 3). Trebouxia would not be placed closely to other green plants, which would form their own monophyletic, separate clade. In our results, we see the separation of Trebouxia from the Viridiplantae (with one exception, the lichen algae Asterochloris, Fig. 1) in our three gene trees, but we found no data that supports the nesting of Trebouxia with lichen-forming fungi deep inside a fungal clade. Therefore, the hypothesis that these are three relatively more recent HGT events involving Trebouxia and only specific lichenized fungal lineages is rejected.
An alternative hypothesis would be that these three HGT events happened by gene transfer from Trebouxia to a fungus. If so, then the fungi (all or some) would be nested inside a monophyletic Viridiplantae. In our results, Trebouxia is not nested within or at the base of the rest of the green plants (Viridiplantae) and neither are the fungal sequences, so our results contradict such a hypothetical scenario.
Instead, the three Trebouxia HGT gene candidates are sister to a monophyletic fungal lineage as a whole (or one node away from the base in the case of oxidoreductase, see Fig. 3), and not placed close to other Viridiplantae. In each case, this supports an ancestral HGT event involving an ancient gene transfer to Trebouxia from an ancestral stem lineage member of the fungi. Our data support that such a scenario must have happened long before the formation of the first lichens, unless the ancestor of all fungi was symbiotic with algae and formed lichens, which is not likely. It is also likely that the HGT for the sulfite efflux enzyme happened before the divergence of Basidiomycota and Ascomycota, since Trebouxia is placed below both of these major fungal groups (see Fig. 1). In the other two HGT candidate trees (Figs. 2 and 3), no Basidiomycotes sequences were similar enough to be included in the alignment, so the timing of the HGT can be interpreted as before or early on in the diversification of the ascomycetes.
Fossil and molecular dating evidence supports the idea that the Trebouxiophyceae lineage had already evolved around 700–900 Mya (Leliaert et al. 2011, 2012), which was at a similar time as the ongoing evolutionary divergence into the major fungal lineages of Ascomycota, Zygomycota, and Basidiomycota, while the diversification of the Ascomycetes happened later than this (Beimforde et al. 2014; Lücking et al. 2009; Prieto and Wedin 2013). This would imply that (1) green algae diversified long before the Ascomycetes, (2) there was a close association (possibly symbiosis) of an ancestral, basal lineage of fungi, and green algae, at least in the Trebouxiophyceae lineage, and, possibly, (3) the ancestral alga that incorporated this fungal gene into its genome could have possibly been (at least hypothetically) pre-disposed to evolve symbiotic association with certain fungi to form lichens.
Despite the intimate nature of the interaction between algae and fungi in general and particularly of the photo- and mycobionts in lichens, it was perhaps surprising to uncover only three gene-encoding fragments that fulfilled our basic criteria for HGT (i.e., high bootstrap support and posterior probability values (≥0.8 and ≥0.95, respectively) for a monophyletic clade formed by Trebouxia and fungal sequences). Moreover, in our blast searches, the most similar sequences to Trebouxia orthologs were not derived from lichen-forming fungi but from Dothideo- and Sordariomycetes, respectively, and this is another piece of information that supports an ancient HGT event involving Trebouxia’s ancestor and ancient fungi. HGT events based on relatively more recent lichen symbiosis might still be uncovered, especially since the complete genome of Trebouxia yet has to be sequenced, assembled, and annotated. Nevertheless, it should be noted that the sequenced 55.3 Mbp is well in the range of the genome of Asterochloris, which is the sister genus to Trebouxia, and has 56.1 Mbp assembled data (http://genome.jgi-psf.org/Astpho1/Astpho1.info.html).
Given the obvious concern for contamination as a possible explanation for these HGT candidate genes, we took the following two precautions to address this issue. First, we used an axenic culture of T. decolorans to generate the Illumina DNA library, thereby eliminating the possibility of bacterial or fungal DNA in the sequenced fraction. Second, to guard against fungal contamination during the sequencing and/or analysis steps, we used PCR to amplify and thereby validate HGT candidate genes using total T. decolorans DNA that was independently prepared and analyzed by AB in Germany; the Illumina sequencing was done in the lab of Debashish Bhattacharya in the USA. All PCR amplification products were sequenced and found to encode identical or near-identical sequences for non-T. decolorans taxa (≥99 % identity) as the HGT candidate. Moreover, the most similar sequences were from Dothideomycetes (for the sulfite efflux pump) and Sordariomycetes (for nitrilase and oxidoreductase). In case of a contamination, the closest sequences should have been derived from representatives of one fungal class only.
Possible function of candidate genes for HGT: The first candidate gene is closely related to a membrane transporter, a sulfite efflux pump belonging to the tellurite-resistance dicarboxylate transporter (TDT) family (Marchler-Bauer et al. 2011). Interestingly, the most closely placed protein to the one from T. decolorans in our phylogenetic tree (Fig. 1) is from another lichen photobiont, Asterochloris, the sister genus of Trebouxia, and lichenized with C. grayi G. Merr. ex Sandst. No homolog of this gene was found in the genome of mycobiont of X. parietina, indicating that the gene transfer did not take place from this lichen fungus to its photobiont, but has happened from a different fungus to this alga (as discussed above). No clues about the transported substances exist, because only very distantly related transporters have thus far been analyzed (Avram and Bakalinsky 1997; Léchenne et al. 2007). Genes from this family are also known from higher plants, where they have been shown to be involved in the stomatal response to CO2, abscisic acid, ozone, light/dark transitions, humidity change, calcium ions, hydrogen peroxide, and nitric oxide (Saji et al. 2008; Vahisalu et al. 2008).
The second candidate gene is a nitrile aminohydrolase, most similar to class-1 nitrilases, which catalyze the hydrolysis of nitriles to carboxylic acids and ammonia, without the formation of “free” amide intermediates (Marchler-Bauer et al. 2013). They are involved in the biosynthesis of natural products and plant hormone metabolism and many enzymes exhibit strong substrate preferences, but because the Trebouxia sequence is not closely related to a sequence of a characterized enzyme, nothing can be stated yet about the possible substrates of this enzyme.
The third candidate gene is an oxidoreductase of which the most similar sequence (EGY18625.1) belongs to the retinol dehydrogenase (retinol-DH), Light dependent Protochlorophyllide (Pchlide) OxidoReductase (LPOR), which has a single domain with a structurally conserved Rossmann fold (Marchler-Bauer et al. 2013). Light-dependent reduction is via NADP-dependent short-chain dehydrogenases/reductases. These enzymes catalyze a wide range of activities including the metabolism of steroids, cofactors, carbohydrates, lipids, aromatic compounds, and amino acids, and act in redox sensing (Marchler-Bauer et al. 2013; http://www.ncbi.nlm.nih.gov/Structure/cdd/cddsrv.cgi?ascbin=8&maxaln=10&seltype=2&uid=212492). Also, in this case, further analysis is needed to shed light on the possible function of this oxidoreductase in the metabolism of T. decolorans.
It is an intriguing hypothesis that the candidate genes for HGT from the fungi into Trebouxia may have important functions in the evolution of the lichen symbiosis. This hypothesis gains support from the fact that these transferred genes have not been found outside symbiotic green algae so far. Nevertheless, it needs to be clearly stated that more data is needed in order to provide sound evidence for this assumption.
In summary, our work suggests that three HGT events involving fungi and a terrestrial alga are likely to have occurred during the early evolutionary history of fungi and green alga (Trebouxiophyceae) evolution. The HGT events appear to be more ancient than the origin of lichen symbiosis between fungi and algae. Our results are intriguing and encourage further detailed HGT analyses involving more taxa from green algal lineages, basal fungi, lichenized fungi, and their genomes. More data will need to be gathered to address many open questions such as the possible roles of these genes in the evolution of the fungi and green algae, the exact timing of HGT between green algae and fungi, and the relationships of acquiring fungal genes into algae and lichen symbiosis evolution.
References
Ahmadjian, V. (1967). A guide to the algae occurring as lichen symbionts: Isolation, culture, cultural physiology and identification. Phycologia, 6, 127–160.
Ahmadjian, V. (1988). The lichen alga Trebouxia: does it occur free-living? Plant Systematics and Evolution, 158, 243–247.
Ahmadjian, V. (1993). The lichen symbiosis. New York City: Wiley. 250 p.
Ahmadjian, V., Brink, J. J., & Shehata, A. I. (1991). Molecular biology of lichens—Search for plasmid DNA and the question of gene movement between bionts. In Y. Yamada, I. Yoshimura, & A. Takahashi (Eds.), Proceedings of international symposium on lichenology 1990 (pp. 2–21). Osaka: Nippon Paint Co., Ltd.
Altincicek, B., Kovacs, J. L., & Gerardo, N. M. (2011). Horizontally transferred fungal carotenoid genes in the two-spotted spider mite Tetranychus urticae. Biology Letters. doi:10.1098/rsbl.2011.0704.44.
Andersson, J. O. (2005). Lateral gene transfer in eukaryotes. Cellular and Molecular Life Sciences, 62, 1182–1197.
Avram, D., & Bakalinsky, A. T. (1997). SSU1 encodes a plasma membrane protein with a central role in a network of proteins conferring sulfite tolerance in Saccharomyces cerevisiae. Journal of Bacteriology, 179, 5971–5974.
Beck, A., & Koop, H. U. (2001). Analysis of the photobiont population in lichens using a single–cell manipulator. Symbiosis, 31, 57–67.
Beck, A., & Mayr, C. (2012). Nitrogen and carbon isotope variability in the green-algal lichen Xanthoria parietina and their implications on mycobiont-photobiont interactions. Ecology and Evolution, 2, 3132–3144.
Beck, A., Friedl, T., & Rambold, G. (1998). Selectivity of photobiont choice in a defined lichen community: Inferences from cultural and molecular studies. New Phytologist, 139, 709–720.
Beimforde, C., Feldberg, K., Nylinder, S., Rikkinen, J., Tuovila, H., Dörfelt, H., Gube, M., Jackson, D. J., Reitner, J., Seyfullah, L. J., & Schmidt, A. R. (2014). Estimating the phanerozoic history of the ascomycota lineages: Combining fossil and molecular data. Molecular Phylogenetics and Evolution. doi:10.1016/j.ympev.2014.04.024.
Belnap, J., Büdel, B., & Lange, O. L. (2001). Biological soil crusts: Characteristics and distribution. Ecological Studies, 150, 3–30.
Boto, L. (2014). Horizontal gene transfer in the acquisition of novel traits by metazoans. Proceedings of the Biological Sciences. doi:10.1098/rspb.2013.2450.
Bubrick, P., Galun, M., & Frensdorff, A. (1984). Observations on free-living Trebouxia de Puymaly and Psuedotrebouxia Archibald, and evidence that both symbionts from Xanthoria parietina (L.) Th. Fr. can be found free-living in nature. New Phytologist, 97, 455–462.
Bubrick, P., Frensdorff, A., & Galun, M. (1985). Proteins from the lichen Xanthoria parietina (L.) Th. Fr. which bind to phycobiont cell walls: isolation and partial purification of an algal-binding protein. Symbiosis, 1, 85–95.
Chan, C. X., Yang, E. C., Banerjee, T., Yoon, H. S., Martone, P. T., et al. (2011). Red-and-green algal monophyly and extensive gene sharing found in a rich repertoire of red algal genes. Current Biology, 21, 328–333.
del Campo, E. M., Casano, L. M., Gasulla, F., & Barreno, E. (2009). Presence of multiple group I introns closely related to bacteria and fungi in plastid 23S rRNAs of lichen-forming Trebouxia. International Microbiology, 12, 59–67.
Doolittle, W. F., Boucher, Y., Nesbo, C. L., Douady, C. J., Andersson, J. O., et al. (2003). How big is the iceberg of which organellar genes in nuclear genomes are but the tip? Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences, 358, 39–57.
Eisenreich, W., Knispel, N., & Beck, A. (2011). Advanced methods for the study of the chemistry and the metabolism of lichens. Phytochemistry Reviews, 10, 445–456.
Etherington, G. J., Dicks, J., & Roberts, I. N. (2005). Recombination analysis tool (RAT): a program for the high-throughput detection of recombination. Bioinformatics, 21, 278–281.
Fitzpatrick, D. A. (2012). Horizontal gene transfer in fungi. FEMS Microbiology Letters, 329, 1–8.
Friedl, T., & Bhattacharya, D. (2002). Origin and evolution of green lichen algae. In J. Seckbach (Ed.), Symbiosis: Mechanisms and model systems (pp. 343–357). Dordrecht: Kluwer Academic Publishers.
Friedl, T., & Büdel, B. (2008). Photobionts. In T. H. Nash (Ed.), Lichen biology (3rd ed., pp. 9–26). Cambridge: Cambridge University Press.
Friedl, T., Besendahl, A., Pfeiffer, P., & Bhattacharya, D. (2000). The distribution of group I introns in lichen algae suggests that lichenization facilitates intron lateral transfer. Molecular Phylogenetics and Evolution, 14, 342–352.
Gogarten, J. P., Doolittle, W. F., & Lawrence, J. G. (2002). Prokaryotic evolution in light of gene transfer. Molecular Biology and Evolution, 19, 2226–2238.
Grube, M., & Hawksworth, D. L. (2007). Trouble with lichen: the reevaluation and reinterpretation of thallus form and fruit body types in the molecular era. Mycological Research, 111, 1116–1132.
Grube, M., Cardinale, M., & Berg, G. (2012). Bacteria and the lichen symbiosis. In B. Hock (Ed.), Fungal associations (pp. 363–372). Berlin: Springer.
Helms, G., Friedl, T., Rambold, G., & Mayrhofer, H. (2001). Identification of photobionts from the lichen family Physciaceae using algal-specific ITS rDNA sequencing. The Lichenologist, 33, 73–86.
Honegger, R. (2012). The symbiotic phenotype of lichen-forming ascomycetes and their endo- and epibionts. In B. Hock (Ed.), Fungal associations (pp. 287–339). Berlin: Springer.
Honegger, R., Edwards, D., & Axe, L. (2013). The earliest records of internally stratified cyanobacterial and algal lichens from the Lower Devonian of the Welsh Borderland. New Phytologist, 197, 264–275.
Joneson, S., Armaleo, D., & Lutzoni, F. (2011). Fungal and algal gene expression in early developmental stages of lichen symbiosis. Mycologia, 103, 291–306.
Keeling, P. J., & Palmer, J. D. (2008). Horizontal gene transfer in eukaryotic evolution. Nature Reviews Genetics, 9, 605–618.
Knoll, A. W. (2014). Paleobiological perspectives on early eukaryotic evolution. Cold Spring Harb Perspect Biol, 6, a016121.
Lappin-Scott, H. M., & Costerton, J. W. (2003). Microbial biofilms (p. 328). Cambridge: Cambridge University Press.
Léchenne, B., Reichard, U., Zaugg, C., Fratti, M., Kunert, J., et al. (2007). Sulphite efflux pumps in Aspergillus fumigatus and dermatophytes. Microbiology, 153, 905–913.
Leliaert, F., Verbruggen, H., & Zechman, F. W. (2011). Into the deep: New discoveries at the base of the green plant phylogeny. Bioessays, 33, 683–692.
Leliaert, F., Smith, D. R., Moreau, H., Herron, M., Verbruggen, H., Delwiche, C. F., & De Clerck, O. (2012). Phylogeny and molecular evolution of the green algae. Critical Reviews in Plant Sciences, 31, 1–46.
Li, W., & Godzik, A. (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics, 22, 1658–1659.
Loftus, B., Anderson, I., Davies, R., Alsmark, U. C., Samuelson, J., et al. (2005). The genome of the protist parasite Entamoeba histolytica. Nature, 433, 865–868.
Lücking, R., Huhndorf, S., Pfister, D. H., Rivas Platas, E., & Lumbsch, H. T. (2009). Fungi evolved right on track. Mycologia, 101, 810–822.
Lutzoni, F., Pagel, M., & Reeb, V. (2001). Major fungal lineages are derived from lichen symbiotic ancestors. Nature, 411, 937–940.
Marcet-Houben, M., & Gabaldon, T. (2010). Acquisition of prokaryotic genes by fungal genomes. Trends in Genetics, 26, 5–8.
Marchler-Bauer, A., Lu, S., Anderson, J. B., Chitsaz, F., Derbyshire, M. K., et al. (2011). CDD: a conserved domain database for the functional annotation of proteins. Nucleic Acids Research. doi:10.1093/nar/gkq1189.
Marchler-Bauer, A., Zheng, C., Chitsaz, F., Derbyshire, M. K., Geer, L. Y., Geer, R. C., Gonzales, N. R., Gwadz, M., Hurwitz, D. I., Lanczycki, C. J., Lu, F., Lu, S., Marchler, G. H., Song, J. S., Thanki, N., Yamashita, R. A., Zhang, D., & Bryant, S. H. (2013). CDD: Conserved domains and protein three-dimensional structure. Nucleic Acids Research, 41(Database issue), D384–52.
Molina, M. C., Muñiz, E., & Vicente, C. (1993). Enzymatic activities of algal-binding protein and its algal cell wall receptor in the lichen Xanthoria parietina. An approach to the parasitic basic of mutualism. Plant Physiology and Biochemistry, 31, 131–142.
Molina, M. C., Bajon, C., Sauvanet, A., Robert, D., & Vicente, C. (1998). Detection of polysaccharides and ultrastructural modification of the photobiont cell wall produced by two arginase isolectins from Xanthoria parietina. Journal of Plant Research, 111, 191–197.
Moran, N. A., & Jarvik, T. (2010). Lateral transfer of genes from fungi underlies carotenoid production in aphids. Science, 328, 624–627.
Moustafa, A., & Bhattacharya, D. (2008). PhyloSort: a user-friendly phylogenetic sorting tool and its application to estimating the cyanobacterial contribution to the nuclear genome of Chlamydomonas. BMC Evolutionary Biology, 8, 6.
Moustafa, A., Beszteri, B., Maier, U. G., Bowler, C., Valentin, K., et al. (2009). Genomic footprints of a cryptic plastid endosymbiosis in diatoms. Science, 324, 1724–1726.
Nash, T. H., III. (2008). Lichen biology. New York City: Cambridge University Press. 486 p.
Nylander, J. A. A., Wilgenbusch, J. C., Warren, D. L., & Swofford, D. L. (2008). AWTY (are we there yet?): a system for graphical exploration of MCMC convergence in Bayesian phylogenetics. Bioinformatics, 24, 581–583.
Peveling, E. (1988). Beziehungen zwischen den Symbiosepartnern in Flechten. Naturwissenschaften, 75, 77–86.
Pilar, F. M. (2012). Horizontal gene transfer in microorganisms (p. 202). Norfolk: Caister Academic Press.
Price, D. C., Chan, C. X., Yoon, H. S., Yang, E. C., Qiu, H., et al. (2012). Cyanophora paradoxa genome elucidates origin of photosynthesis in algae and plants. Science, 335, 843–847.
Prieto, M., & Wedin, M. (2013). Dating the diversification of the major lineages of Ascomycota (Fungi). PLoS ONE, 8(6), e65576.
Rambaut, A., Drummond, A.J. (2007). Tracer v1.5 [computer program]. Website http://beast.bio.ed.ac.uk/Tracer.
Richards, T. A., Soanes, D. M., Foster, P. G., Leonard, G., Thornton, C. R., et al. (2009). Phylogenomic analysis demonstrates a pattern of rare and ancient horizontal gene transfer between plants and fungi. Plant Cell, 21, 1897–1911.
Richards, T. A., Soanes, D. M., Jones, M. D. M., Vasieva, O., Leonard, G., Paszkiewicz, K., Foster, P. G., Hall, N., & Talbot, N. J. (2011). Horizontal gene transfer facilitated the evolution of plant parasitic mechanisms in the oomycetes. Proceedings of the National Academy of Sciences of the United States of America, 108, 15258–15263.
Richardson, A. O., & Palmer, J. D. (2007). Horizontal gene transfer in plants. Journal of Experimental Biology, 58, 1–9.
Ronquist, F., & Huelsenbeck, J. P. (2003). MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics, 19, 1572–1574.
Saji, S., Bathula, S., Kubo, A., Tamaoki, M., Kanna, M., et al. (2008). Disruption of a gene encoding C4-dicarboxylate transporter-like protein increases ozone sensitivity through deregulation of the stomatal response in Arabidopsis thaliana. Plant and Cell Physiology, 49, 2–10.
Schmidt, H. A., Strimmer, K., Vingron, M., & von Haeseler, A. (2002). TREE-PUZZLE: Maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics, 18, 502–504.
Schmitt, I., & Lumbsch, H. T. (2009). Ancient horizontal gene transfer from bacteria enhances biosynthetic capabilities of fungi. PLoS ONE, 4, e4437.
Schwendener, S. (1867). Über die wahre Natur der Flechten. Verh Schweiz Naturf Ges, 51, 88–90.
Seaward, M. R. D. (2008). Environmental role of lichens. In T. H. Nash III (Ed.), Lichen biology (2nd ed., pp. 274–298). New York City: Cambridge University Press.
Shimodaira, H., & Hasegawa, M. (1999). Multiple comparisons of loglikelihoods with applications to phylogenetic inference. Molecular Biology and Evolution, 16, 1114–1116.
Sing, R. S., & Walia, A. K. (2014). Characteristics of lichen lectins and their role in symbiosis. Symbiosis. doi:10.1007/s13199-014-0278-y.
Slot, J. C., & Hibbett, D. S. (2007). Horizontal transfer of a nitrate assimilation gene cluster and ecological transitions in fungi: A phylogenetic study. PLoS ONE, 2, e1097.
Stamatakis, A. (2006). RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics, 22, 2688–2690.
Stamatakis, A., Hoover, P., & Rougemont, J. (2008). A rapid bootstrap algorithm for the RAxML Web Servers. Systematic Biology, 57, 758–771.
Stanke, M., & Morgenstern, B. (2005). Augustus: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Research, 33, W465–W467.
Strimmer, K., & Rambaut, A. (2002). Inferring confidence sets of possibly mis-specified gene trees. Proceedings of the Royal Society of London, Biological Sciences, 269, 137–142.
Taylor, T. N., Hass, H., Remy, W., & Kerp, H. (1995). The oldest fossil lichen. Nature, 378, 244.
Taylor, T. N., Hass, H., Remy, W., & Kerp, H. (1997). A cyanolichen from the lower Devonian Rhynie Chert. American Journal of Botany, 84, 992–1004.
Trembley, M. L., Ringli, C., & Honegger, R. (2002). Morphological and molecular analysis of early stages in the resynthesis of the lichen Baeomyces rufus. Mycological Research, 106, 768–776.
Tschermak-Woess, E. (1978). Myrrnecia reticulata as a phycobiont and free-living - free-living Trebouxia- the problem of Stenocybe septata. The Lichenologist, 10, 69–79.
Tschermak-Woess, E. (1988). New and known taxa of Chlorella (Chlorophyceae): occurrence as lichen phycobionts and observations on living dictyosomes. Plant Systematics and Evolution, 159, 123–139.
Vahisalu, T., Kollist, H., Wang, Y. F., Nishimura, N., Chan, W. Y., et al. (2008). SLAC1 is required for plant guard cell S-type anion channel function in stomatal signalling. Nature, 452, 487–491.
Yoon, H. S., Hackett, J. D., Ciniglia, C., Pinto, G., & Bhattacharya, D. (2004). A molecular timeline for the origin of photosynthetic eukaryotes. Molecular Biology and Evolution, 21, 809–818.
Yoon, H. S., Price, D. C., Stepanauskas, R., Rajah, V. D., Sieracki, M. E., et al. (2011). Single cell genomics reveals trophic interactions and evolutionary history of uncultured protists. Science, 332, 714–717.
Yuan, X., Xiao, S., & Taylor, T. N. (2005). Lichen-like symbiosis 600 million years ago. Science, 308, 1017–1020.
Acknowledgments
We sincerely thank Debashish Bhattacharya and Dana Price (both Rutgers) for crucial help with genome sequencing, data analysis, setting up the web page at http://dbdata.rutgers.edu/data/Trebouxia/ and long, fruitful, and encouraging discussions. Divino Rajah (Rutgers) is gratefully acknowledged for technical support. Paul Dyer and the Joint Genome Institute are thanked for access to the Xanthoria parietina fungal genome, Daniele Armaleo and the Joint Genome Institute are thanked for access to the Asterochloris sp. algal genome and Katherine McMahon and the Joint Genome Institute are thanked for access to the Cladonia grayi fungal genome. This work was generously supported by Illumina, Inc., who provided sequencing reagents to the Genome Cooperative at Rutgers University (http://dblab.rutgers.edu/genome_cooperative/) and the German Science Foundation (BE3825/2-1 to AB). PKD thanks the Ministerio de Ciencia e Innovación (Spain) for financial support (CGL2013-42498-P, CGL2010-21646/BOS) and MCM acknowledges support by a research grant from Universidad Rey Juan Carlos (Estancias Breves de Investigación) as Visiting Scientist to Rutgers University. NZ thanks Rutgers University for startup funds. LS was funded by US Department of Agriculture award USDA/NJAES-NJ17112.
Author information
Authors and Affiliations
Corresponding author
Additional information
Research experiments were done in the labs of the Botanische Staatssammlung München and Rutgers University (see M & M for details)
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplement S1
Phylogenetic tree derived from Bayesian analysis of the alignment of the sequences from the sulfite efflux pump/tellurite-resistance dicarboxylate transporter (TDT) family. Fungal sequences (Fungi) are marked in blue, green plants (Viridiplantae) in green, red algae (Rhodophyta) in red, Archaea sequences in purple, and all bacteria in black. The Trebouxia sequence is in bold text, and bolded branches are supported by posterior probability values ≥ 0.90 and bootstrap values ≥ 0.8 (with exact numbers marked by each branch). (EPS 10036 kb)
Supplement S2
Phylogenetic tree derived from Bayesian analysis of the alignment of the sequences from the class-1 nitrilase / cyanide hydratase (CH) family. Fungal sequences (Fungi) are marked in blue, green plants (Viridiplantae) in green, red algae (Rhodophyta) in red, animal (Metazoa) sequences in brown, other eukaryotes in pink, Archaea sequences in purple, and all bacteria in black. The Trebouxia sequence is in bold text, and bolded branches are supported by posterior probability values ≥ 0.90 and RAxML bootstrap values ≥ 0.8 (with exact numbers marked by each branch). (EPS 9882 kb)
Supplement S3
Phylogenetic tree derived from Bayesian analysis of the alignment of the sequences from the oxidoreductase/retinol dehydrogenases family. Fungal sequences (Fungi) are marked in blue, green plants (Viridiplantae) in green, animal (Metazoa) sequences in brown, other eukaryotes in pink, and all bacteria are in black. The Trebouxia sequence is in bold text, and bolded branches are supported by posterior probability values ≥ 0.90 and RAxML bootstrap values ≥ 0.8 (with exact numbers marked by each branch). (EPS 11106 kb)
Rights and permissions
About this article

Cite this article
Beck, A., Divakar, P.K., Zhang, N. et al. Evidence of ancient horizontal gene transfer between fungi and the terrestrial alga Trebouxia . Org Divers Evol 15, 235–248 (2015). https://doi.org/10.1007/s13127-014-0199-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13127-014-0199-x