
Abstract
A large number of studies have reported that the prevalence of beta thalassemia carriers in India varies by ethnic groups. The objective of this study was to conduct a systematic review of the published studies and conduct a meta-analysis to determine the prevalence of beta thalassaemia carriers in India. A PubMed database search using keywords “beta thalassaemia AND India” identified 1088 articles of which 69 articles were included in the review. Studies using diagnostic tests and methods recommended by the International Council for Standardization in Haematology were used for calculation of pooled prevalence. Pooled prevalence was calculated using a random effects model using Review Manager version 5.3. Studies had screened five categories of populations, that is, the general population; tribal groups, communities not belonging to tribal groups, persons with anemia, and persons referred with a suspicion of hemoglobinopathy. This heterogeneity contributed to a high pooled prevalence of beta thalassemia carriers of 8.23% (95% CI 7.36–9.10). Sub-group analysis however yielded 3.74% (95% CI 2.52–4.97) pooled prevalence of beta thalassemia carriers in the general population. It was 4.6% (95% CI 3.2–6.2) among tribal groups. Quality of prevalence studies was limited by methodological issues including non-random sampling methods, heterogeneity of population types screened, and lack of use of recommended diagnostic cut-offs. Prevalence of beta thalassemia carriers was similar in tribal populations and the general population, indicating the need to further investigate the prevalence of beta thalassemia carriers in tribal groups.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Beta thalassemias are genetic disorders with quantitative deficiency in the synthesis of the beta globin chains of hemoglobin. Patients with homozygous beta thalassaemia, referred to as beta thalassaemia major, are chronically anemic, are transfusion-dependent, and have a low life expectancy when optimal care is unavailable. Persons heterozygous for beta thalassaemia, referred to as carriers, are asymptomatic and mildly anemic (Galanello and Origa 2010). Magnitude of beta thalassaemia is obtained by measuring the prevalence of beta thalassaemia carriers (Hickman et al. 1999).
Estimates indicate that the global carrier prevalence ranges between 0.5 and 20% (Weatherall et al. 2006). Beta thalassemias are considered to be widespread particularly in the Indian sub-continent by virtue of its large population (Weatherall and Clegg 2001; Modell and Darlison 2008). There is no national estimate on the prevalence of beta thalassaemia carriers, although there are several studies in smaller geographical locations and defined populations to determine the prevalence of beta thalassaemia carriers.
These studies have examined diverse populations. Some studies have included persons from villages or towns, have been conducted at antenatal clinics, have included pregnant women, and have recruited students from academic institutions. There are several studies that have measured the prevalence of beta thalassemia carriers among tribal groups and among specific communities. Yet other studies have tested for beta thalassaemia carrier status among persons with anemia, patients suspected with hemoglobinopathies, or extended family members of patients with beta thalassaemia major (Jain et al. 1981; Jain et al. 1983; Choubisa 1985; Rao and Gorakshakar 1990; Choubisa 1991; Manglani et al. 1997; Balgir et al. 1999; Ambekar et al. 2001; Gajra et al. 2002; Ghosh et al. 2002; Choubisa et al. 2004; Chhotray et al. 2004; Balgir 2005a, b; Choubisa 2006; Sinha et al. 2006; Jawahirani et al. 2007; Colah et al. 2008; Mulchandani et al. 2008; Gupta et al. 2009; Kumar and Tamhankar 2009; Saraswathy 2009; Munshi et al. 2009; Choubisa 2009; Sachdev et al. 2010; Madan et al. 2010; Colah et al. 2010; Balgir 2010; Rao et al. 2010; Chandrashekar and Soni 2011; Dolai et al. 2012; Jain et al. 2012; Parthasarthy 2012; Patel et al. 2012; Bhukhanvala et al. 2012; Achoubi et al. 2012; Kulkarni et al. 2013; Philip et al. 2013; Rakholia and Chaturvedi 2013; Mohanty et al. 2013; Tiwari et al. 2013; Bhukhanvala et al. 2013; Baxi et al. 2013; Piplani et al. 2013; Baruah et al. 2014; Patel et al. 2014; Purohit et al. 2014; Sahoo et al. 2014; Verma et al. 2014; Chatterjee et al. 2015; Choudhuri et al. 2015; Mukhopadhyay et al. 2015; Nagar et al. 2015; Mohanty et al. 2015; Mondal and Mandal 2016; Teli et al. 2016).
The reported prevalence of beta thalassaemia carriers ranged from 0.2 to 21.8% (Munshi et al. 2009; Achoubi et al. 2012). Multicentric studies reported carrier prevalence estimates of 2.8 to 4.04% (Madan et al. 2010; Mohanty et al. 2013). Colah et al. 2010 has reported thalassaemia carrier frequencies by district, for the states of Gujarat and Maharashtra, and reported carrier frequencies ranging from 0.7 to 9.5% (Colah et al. 2010). The differences in the reported prevalence estimates between studies could be inherent population characteristics or methodological issues, such as use of different screening and diagnostic tests and by the use of different diagnostic cut-offs for HbA2 levels (ranging from 3.0 to 4.5%). The purpose of this study was to systematically review these studies and conduct a meta-analysis to predict the number of beta thalassemia carriers in India.
Methods
This systematic review and meta-analysis followed the Preferred Reporting Items for Systematic Reviews and Meta-Analysis reporting guideline (Page et al. 2021).
Electronic literature search
The PubMed database (MEDLINE) was searched using the search terms (“beta thalassaemia”[All Fields] OR “beta-thalassemia”[MeSH Terms] OR “beta-thalassemia” [All Fields] OR (“beta”[All Fields] AND “thalassemia”[All Fields]) OR “beta thalassemia”[All Fields]) AND (“India”[MeSH Terms] OR “India”[All Fields]) in November 2022 which elicited 1088 articles. There were no restrictions on date of publication. The citations of eligible articles were screened to identify articles for potential inclusion in the review.
Study selection
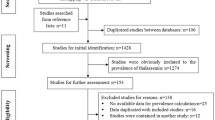
All articles were screened for the possibility of extracting data on the number of beta thalassaemia carriers. Studies reporting the methodology in sufficient detail to extract relevant data were included. Studies were included in the review if they fulfilled the following criteria: (1) reported data on number of beta thalassaemia carriers and (2) were conducted in India. Studies were excluded if they were published before 1978 (as the first guideline for HbA2 cut-off for carrier diagnosis had been issued in this year by the International Council for the Standardization of Haematology (ICSH) (Stephens et al. 2012). Mutation analysis studies, studies evaluating the sensitivity and specificity of diagnostic tests, studies screening persons post-blood transfusion, micromapping studies, and studies presenting duplicated data were excluded. Finally, 69 studies were selected for the review (Fig. 1). SD identified articles for extraction which was independently verified by AK. Any disputes were resolved through discussion.

Selection of studies for review (PRISMA)
Data extraction
A data extraction format was designed in MS Excel to extract data on the following study variables: study setting, geographical location of study, type of population characterized, description of population characterized, sampling method, sample size, study design, screening and diagnostic techniques used, diagnostic cut-offs used, informed consent administered, and ethics committee approval. The primary outcome was the number of beta thalassaemia carriers. The prevalence estimates of each study included in the review were verified by recalculating the prevalence from the numerator (number of beta thalassaemia carriers) and the denominator (total number of individuals screened). Quality of data was assessed by comparison of study designs, sampling methods, and whether screening and diagnostic methods used the case definitions as prescribed.
Screening for hemoglobinopathies primarily involves a hemogram used to detect microcytosis or a NESTROFT (Naked Eye Single Tube Red cell Osmotic Fragility Test) which is usually used in resource-limited or field-based settings (Traeger-Synodinos et al. 2014; Ministry of Health and Family Welfare. Government of India 2016). Diagnosis for hemoglobinopathies is done for samples with microcytosis by quantification of HbA2 by electrophoresis, chromatography, HPLC, capillary and Hb electrophoresis, or/and DNA analysis using PCR-based methods, sequencing, reverse dot blot analysis, and microarrays (Traeger-Synodinos et al. 2014). The ICSH and the European Molecular Genetics Quality Network (EMQN) state that persons with HbA2 > 3.5% should be diagnosed as carriers, and those in the range between 3.5 and 4.0% should be screened further using DNA analysis for silent beta thalassemia mutations, severe iron deficiency, or other hemoglobin variants (International Committee for Standardization in Haematology 1978; Stephens et al. 2012; Traeger-Synodinos et al. 2014).
Data analysis
Prevalence was calculated as the number of beta thalassaemia carriers per 100 individuals screened. Pooled prevalence was calculated in Review Manager (version 5.3) using the inverse variance method reported by Allagh et al. (2015 and Bhide and Kar (2018). A random effects model was used for meta-analysis due to the high heterogeneity between the studies (I2 > 95%, p < 0.05) and depicted using a forest plot. Prior to meta-analysis, standard error (SE) for each prevalence estimate was calculated using the formula SE = √p(1-p)/n where p = proportion of individuals diagnosed with beta thalassaemia trait and n = total sample size for the study.
Results
Search resultss
The search strategy identified 1088 articles which were published between 1978 and 2022. Abstracts of these articles were screened, and a total of 69 articles were identified to be eligible for inclusion in the review (Fig. 1).
Study characteristics
The sample size of studies ranged from 100 to 287,258 individuals. Studies were reported from across the country, with 32% (22/69) from the eastern region (Balgir et al. 1999; Gajra et al. 2002; Chhotray et al. 2004; Balgir 2005a, b, 2010; Dolai et al. 2012; Jain et al. 2012; Mondal et al. 2012; Baruah et al. 2014; Purohit et al. 2014; Sahoo et al. 2014; Chatterjee et al. 2015; Choudhuri et al. 2015; Mukhopadhyay et al. 2015; Nagar et al. 2015; Mondal and Mandal 2016; Teli et al. 2016), 25% (17/69) from western India (Rao and Gorakshakar 1990; Manglani et al. 1997; Ambekar et al. 2001; Ghosh et al. 2002; Jawahirani et al. 2007; Colah et al. 2008; Mulchandani et al. 2008; Colah et al. 2010; Patel et al. 2012; Bhukhanvala et al. 2012; Philip et al. 2013; Rakholia and Chaturvedi 2013; Tiwari et al. 2013; Bhukhanvala et al. 2013; Purohit et al. 2014; Mohanty et al. 2015), 16% (11/69) from central (Jain et al. 1981; Jain et al. 1983; Choubisa 1985; Choubisa 1991; Choubisa et al. 2004; Choubisa 2006; Gupta et al. 2009; Tamhankar et al. 2009; Baxi et al. 2013), 13% (9/69) studies from the north (Sinha et al. 2006; Saraswathy 2009; Sachdev et al. 2010; Rao et al. 2010; Verma et al. 2014), 7% (5/69) from south India (Munshi et al. 2009; Chandrashekar and Soni 2011; Kulkarni et al. 2013), and 3% (2/69) studies from the northeast (De et al. 2006; Achoubi et al. 2012). There were 3/69 (4%) multicentric studies (Madan et al. 2010; Mohanty et al. 2013), and one study had not mentioned the location (Parthasarthy 2012) (Table 1).
Population sub-groups
The included studies surveyed populations with different levels of risk of being carriers of beta thalassemia, that is, general population, endogamous populations with tribal ethnicity, non-tribal populations practicing endogamy, patients with anemia, and persons clinically referred on suspicion of being a carrier of a hemoglobinopathy. Due to this variation, pooled prevalence estimates were made based on these categories.
Description of screening and diagnostic tests used
The screening tests used in the studies were clinical suspicion, NESTROFT, peripheral smear, and hemogram. The diagnostic tests used by the studies were electrophoresis (agar or cellulose acetate), high-performance liquid chromatography (HPLC), and molecular analysis (ARMS PCR, sequencing).
There were 65/69 (93%) studies that had used a screening procedure prior to diagnosis, of which 42/65 (63%) studies had used hemogram (Balgir et al. 1999; Ghosh et al. 2002; Balgir 2005a, b, 2010; De et al. 2006; Gupta et al. 2009; Tamhankar et al. 2009; Sachdev et al. 2010; Madan et al. 2010; Colah et al. 2010; Rao et al. 2010; Chandrashekar and Soni 2011; Dolai et al. 2012; Jain et al. 2012; Mondal et al. 2012; Parthasarthy 2012; Patel et al. 2012; Bhukhanvala et al. 2012, 2013; Philip et al. 2013; Mohanty et al. 2013; Tiwari et al. 2013; Baxi et al. 2013; Baruah et al. 2014; Chatterjee et al. 2015; Choudhuri et al. 2015; Mukhopadhyay et al. 2015; Nagar et al. 2015; Mondal and Mandal 2016; Teli et al. 2016), 9/65 (14%) studies had used NESTROFT (Jain et al. 1981; Ambekar et al. 2001; Jawahirani et al. 2007; Colah et al. 2008; Saraswathy 2009; Munshi et al. 2009; Achoubi et al. 2012; Kulkarni et al. 2013; Rakholia and Chaturvedi 2013), and 2/65 (4%) studies had examined a peripheral smear (Jain et al. 1983; Choubisa 1985) to screen for beta thalassaemia carriers. There were 13/65 (19%) studies that had used a combination of methods of NESTROFT, hemogram, and peripheral smear examination (Manglani et al. 1997; Gajra et al. 2002; Choubisa et al. 2004; Chhotray et al. 2004; Choubisa 2006; Sinha et al. 2006; Mulchandani et al. 2008; Choubisa 2009; Verma et al. 2014; Mohanty et al. 2015) for screening (Table 1). Of the 69 studies, 68 (97%) studies had reported the type of diagnostic test used. Of these, 23/68 (34%) studies had done HbA2 analysis by electrophoresis (Jain et al. 1981; Jain et al. 1983; Choubisa 1985; Rao and Gorakshakar 1990; Choubisa 1991; Manglani et al. 1997; Balgir et al. 1999; Ghosh et al. 2002; Choubisa et al. 2004; Balgir 2005a, b; Choubisa 2006; Sinha et al. 2006; Jawahirani et al. 2007; Mulchandani et al. 2008; Choubisa 2009; Madan et al. 2010; Balgir 2010; Jain et al. 2012; Rakholia and Chaturvedi 2013; Tiwari et al. 2013), 30/68 (41%) studies had done HbA2 analysis by HPLC (Gupta et al. 2009; Sachdev et al. 2010; Colah et al. 2010; Rao et al. 2010; Chandrashekar and Soni 2011; Dolai et al. 2012; Mondal et al. 2012; Parthasarthy 2012; Patel et al. 2012; Philip et al. 2013; Mohanty et al. 2013, 2015; Baruah et al. 2014; Verma et al. 2014; Chatterjee et al. 2015; Choudhuri et al. 2015; Mukhopadhyay et al. 2015; Mondal and Mandal 2016; Teli et al. 2016), and one study (Sahoo et al. 2014) had used ARMS PCR for carrier diagnosis. There were 12/68 (17%) studies that had used mutation analysis by ARMS PCR for carrier detection in combination with HPLC and electrophoresis methods (Gajra et al. 2002; Chhotray et al. 2004; De et al. 2006; Saraswathy 2009; Tamhankar et al. 2009; Munshi et al. 2009; Achoubi et al. 2012; Baxi et al. 2013; Purohit et al. 2014; Nagar et al. 2015), whereas 6/68 (9%) studies had used a combination of HPLC and cellulose electrophoresis methods for carrier detection (Ambekar et al. 2001; Colah et al. 2008; Patel et al. 2012; Bhukhanvala et al. 2012, 2013) (Table 1).
Prevalence of beta thalassaemia carriers
Group I: general population
There were 28/69 studies which had screened 703,615 persons, of which 33,951 were diagnosed as beta thalassaemia carriers (Table 1). The population included students from academic institutions (n = 4, 14%) (Tamhankar et al. 2009; Madan et al. 2010), pregnant women from antenatal clinics (n = 11, 39%) (Gajra et al. 2002; Sinha et al. 2006; Colah et al. 2008; Kulkarni et al. 2013; Philip et al. 2013; Tiwari et al. 2013; Bhukhanvala et al. 2013; Baxi et al. 2013; Patel et al. 2014; Choudhuri et al. 2015), and individuals selected from community settings (n = 4, 14%) (Choubisa 1991, 2006; Bhukhanvala et al. 2012; Achoubi et al. 2012), and 9/29 (31%) studies had sampled the population to be screened from multiple sources, i.e., community, healthcare, and academic institutions (Choubisa 1991, 2006; Colah et al. 2010; Dolai et al. 2012; Jain et al. 2012; Patel et al. 2012; Mohanty et al. 2013; Sahoo et al. 2014; Chatterjee et al. 2015; Nagar et al. 2015) Studies screening the general population had larger sample sizes as compared to studies screening other population types. Of these, 15/28 (54%) studies were eligible (Choubisa 1991, 2006; Gajra et al. 2002; Tamhankar et al. 2009; Madan et al. 2010; Jain et al. 2012; Patel et al. 2012; Bhukhanvala et al. 2012, 2013; Achoubi et al. 2012; Baxi et al. 2013; Sahoo et al. 2014; Nagar et al. 2015). They included 197,735 persons of which 3721 were beta thalassaemia carriers. Pooled prevalence of beta thalassaemia carriers in the general population was calculated to be 3.74% (95% CI 2.52–4.97) (Fig. 2a).

Pooled prevalence of beta thalassaemia carriers in a general population, b tribal communities, c communities, d persons with anemia, e persons with suspicion of hemoglobinopathy
Group II: tribal communities
There were 14 studies that had screened a total of 24,287 individuals from 41 communities, and 973 persons were diagnosed as beta thalassaemia carriers (Table 1). The pooled prevalence of beta thalassaemia carriers was calculated from 8/14 (57%) eligible studies which had screened 4650 persons of which 263 were beta thalassaemia carriers (Jain et al. 1981; Jain et al. 1983; Choubisa et al. 2004; Balgir 2005a; Choubisa 2009; Balgir 2010; Purohit et al. 2014) Pooled prevalence of beta thalassaemia carriers in the tribal communities was estimated to be 4.68% (95% CI 3.17–6.19) (Fig. 2b).
Group III: communities not belonging to tribal groups
Members of ethnic communities were targeted for screening either because there was a purported higher prevalence of beta thalassaemia amongst the members or they practiced endogamy, i.e., marriage within the community. There were a total of 9 eligible studies that screened 16,943 individuals from five communities (Table 1). There were 4/9 (36%) studies that had screened only Sindhis (n = 2769 individuals) (Manglani et al. 1997; Jawahirani et al. 2007; Mulchandani et al. 2008; Saraswathy 2009; Rakholia and Chaturvedi 2013) because beta thalassemias are considered to be prevalent in this community. The other communities screened were different castes and sub-castes of Gujaratis (n = 5003), Muslims (n = 4870), scheduled castes (n = 1895), Punjabis (n = 622), Tharu (n = 493), and Maharashtrians (n = 109). The pooled prevalence was calculated from 5/9 (56%) eligible studies which had screened 14,231 persons of which 799 were carriers. The pooled prevalence of beta thalassemia carriers in communities not belonging to tribal groups is 7.74% (95% CI 4.39–11.08) (Fig. 2c). This pooled estimate has to be considered with caution since pooling data from these different communities themselves increases heterogeneity.
Group IV: persons with anemia
Studies in this group had screened patients with anemia. There were 13 studies that had screened 59,846 patients with anemia, of which 5630 were beta thalassaemia carriers (Chhotray et al. 2004; Balgir 2005b; Gupta et al. 2009; Tamhankar et al. 2009; Munshi et al. 2009; Parthasarthy 2012; Piplani et al. 2013; Baruah et al. 2014; Verma et al. 2014; Teli et al. 2016) (Table 1). The pooled prevalence of beta thalassaemia carriers was calculated from 5/13 (39%) eligible studies which had screened 3917 persons of which 644 were beta thalassaemia carriers. The pooled prevalence of beta thalassaemia carriers in persons with anemia was calculated to be 14.1% (95% CI 9.45–18.76) (Fig. 2d).
Group V: persons with suspicion of hemoglobinopathy
This group included studies that had screened individuals with a high index of suspicion such as extended family members or patients suspected with hemolytic anemia. There were 11 studies that had screened 131,176 individuals with a suspicion of hemoglobinopathy, of which 7630 were beta thalassaemia carriers (Ambekar et al. 2001; Sachdev et al. 2010; Rao et al. 2010; Chandrashekar and Soni 2011; Jain et al. 2012; Mondal et al. 2012; Philip et al. 2013; Verma et al. 2014; Mukhopadhyay et al. 2015; Mondal and Mandal 2016). The pooled prevalence of beta thalassaemia carriers was calculated from 3/12 (25%) eligible studies, which had screened 2772 persons of which 771 were beta thalassaemia carriers (Chhotray et al. 2004; Balgir 2005b; Tamhankar et al. 2009; Munshi et al. 2009; Parthasarthy 2012). Thus, the pooled prevalence of beta thalassaemia carriers in persons with suspicion of hemoglobinopathy was estimated to be 38.58% (95% CI 10.53–66.63) (Fig. 2e).
It is noteworthy that when all studies are included including 38/69 (55%) studies not using the recommended diagnostic cut-offs, the pooled prevalence of beta thalassaemia carriers was 8.23% (95% CI 7.36–9.10). Figure 3 shows that while few regions of the country had multiple studies reporting data for different population sub-groups, there was no reliable data for other parts of the country.

Reported prevalence of beta thalassaemia carriers in India in the general population (G); tribal communities (T); ethnic communities not belonging to tribal groups I; persons with anemia (A); persons with suspicion of a hemoglobinopathy (H)
Discussion
The systematic review identified that there were more than a thousand published studies on beta thalassemia in India. However, only a limited number were prevalence studies reporting the magnitude of beta thalassemia carriers. The meta-analyses identified methodological issues that could influence prevalence estimates. The first methodological issue was that studies had been conducted in diverse populations, with different levels of risk. For example, studies conducted among school children would represent the true prevalence in the general population, while prevalence studies from tribal communities would have a higher prevalence due to endogamy and known high prevalence of the circulating allele. Similarly, patients with chronic anemia referred for beta thalassaemia testing would represent a biased sample and therefore may represent higher prevalence.
The second source of possible error was the lack of random sampling or lack of description of the method by which the sample had been drawn. Only 17% of studies had used random sampling methods, and 85% of studies either had not mentioned the type of sampling method used or had provided insufficient information to decipher the method used. The third methodological issue that could influence pooled prevalence estimates was the use of different screening tests and different diagnostic cut-offs used for HbA2 analysis, in lieu of those recommended by ICSH and EMQN. Such sources of error were evident in the meta-analysis, as only half of the studies in each sub-group were eligible for analysis.
The meta-analysis circumvented these sources of error by conducting sub-group analyses and by including only those studies that reported using the recommended diagnostic cut-offs. Without this, the pooled prevalence estimate was inordinately high at over 8%. The results of the meta-analysis confirm the high prevalence of beta-thalassemia carriers in the general population (3.7%). These estimates have significant public health implications. Using a 4% carrier prevalence and the method of Hickman et al. 1999, an estimated 8740 children with beta thalassaemia major may be born each year in India, in the absence of preventive interventions. An equivalent prevalence of 4% in tribal communities is indicative of the need to further investigate beta thalassaemia prevalence in these populations. It is important to point out that eligible studies, measuring the prevalence of beta thalassemia in tribal populations, were limited. It is noteworthy that a program for the elimination of sickle cell disease has been launched recently in India (Ministry of Health and Family Welfare. Government of India 2023). The results of this analysis indicate that beta thalassemia may be as widespread as sickle cell disease, and it may be relevant to increase the scope of this project to include all hemoglobinopathies common in India.
Strengths and limitations of the study
The limitation of this study is that only a single database was used to search for studies. However, references of all articles were scanned in order to identify possible studies published in local journals and articles not indexed in the PubMed database.
Conclusions
Despite a large body of literature on beta thalassemia prevalence in India, this study identified several methodological issues that could influence prevalence estimates. These include unclear methodologies, non-random samples, small size of studies, screening of heterogeneous population groups, and not using internationally recommended diagnostic cut-offs. Data from 69 studies identified approximately 4% prevalence of beta thalassemia carriers in the general population. A similar high prevalence of beta thalassemia among tribal population groups indicates the need for including screening, prevention, and care for beta thalassemia in the ongoing project for elimination of sickle cell disease in India. In light of the high prevalence of beta thalassemia carriers in India, methodologically rigorous studies are needed to assist public health decision-making for prevention and care.
Data Availability
Not applicable.
References
Achoubi N, Asghar M, Saraswathy KN, Murry B (2012) Prevalence of β-thalassemia and hemoglobin E in two migrant populations of Manipur, North East India. Genet Test Mol Biomarkers 16:1195–1200. https://doi.org/10.1089/gtmb.2011.0373
Allagh KP, Shamanna BR, Murthy GVS, Ness AR, Doyle P, Neogi SB, Pant HB (2015) Birth prevalence of neural tube defects and orofacial clefts in India: a systematic review and meta-analysis. PLoS ONE 10:e0118961. https://doi.org/10.1371/journal.pone.0118961
Ambekar SS, Phadke MA, Balpande DN, Mokashi GD, Khedkar VA (2001) The prevalence and heterogeneity of beta thalassemia mutations in the Western Maharashtra Population : a hospital based study. Indian J Hum Genet 1:219–223
Balgir RS (2005a) The spectrum of haemoglobin variants in two scheduled tribes of Sundargarh district in north-western Orissa, India. Ann Hum Biol 32:560–573. https://doi.org/10.1080/03014460500228741
Balgir RS (2005b) Spectrum of hemoglobinopathies in the state of Orissa, India: a ten years cohort study. J Assoc Physicians India 53:1021–1026
Balgir RS (2010) Genetic diversity of hemoglobinopathies, G6PD deficiency, and ABO and Rhesus blood groups in two isolates of a primitive Kharia Tribe in Sundargarh District of Northwestern Orissa, India. J Community Genet 1:117–123. https://doi.org/10.1007/s12687-010-0016-y
Balgir RS, Murmu B, Dash BP (1999) Hereditary hemolytic disorders among the Ashram school children in Mayurbhanj district of Orissa. J Assoc Physicians India 47:987–990
Baruah MK, Saikia M, Baruah A (2014) Pattern of hemoglobinopathies and thalassemias in upper Assam region of North Eastern India : high performance liquid chromatography studies in 9000 patients. Indian J Pathol Microbiol 57:236–243. https://doi.org/10.4103/0377-4929.134680
Baxi A, Manila K, Kadhi P, Heena B (2013) Carrier screening for β thalassemia in pregnant Indian women: experience at a single center in Madhya Pradesh. Indian J Hematol Blood Transfus 29:71–74. https://doi.org/10.1007/s12288-012-0165-8
Bhargava M, Kumar V, Pandey H, Singh V, Misra V, Gupta P (2020) Role of hematological indices as a screening tool of beta thalassemia trait in Eastern Uttar Pradesh: an institutional study. Indian J Hematol Blood Transfus 36:719–724. https://doi.org/10.1007/s12288-020-01282-z
Bhide P, Kar A (2018) A national estimate of the birth prevalence of congenital anomalies in India: systematic review and meta-analysis. BMC Pediatr 18:1–10. https://doi.org/10.1186/s12887-018-1149-0
Bhukhanvala DS, Sorathiya SM, Shah AP, Patel AG, Gupte SC (2012) Prevalence and hematological profile of β-thalassemia and sickle cell anemia in four communities of Surat city. Indian J Hum Genet 18:167–171. https://doi.org/10.4103/0971-6866.100752
Bhukhanvala DS, Sorathiya SM, Sawant P, Colah R, Ghosh K, Gupte SC (2013) Antenatal screening for identification of couples for prenatal diagnosis of severe hemoglobinopathies in Surat, South gujarat. J Obstet Gynaecol India 63:123–127. https://doi.org/10.1007/s13224-012-0271-4
Chandrashekar V, Soni M (2011) Hemoglobin disorders in South India. ISRN Hematol 748939:1–6. https://doi.org/10.5402/2011/748939
Chatterjee T, Chakravarty A, Chakravarty S (2015) Population screening and prevention strategies for thalassemias and other hemoglobinopathies of Eastern India : experience of 18,166 cases. Hemoglobin 39:384–388. https://doi.org/10.3109/03630269.2015.1068799
Chhotray GP, Dash BP, Ranjit M (2004) Spectrum of hemoglobinopathies in Orissa, India. Hemoglobin 28:117–122. https://doi.org/10.1081/HEM-120034244
Choubisa SL (1985) Erythrocyte glucose-6-phosphate dehydrogenase deficiency & thalassemic genes in scheduled castes of Rajasthan. Indian J Med Res 82:554–558
Choubisa SL (1991) Abnormal haemoglobins, thalassaemia and G-6-PD enzyme deficiency in Rajasthan (western-India). Haematologia (budap) 24:153–165
Choubisa SL (2006) Erythrocyte mutant genes in inhabitants of arid-environment of Western Rajasthan (India). Indian J Phys Anthropol Hum Genet 25:15–30
Choubisa SL (2009) Sickle cell haemoglobin, thalassaemia and G-6-PD enzyme deficiency genes in Garasiya tribe inhabited malaria endemic areas of Sirohi District, Rajasthan (India). J Commun Dis 41:13–18
Choubisa SL, Choubisa D, Choubisa L (2004) Erythrocyte genetic disorders in inhabitants of Aravali Hilly-Environment of Rajasthan (India). Indian J Phys Anthropol Hum Genet 23:145–159
Choudhuri S, Sen A, Ghosh MK, Misra S (2015) Effectiveness of prenatal screening for hemoglobinopathies in a developing country. Hemoglobin 39:380–383. https://doi.org/10.3109/03630269.2014.1003564
Colah R, Surve R, Wadia M, Solanki P, Mayekar P, Thomas M, Gorakshakar A, Dastur A, Mohanty D (2008) Carrier screening for β -thalassemia during pregnancy in India : a 7-year evaluation. Genet Test 12:181–186
Colah R, Gorakshakar A, Phanasgaonkar S, D’Souza E, Nadkarni A, Surve R, Sawant P, Master D, Patel R, Ghosh K, Mohanty D (2010) Epidemiology of β-thalassaemia in Western India: mapping the frequencies and mutations in sub-regions of Maharashtra and Gujarat. Br J Haematol 149:739–747. https://doi.org/10.1111/j.1365-2141.2010.08131.x
De M, Halder A, Podder S, Sen R, Chakrabarty S, Sengupta B, Chakraborty T, Das U, Talukder G (2006) Anemia and hemoglobinopathies in tribal population of Eastern and North-eastern India. Hematology 11:371–373. https://doi.org/10.1080/10245330600840180
Dolai TK, Dutta S, Bhattacharyya M, Ghosh MK (2012) Prevalence of hemoglobinopathies in rural Bengal, India. Hemoglobin 36:57–63
Gajra B, Chakraborti S, Sengupta B (2002) Prenatal Diagnosis of thalassaemias. Int J Hum Genet 2:173–178
Galanello R, Origa R (2010) Beta-thalassemia. Orphanet J Rare Dis 5:1–15. https://doi.org/10.1186/1750-1172-5-11
Ghosh K, Mukherjee MB, Shankar U, Kote SL, Nagtilak SB, Kolah RB, Surve RR, Tamankar AA, Sukumar S, Mohanty (2002) Clinical examination and hematological data in asymptomatic & apparently healthy school children in a boarding school in a tribal area. Indian J Public Health 46:61–65
Gosavi M, Chavan R, Bellad MB (2021) NESTROFT—A Cost-Effective Mass Screening Tool for the Detection of β-Thalassemia Carrier Status in Anemic Pregnant Women: A Step Toward Reducing the National Disease Burden. J Lab Physicians 13:368–373. https://doi.org/10.1055/s-0041-1732493
Gupta PK, Kumar H, Kumar S, Jaiprakash M (2009) Cation exchange high performance liquid chromatography for diagnosis of haemoglobinopathies. Med J Armed Forces India 65:33–37
Hickman M, Modell B, Greengross P, Chapman C, Layton M, Falconer S, Davies SC (1999) Mapping the prevalence of sickle cell and beta thalassaemia in England: estimating and validating ethnic-specific rates. Br J Haematol 104:860–867
International Committee for Standardization in Haematology (1978) Recommendations for selected methods for quantitative estimation of Hb A2 and for Hb A2 reference preparation. Br J Haematol 38:573–578
Jain R, Mehta N, Joshi K, Gupta O, Andrew A (1981) Sickle cell trait, thalassemia and glucose-6-phosphate dehydrogenase deficiency in Bhil tribe of southern Rajasthan. Indian J Med Res 73:548–553
Jain R, Andrew A, Choubisa SL (1983) Sickle cell & thalassaemic genes in the tribal population of Rajasthan. Indian J Med Res 78:836–840
Jain BB, Roy RN, Ghosh S, Ghosh T, Banerjee U, Bhattacharya SK (2012) Screening for thalassemia and other hemoglobinopathies in a tertiary care hospital of West Bengal : implications for population screening. Indian J Public Health 56:297–300. https://doi.org/10.4103/0019-557X.106419
Jawahirani A, Mamtani M, Das K, Rughwani V, Kulkarni H (2007) Prevalence of beta-thalassaemia in subcastes of Indian Sindhis: Results from a two-phase survey. Public Health 121:193–198. https://doi.org/10.1016/j.puhe.2006.10.017
Kulkarni P, Masthi NRR, Niveditha S, Suvarna R (2013) The prevalence of the beta thalassemia trait among the pregnant women who attended the ANC Clinic in a PHC, by using the NESTROF test in Bangalore, Karnataka. J Clin Diagnostic Res 7:1414–1417. https://doi.org/10.7860/JCDR/2013/5286.3149
Kumar R, Tamhankar PM (2009) A novel beta-globin mutation ( HBB : c . 107A > G ; or codon 35 β ( A → G )) at alpha – beta chain interfaces. 1269–1271 . https://doi.org/10.1007/s00277-009-0760-4
Madan N, Sharma S, Sood SK, Colah R, Bhatia LHM (2010) Frequency of β-thalassemia trait and other hemoglobinopathies in northern and western India. Indian J Hum Genet 16:16–25. https://doi.org/10.4103/0971-6866.64941
Maji SK, Dolai TK, Pradhan S, Maity A, Mandal S, Mondal T, Manna S, Mandal PK (2020) Implications of Population screening for thalassemias and hemoglobinopathies in rural areas of West Bengal, India: report of a 10-year study of 287,258 cases. Hemoglobin 44:432–437. https://doi.org/10.1080/03630269.2020.1831530
Manglani M, Lokeshwar MR, Vani VG, Bhatia N, Mhaskar V (1997) ’NESTROFT’–an effective screening test for beta thalassemia trait. Indian Pediatr 34:702–707
Ministry of Health and Family Welfare. Government of India (2016) Prevention and control of hemoglobinopathies in India - thalassemias, sickle cell disease and other variant hemoglobins
Ministry of Health and Family Welfare. Government of India (2023) Guidelines for national programme for prevention & management of sickle cell disease. New Delhi
Modell B, Darlison M (2008) Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ 2008:480–487. https://doi.org/10.2471/BLT.06.036673
Mohanty D, Colah RB, Gorakshakar AC, Patel RZ, Master DC, Mahanta J, Sharma SK, Chaudhari U, Ghosh M, Das S, Britt RP, Singh S, Ross C, Jagannathan L, Kaul R, Shukla DK, Muthuswamy V, Colah RB, Gorakshakar a C, Patel RZ, Master DC, Mahanta J, Sharma SK, Chaudhari U, Ghosh M, Das S, Britt RP, Singh S, Ross C, Jagannathan L, Kaul R, Shukla DK, Muthuswamy V (2013) Prevalence of β-thalassemia and other haemoglobinopathies in six cities in India: a multicentre study. J Community Genet 4:33–42. https://doi.org/10.1007/s12687-012-0114-0
Mohanty D, Mukherjee MB, Colah RB, Wadia M, Ghosh K, Chottray GP, Jain D, Italia Y, Ashokan KS, Kaul R, Shukla DK, Muthuswamy V (2015) Spectrum of hemoglobinopathies among the primitive tribes: a multicentric study in India. Asia-Pacific J Public Heal 27:NP562–71 .https://doi.org/10.1177/1010539513480231
Mondal SK, Mandal S (2016) Prevalence of thalassemia and hemoglobinopathy in eastern India : a 10 year high performance liquid chromatography study of 119, 336 cases. Asian J Transfus Sci 10:105–110. https://doi.org/10.4103/0973
Mondal B, Maiti S, Biswas BK, Ghosh D, Paul S (2012) Prevalence of hemoglobinopathy, ABO and rhesus blood groups in rural areas of West Bengal. J Res Med Sci 17:772–776
Mukhopadhyay D, Saha K, Sengupta M, Mitra S, Datta C, Mitra PK (2015) Spectrum of hemoglobinopathies in West Bengal, India : a CE-HPLC study on 10407 Subjects. Indian J Hematol Blood Transfus 31:98–103. https://doi.org/10.1007/s12288-014-0373-5
Mulchandani DV, Fulare MB, Zodpey SP, Vasudeo ND (2008) Prevalence and some epidemiological factors of beta thalassaemia trait in Sindhi community of Nagpur City, India. Indian J Public Health 52:11–15
Munshi A, Anandraj MPJS, Joseph J, Shafi G, Anila a N, Jyothy A (2009) Inherited hemoglobin disorders in Andhra Pradesh, India: a population study. Clin Chim Acta 400:117–9. https://doi.org/10.1016/j.cca.2008.10.025
Nagar R, Sinha S, Raman R (2015) Haemoglobinopathies in eastern Indian states : a demographic evaluation. J Community Genet 6:1–8. https://doi.org/10.1007/s12687-014-0195-z
Nigam N, Kushwaha R, Yadav G et al (2020) A demographic prevalence of β Thalassemia carrier and other hemoglobinopathies in adolescent of Tharu population. Journal of Family Medicine and Primary Care 9(8):4305–4310. https://doi.org/10.4103/jfmpc.jfmpc_879_20
Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, Shamseer L, Tetzlaff JM, Akl EA, Brennan SE, Chou R, Glanville J, Grimshaw JM, Hróbjartsson A, Lalu MM, Li T, Loder EW, Mayo-Wilson E, McDonald S, McGuinness LA, Stewart LA, Thomas J, Tricco AC, Welch VA, Whiting P, Moher D (2021) The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. Syst Rev 10:1–11. https://doi.org/10.1186/s13643-021-01626-4
Parthasarthy V (2012) A search for beta thalassemia trait in India. Turkish J Hematol 29:427–429. https://doi.org/10.5505/tjh.2012.21703
Patel AG, Shah AP, Sorathiya SM, Gupte SC (2012) Hemoglobinopathies in South Gujarat population and incidence of anemia in them. Indian J Hum Genet 18:294–298. https://doi.org/10.4103/0971-6866.107979
Patel GM, Parmar A, Zalavadiya D, Talati K (2021) Tackling the menace of anemia and hemoglobinopathies among young adults – Conceptualizing university-level screening. Indian J Community Med 46(1):117–10. https://doi.org/10.4103/ijcm.IJCM_329_20
Patel AP, Patel RB, Patel SA, Vaniawala SN, Patel S, Shrivastava NS, Sharma NP, Jayendrasinh V, Parmar PH, Naik MR (2014) β –thalassemia mutations in Western India : outcome of prenatal diagnosis in a hemoglobinopathies project. Hemoglobin 38:329–334. https://doi.org/10.3109/03630269.2014.951889
Philip J, Sarkar RS, Kushwaha N (2013) Microcytic hypochromic anemia : should high performance liquid chromatography be used routinely for screening anemic and antenatal patients ? Indian J Pathol Microbiol 56:109–113. https://doi.org/10.4103/0377-4929.118699
Piplani S, Manan R, Lalit M, Manjari M, Bhasin T, Bawa J (2013) NESTROFT – a valuable, cost effective screening test for beta thalassemia trait in North Indian Punjabi Population. J Clin Diagn Res 7:2784–2787. https://doi.org/10.7860/JCDR/2013/6834.3759
Purohit P, Dehury S, Patel S, Patel DK (2014) Prevalence of deletional alpha thalassemia and sickle gene in a tribal dominated malaria endemic area of Eastern India. ISRN Hematol 745245. https://doi.org/10.1155/2014/745245
Rakholia R, Chaturvedi P (2013) Prevalence of β thalassemia carrier state in Sindhi community of Wardha and evaluation of risk factors for β thalassemia trait. Niger J Clin Pract 16:375–380
Rao VR, Gorakshakar AC (1990) Sickle cell hemoglobin, beta-thalassemia and G6PD deficiency in tribes of Maharashtra, India. Gene Geogr 4:131–134
Rao S, Kar R, Gupta SK, Chopra A, Saxena R (2010) Spectrum of haemoglobinopathies diagnosed by cation exchange-HPLC & modulating effects of nutritional deficiency anaemias from north India. Indian J Med Res 132:513–519
Ray GK, Jena RK (2019) Spectrum of hemoglobinopathies: a new revelation in a tertiary care hospital of Odisha. Indian J Hematol Blood Transfus 35:513–517. https://doi.org/10.1007/s12288-018-1052-8
Sabitha Rani SS, Vamshidhar IS, Bangaru S, John NA, John J (2022) A study of spectrum of sickle cell anemia and thalassemia in a teaching institute of South India. Niger J Clin Pract 25(4):490–495. https://doi.org/10.4103/njcp.njcp_1742_21
Sachdev R, Dam AR, Tyagi G (2010) Detection of Hb variants and hemoglobinopathies in Indian population using HPLC: report of 2600 cases. Indian J Pathol Microbiol 53:57–62. https://doi.org/10.4103/0377-4929.59185
Sahoo SS, Biswal S, Dixit M (2014) Distinctive mutation spectrum of the HBB gene in an urban eastern Indian population.pdf. Hemoglobin 38:33–38
Saraswathy KN (2009) b-thalassaemia among the Sindhi community in Delhi. Natl Med J India 22:334
Sinha M, Panigrahi I, Shukla J, Khanna A, Saxena R (2006) Spectrum of anemia in pregnant Indian women and importance of antenatal screening. Indian J Pathol Microbiol 49:373–375
Sonkawade ND, Kinikar AA, Kulkarni RK, Dawre RM, Valvi CT, Kamath PA (2022) Screening of Extended Family Members of Thalassemia Major Children as a Thalassemia Preventive Strategy. Ethiop J Health Sci 32:1203–1210. https://doi.org/10.4314/ejhs.v32i6.18
Stephens AD, Angastiniotis M, Baysal E, Chan V, Fucharoen S, Giordano PC, Hoyer JD, Mosca A, Wild B (2012) ICSH recommendations for the measurement of Haemoglobin A2. Int J Lab Hematol 34:1–13. https://doi.org/10.1111/j.1751-553X.2011.01368.x
Tamhankar PM, Agarwal S, Arya V, Kumar R, Gupta UR, Agarwal SS (2009) Prevention of homozygous beta thalassemia by premarital screening and prenatal diagnosis in India. Prenat Diagn 29:83–88. 10.1002/pd
Teli AB, Deori R, Saikia SP, Pathak K, Panyang R, Rajkakati R (2016) β –Thalassaemia and its co-existence with haemoglobin E and haemoglobin S in Upper Assam Region of North Eastern India : a hospital based study. J Clin Diagnostic Res 10:1–4. https://doi.org/10.7860/JCDR/2016/16265.7675
Tiwari M, Kotwal J, Kotwal A, Mishra P, Dutta V, Chopra S (2013) Correlation of haemoglobin and red cell indices with serum ferritin in Indian women in second and third trimester of pregnancy. Med J Armed Forces India 69:31–36. https://doi.org/10.1016/j.mjafi.2012.07.016
Traeger-Synodinos J, Harteveld CL, Old JM, Petrou M, Galanello R, Giordano P, Angastioniotis M, De SB, Henderson S, May A (2014) EMQN best practice guidelines for molecular and haematology methods for carrier identification and prenatal diagnosis of the haemoglobinopathies. Eur J Hum Genet 23:426–437. https://doi.org/10.1038/ejhg.2014.131
Tripathi P, Kumar R, Agarwal S (2018) Spectrum and hematological profile of hereditary anemia in North Indians: SGPGI experience. Intractable Rare Dis Res 7:258–263. https://doi.org/10.5582/irdr.2018.01093
Verma S, Talukdar B, Gupta R (2014) ‘ Reflex ’ HPLC testing as a screening modality for variant hemoglobins : A pilot study of 1310 cases at a pediatric referral hospital. Hematology 19:299–303
Weatherall DJ, Clegg JB (2001) Inherited haemoglobin disorders : an increasing global health problem. Bull World Health Organ 79:704–712
Weatherall D, Akinyanju O, Fucharoen S, Olivieri N, Musgrove P (2006) Inherited disorders of hemoglobin. In: Jamison DT, Breman JG, Measham AR, Alleyne G, Claeson M, Evans DB, Jha P, Mills A, Musgrove P (eds) Disease Control Priorities in Developing Countries, 2nd edn. Oxford University Press, The International Bank for Reconstruction and Development / The World Bank; New York, pp 664–680
Acknowledgements
SD gratefully acknowledges the receipt of an INSPIRE fellowship from the Department of Science and Technology, Government of India.
Funding
INSPIRE fellowship from the Department of Science and Technology, Ministry of Science and Technology, Government of India to SD.
Author information
Authors and Affiliations
Contributions
SD designed the study, conducted the literature review, analysed the data and drafted the manuscript. AK conceptualized the study, supervised the search, selection of data and data analyses, wrote the final draft of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
Secondary data analysis, and therefore did not require review of the Institutional Ethics Committee.
Consent to participate
Not applicable.
Conflict of interest
Sumedha Dharmarajan and Anita Kar declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sumedha, D., Anita, K. Prevalence of beta thalassemia carriers in India: a systematic review and meta-analysis. J Community Genet 14, 527–541 (2023). https://doi.org/10.1007/s12687-023-00683-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12687-023-00683-7