
Abstract
Hunan province located in the south of China has a high incidence of haemoglobinopathies. In the present study, we surveyed the accurate population frequency data of the local population in Changsha city of Hunan province in China. The data includes the carrying rate, gene mutation types and their distribution features for thalassaemia. In total, 7500 consecutive samples from five geographical areas of Changsha were analysed for both haematological and molecular parameters. There was a high prevalence of carriers of \(\upalpha \)-thalassaemia (2.57%), \(\upbeta \)-thalassaemia (1.9%) and both \(\upalpha \)-thalassaemia and \(\upbeta \)-thalassaemia (0.08%). Overall, 4.54% of the population in this area represented heterozygous carriers of \(\upalpha \)-thalassaemia and \(\upbeta \)-thalassaemia. The mutation spectrum of \(\upalpha \)-thalassaemia and \(\upbeta \)-thalassaemia and its haematological characterization were fully described for this area. The present study is the first to report the prevalence of thalassaemia in Hunan province population. Both \(\upalpha \)-thalassaemia and \(\upbeta \)-thalassaemia carriers are widely distributed in Changsha. The knowledge gained from the present study will allow for an estimation of the projected number of pregnant women at risk for thalassaemia, and the design of a screening strategy for the control of thalassaemia in Changsha.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Thalassaemia is the most common autosomal recessive disorder worldwide (Joly et al. 2014; Sabath et al. 2015). Thalassaemia has two main forms, alpha (\(\upalpha \))-thalassaemia and beta (\(\upbeta \))-thalassaemia (Bank 2015). The phenotype of \(\upalpha \)-thalassaemia depends upon the degree of \(\upalpha \)-globin chain deficiency relative to \(\upbeta \)-globin production (Liebhaber and Kan 1983; Piel and Weatherall 2014). The Bart’s hydrops fetalis syndrome is characterized by severe intrauterine anaemia, and is lethal in utero or soon after birth, due to a homozygous \(\upalpha ^{0}\) thalassaemia mutation that results in the loss of all four globins (Chen et al. 2015). Haemoglobin H disease is an intermediate form, having moderately severe, but variable anaemia that is caused by the loss of three functional genes that mostly combine to form an \(\upalpha ^{+}\) thalassaemia (2\(\upalpha \)/ or \(\upalpha ^{\mathrm{T}}\upalpha \)/) and an \(\upalpha ^{0}\) thalassaemia defect (Chen et al. 2000). \(\upbeta \)-Thalassaemia is caused by the absence (\(\upbeta ^{0})\) or ineffective (\(\upbeta ^{+})\) synthesis of \(\upbeta \)-globin chains (Joly et al. 2014). Thalassaemia major involves the inheritance of two mutant \(\upbeta \)-globin alleles and is a severe transfusion-dependent anaemia. If left untreated with regular blood transfusions, thalassaemia major can lead to death within the first year of life (Van de Velde et al. 2004). To date, Over 200 different \(\upbeta \)-globin mutations have been identified, with the majority being single nucleotide substitutions, deletions, or insertions of nucleotides leading to a frameshift; rarely does \(\upbeta \)-thalassaemia results from a gross gene deletion (Higgs 1993; Colah et al. 2010; Bank 2015).

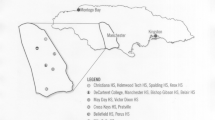
Location of Changsha city in Hunan province and the five study regions including Liuyang county (east), Changsha county (middle east), Changsha district (middle), Wangcheng (middle west) and Ningxiang (west).
Thalassaemia is most prevalent in developing countries. Approximately 5% of the global populations are carriers of thalassaemia, and more than half of these cases occur in Southeast Asia (Colah et al. 2010). In China, there is a high population frequency of thalassaemia in the southern regions of the Yangtze River, particularly in the three most southerly provinces of Guangdong, Guangxi and Hainan (Shi et al. 2011). Hunan province is located at the south bank of the middle reaches of the Yangtze River, adjacent to the Guangdong and Guangxi provinces. In a previous report, the incidence of thalassaemia in Shenzhen inhabitants who migrated from Hunan province was 4.18% (\(\upalpha \)-thalassaemia, 2.15%; \(\upbeta \)-thalassaemia, 2.03%), suggesting that thalassaemia is highly prevalent in the Hunan populations (Li et al. 2005; Huang et al. 2006). However, little is known about the prevalence of thalassaemia or the molecular characteristics of the inhabitants of Changsha in Hunan province.
In the present study, we performed a large-scale prenatal screening of \(\upalpha \)-thalassaemia and \(\upbeta \)-thalassaemia in 7500 samples from Hunan province using haematological and molecular analyses. We aimed to determine the prevalence of thalassaemia and the molecular characteristics of the inhabitants of Changsha to provide an innovative strategy for carrier screening of pregnant couples, genetic counselling, and prenatal diagnosis.
Materials and methods
Population samples
The study population included 7500 individuals (5584 women and 1184 men) from five regional Maternal and Child Health Hospitals (1500 specimens were collected in each hospital) who attended a clinic for prenatal testing in Changsha between May 2014 and May 2015. These regions—Liuyang county (east), Changsha county (middle east), the Changsha district (middle), Wangcheng (middle west) and Ningxiang (west)—have the largest populations of Changsha city (figure 1). All the samples used in the study were of Han Chinese and Changsha descents. This study was conducted in accordance with the declaration of Helsinki. This study was conducted with approval from the Ethics Committee of Changsha Maternal and Child Health Hospital. Written informed consent was obtained from all participants’ guardians.
Screening strategy and experimental analysis

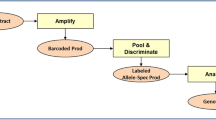
Diagnostic flowchart for the detection of Hb disorders in this study. The positive sample numbers detected at each step are indicated in the left most position. In the molecular screening step, Gap-PCR was used for genotyping \(-\upalpha 3.7\)/ and \(-\upalpha 4.2\)/ deletions and the —SEA/ mutation, respectively. In the confirmative test step, Gap-PCR was used to type the common known \(\upalpha \)-thalassaemia deletions or \(\delta \upbeta \)-thalassaemia/HPFH deletions; MLPA method was used to detect unknown gross deletions in the \(\upalpha \)- or \(\upbeta \)-globin gene cluster; RDB was used to detect known point mutations causing \(\upalpha \)- or \(\upbeta \)-thalassaemia; DNA sequencing was used to identify novel or rare point mutations in the entire \(\upalpha 1\)- and \(\upalpha 2\)- or \(\upbeta \)-globin gene. The detailed information on the primer design and PCR/DHPLC conditions are available. In addition, DNA sequencing was used to characterize point mutations in the \(\upalpha \)-, \(\upbeta \)- or \(\delta \)-globin gene, in all the samples that contained a Hb band that migrated abnormally. All individuals with \(\upbeta \)-thalassaemia were analysed to determine whether they had coinherited any one of the six \(\upalpha \)-thalassaemia defects (—SEA/, –\(\upalpha 3.7\)/, –\(\upalpha 4.2/, \upalpha \hbox {WS}\upalpha /, \upalpha \hbox {QS}\upalpha \)/ and \(\upalpha \hbox {CS}\upalpha \)/) that are common among the Chinese.
The diagnostic flowchart used in this study is illustrated in figure 2. We designed a strategy that combined phenotypic screening and genotyping (Zeng and Huang 2001; Xu et al. 2004; Xiong et al. 2010). All 7500 participants were screened for the presence of defects using two procedures. We used a haematological screening protocol to detect all the suspected subjects who had haematological phenotypes based on full blood counts (FBCs) and haemoglobin test as described previously (Daniel 2007). We found 799 positive samples, and all positive samples were further characterized using a molecular diagnosis, as described previously (Chatterjee et al. 2014) and guided by blood analysis to identify three different types of haemoglobin disorders. We found 172 \(\upalpha \)-thalassaemia carriers, 143 \(\upbeta \)-thalassaemia carriers, six \(\upalpha \)-thalassaemia compound \(\upbeta \)-thalassaemia carriers based on this confirmative test. For those subjects who had no positive haematological phenotypes, we directly used a DNA-based molecular screening protocol to detect \(\upalpha \)-thalassaemia silent carriers (Atanasovska et al. 2012). A total of 21 such carriers were detected using this protocol. The criteria that indicated the possibility of heterozygosity for various thalassaemia types were based on previous studies (Yang et al. 1989; Jia et al. 2004).
Haematological analysis
Haematological screening was performed on all specimens in the Child Health Hospitals of Changsha. A total of 2 mL of peripheral blood was collected into a K3-EDTA tube and used for peripheral blood counts and red blood cell indices according to standard laboratory procedures for the F820 type blood cell analysis system (CIS Company, Chiba, Japan). Subjects with low red blood cell mean corpuscular volume (MCV) values (80 fl) or low mean corpuscular haemoglobin (MCH) values (27 pg) were considered possible thalassaemia carriers and deeply analysed using a high speed automatic electrophoresis analytic system (Sebia, France) to assess the concentration of haemoglobin (Hb) A, A2 and F, and any abnormal haemoglobin. The carrier state for phenotypic \(\upbeta \)-thalassaemia or \(\upbeta \)-thalassaemia compounded with \(\upalpha \)-thalassaemia was detected in samples in which both the concentrations of Hb A2 exceeded 3.5%, and the MCV was lower than 80 fl. A low concentration of Hb A2 (<2.5%) should be present in \(\upalpha \)-thalassaemia carriers.
DNA analysis
The molecular diagnosis of \(\upalpha \)-thalassaemia and \(\upbeta \)-thalassaemia was performed on specimens that have a high risk of being possible thalassaemia carriers. For these samples, genomic DNA was extracted from peripheral blood leukocytes using the DNA Blood Mini Kit (Yi Sheng Tang China Shengzhen). The three known \(\upalpha \)-thalassaemia deletions —SEA, –a3.7 and –a4.2, the three \(\upalpha \)-thalassaemia mutations (Hb Constant Spring (HBA2: c.427T.C), Hb WS (HBA2: c.369C>G) and Hb Quong Sze (HBA2: c.377T.C)), and 17 known \(\upbeta \)-thalassaemia mutations that are most commonly observed in Chinese populations (HBB: c.279A.G, HBB: c.278A.G, HBB: c.250A.C, HBB: c.2T.G, HBB: c.45_46insG, HBB: c.52A.T, HBB: c.79G.A, HBB: c.84_85insC, HBB: c.94delC, HBB: c.92 + 1G.T, HBB: c.92+ 1G.A, HBB: c.92 + 5G.C, HBB: c.126_129delCTTT, HBB: c.130G.T, HBB: c.216_217insA, HBB: c.316–197C.T, HBB: c.79G>A) were analysed using a commercial thalassaemia reverse dot blot (RDB) gene chip (Yi Sheng Tang China Shengzhen). Sequencing was performed for the specimens that were not phenotyped using the reagent kit for detecting an abnormal blood phenotype.
Statistical analysis
Statistical analysis was conducted with SPSS 19.0 statistical software. The prevalence of different thalassaemia alleles was calculated using the standard Hardy–Weinberg formula. All statistical tests were two-tailed test, and the data were deemed significantly different when \(P~<~0.05\).
Results
The details of the survey of population are shown in table 1. Of the 7500 participants who received haematological screening, 10.7% (799/7500) were microcytosis carriers (MCV < 80 fl or MCH < 27 pg). Of these, 643 carriers exhibited \(\upalpha \)-thalassaemia with a Hb A2 concentration of <2.5% based on haemoglobin electrophoresis. In addition, 156 carriers presented with an Hb A2 concentration that was >3.5%; these were considered as carriers of \(\upalpha \)-thalassaemia or \(\upbeta \)-thalassaemia compounded with \(\upalpha \)-thalassaemia. We also used DNA sequencing to characterize point mutations in the \(\upalpha \)-, \(\upbeta \)- or \(\delta \)-globin gene. We identified a total of 341 cases as carriers, of which 193 (2.57%) had \(\upalpha \)-thalassaemia, 143 (1.92%) had \(\upbeta \)-thalassaemia and 6 (0.08%) had both \(\upalpha \)-thalassaemia and \(\upbeta \)-thalassaemia (table 1). A total of 193 samples were analysed for the most common three \(\upalpha \)-thalassaemia deletions (—SEA, a3.7 and a4.2) using Gap PCR. The other three nondeletion types of \(\upalpha \)-thalassaemia mutations (Hb Constant Spring, Hb Quong Sze and Hb Westmead) were detected using an RDB gene chip (table 2). A total of 156 microcytosis carriers were deeply genotyped for both \(\upalpha \)-thalassaemia and \(\upbeta \)-thalassaemia, the latter were detected using an RDB gene chip for analysis of 17 common mutations in Chinese populations. If we could identify a mutation, we then characterized the sample using DNA sequencing. We found that the prevalence of thalassaemia mutations in the Changsha population is 4.55% (table 3).
Discussion
Thalassaemia is common in southern China, especially in the Guangxi province (Yin et al. 2011). The screening of severe determinants of thalassaemia is critically important for the management and control of thalassaemia. The management and control of thalassaemia remain unsatisfactory and suboptimal in China when compared to other countries where the disease is prevalent.
No previous study has investigated thalassaemia in the city of Changsha in Hunan province of China. To estimate the future burden of this disease and the requirements for its control in Changsha, the city has begun screening for thalassaemia using premarital pregnancy tests and gene screening because epidemiological studies of the disease indicate a high incidence in similar ‘poor’ areas, such as Sichuan and Guizhou (Cai et al. 2010; Huang et al. 2013). Previous studies have suggested that thalassaemia is only highly prevalent in the cities of Guangdong and Guangxi; however, large-scale thalassaemia epidemiological survey data has not been collected in Hunan Changsha regions. Our study reveals the actual rate of the population of gene mutation spectrum carriers in Changsha, and characteristics of the thalassaemia gene mutation types. Our results show that the prevalence of thalassaemia mutations in the Changsha population is 4.55% (table 3). From table 3, the rate of thalassaemia is different between the five districts of Changsha, Liuyang and the urban area of the city, the highest and the Wangcheng district, the lowest. Regional features and population mobility may account for the differences. Liuyang is located at the northeast of the city and neighbours with Jiangxi province, leading to high movement of population, while the urban area of Changsha is where the capital of the province located and the movement of population is naturally high. High population movement results in the increase of thalassaemia rate. In contrast, the Wangcheng district is located at the north of the city and its population movement is relatively low.
Based on our results on the epidemiological regularity of thalassaemia, a series of effective screening programmes for the region can be established. The Chinese Center for Disease Control and Prevention sought to establish the molecular basis of thalassaemia to provide first-hand data for its prevention, and determine the prevalence of thalassaemia in high-risk areas, such as Changsha. To reduce the frequency of thalassaemia in subsequent generations in Changsha, our results support the implementation of programmes to screen for carriers of the common severe determinants of thalassaemia.
References
Atanasovska B., Bozhinovski G., Chakalova L., Kocheva S., Karanfilski O. and Plaseska-Karanfiska D. 2012 Molecular diagnostics of \(\beta \)-thalassemia. Balkan J. Med. Genet. 15, 61–65.
Bank A. 2015 The thalassemia syndromes. Blood 51, 369–384.
Cai P., Jiang X. U. and Yan H. U. 2010 An epidemiological investigation and management of the first a/h1n1 (swine flu) outbreak case in college, in sichuan. Mod. Prev. Med. 120, 83–84.
Chatterjee R., Bajoria R., Shah F. T., Porter J. B. and Fedele S. 2014 High index of suspicion for early diagnosis of alendronate-induced stage zero osteonecrosis of jaw in thalassaemia major. Br. J. Haematol. 166, 292–294.
Chen F. E., Ooi C., Ha S. Y., Cheung B. M. Y., Todd D., Liang R. et al. 2000 Genetic and clinical features of hemoglobin H disease in Chinese patients. N. Engl. J. Med. 343, 544–550.
Chen M., Loh S. F., Yu S. L., Nair S., Tan H. H., Nadarajah S. et al. 2015 Rapid and reliable preimplantation genetic diagnosis of common Hb Bart’s hydrops fetalis syndrome and Hb H disease determinants using an enhanced single-tube decaplex PCR assay. Am. J. Hematol. 90, E194–E196.
Colah R., Gorakshakar A. and Nadkarni A. 2010 Global burden, distribution and prevention of \(\beta \)-thalassemias and hemoglobin E disorders. Expert Rev. Hematol. 3, 103–117.
Daniel Y. 2007 Haemoglobinopathy diagnostic tests: blood counts, sickle solubility test, haemoglobin electrophoresis and high-performance liquid chromatography. In Practical management of haemoglobinopathies (ed. I. E. Okpala), pp. 10–19. Blackwell Publishing Ltd, Oxford, UK (doi: 10.1002/9780470988398.ch2).
Higgs D. R. 1993 The thalassaemia syndromes. Q. J. Med. 86, 559–564.
Huang S. W., Liu X. M., Li G. F., Su L., Wu X. and Wang R. L. 2013 Spectrum of \(\beta \)-thalassemia mutations in Guizhou Province, PR China, including first observation of codon 121 (GAA\({>}\)TAA) in Chinese population. Clin. Biochem. 46, 1865–1868.
Huang Y., Qu X., Zhang L., Yu Y. and Wu S. 2006 The incidence of \(\beta \)-thalassemia and analysis of \(\beta \)-thalassemia gene mutation in children in guangzhou region. J. Clin. Hematol. 6, 355–357.
Jia S. Q., Li J., Mo Q. H., Liao C., Li L. Y. and Xu X. M. 2004 Alpha0 thalassaemia \(\beta \)-thalassemia as a result of a novel 11.1 kb deletion eliminating both of the duplicated alpha globin genes. J. Clin. Pathol. 57, 164–167.
Joly P., Pondarre C. and Badens C. 2014 Beta-thalassemias: molecular, epidemiological, diagnostical and clinical aspects. Ann. Biol. Clin. (Paris) 72, 639–668.
Li M., Nie L. P., Tao X. M., Li Y. Z., Chen X. Z., Zheng G. Q. et al. 2005 Frequency and genotypes of thalassemia in Shenzhen area. J. Trop. Med. 3, 301–303.
Liebhaber S. A. and Kan Y. W. 1983 alpha-thalassemia caused by an unstable alpha-globin mutant. J. Clin. Invest. 71, 461–466.
Piel F. B. and Weatherall D. J. 2014 The \(\alpha \)-thalassemias. N. Engl. J. Med. 371, 1908–1916.
Sabath D. E., Bender M. A., Sankaran V. G., Vamos E., Kentsis A., Yi H. S. et al. 2015 Characterization of deletions of the HBA and HBB loci by array comparative genomic hybridization. J. Mol. Diagn. 18, 92–99.
Shi X. N., Chu J. Y. and Yang Z. Q. 2011 Genotype distribution and characteristics of beta-thalassemia of different province in China. Med. Recapitulate 4, 495–497.
Van de Velde H., Georgiou I., De Rycke M., Schots R., Sermon K., Lissens W. et al. 2004 Novel universal approach for preimplantation genetic diagnosis of beta-thalassaemia in combination with HLA matching of embryos. Hum. Reprod. 19, 700–708.
Yang K. G., Kutlar F., George E., Wilson J. B., Kutlar A., Stoming T. A. et al. 1989 Molecular characterization of \(\beta \)-thalassemia mutations in malay patients with HbE-\(\beta \)-globi–gene mutations in Malay patients with HbE-\(\beta \)-thalassaemia and thalassaemia major. Br. J. Haematol. 72, 73–80.
Yin X. L., Wu Z. K., He Y. Y., Zhou T. H., Zhou Y. L. and Zhang X. H. 2011 Treatment and complications of thalassemia major in Guangxi, Southern China. Pediatr. Blood Cancer 57, 1174–1178.
Xiong F., Sun M., Zhang X., Cai R., Zhou Y., Lou J. et al. 2010 Molecular epidemiological survey of haemoglobinopathies in the Guangxi Zhuang autonomous region of southern China. Clin. Genet. 78, 139–148.
Xu X. M., Zhou Y. Q., Luo G. X., Liao C., Zhou M., Chen P. Y. et al. 2004 The prevalence and spectrum of alpha and beta thalassaemia in Guangdong Province: implications for the future health burden and population screening. J. Clin. Pathol. 57, 517–522.
Zeng Y. and Huang S. 2001 The studies of hemoglobinopathies and thalassemia in China-the experiences in Shanghai Institute of Medical Genetics. Clin. Chim. Acta 313, 107–111.
Author information
Authors and Affiliations
Corresponding author
Additional information
He J., Zeng H., Zhu L., Li H., Shi L. and Hu L. 2017 Prevalence and spectrum of thalassaemia in Changsha, Hunan province, China: discussion of an innovative screening strategy. J. Genet. 96, xx–xx
Corresponding editor: Rajiva Raman
Rights and permissions
About this article

Cite this article
He, J., Zeng, H., Zhu, L. et al. Prevalence and spectrum of thalassaemia in Changsha, Hunan province, China: discussion of an innovative screening strategy. J Genet 96, 327–332 (2017). https://doi.org/10.1007/s12041-017-0779-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12041-017-0779-6