
Abstract
We report on the structure, the chemisorption and the electrocatalytic properties of multilayer (2.2, 3.3 and 5.5 physical monolayers) Pt films deposited on W(111) elaborated by molecular beam epitaxy. The Pt/W(111) surfaces were characterized by low-energy electron diffraction (LEED), X-ray photoelectron spectroscopy (XPS) and cyclic voltammetry (CV). Pronounced changes of the surface reactivity were noticed as the Pt coverage is decreased. In particular, the affinity for under-potentially deposited hydrogen (Hupd) and hydroxyl (OHads) species and the ability to electrooxidize a monolayer of COads were depreciated in agreement with strain and ligand effects.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The chemisorption and catalytic properties of a metal surface depend on its electronic [1, 2] and structural properties [3–5]. In that respect, bimetallic surfaces attract tremendous research interest because they offer the opportunity to tailor the selectivity, the catalytic activity and the stability of a given metal. Besides ‘ensemble effects’ [6], the modifications of the catalytic properties of a bimetallic surface are attributed to the combination of strain and ligand effects [7–16]. The strain effect arises from the compressed or expanded arrangement of the surface metal atoms and consequently increases or decreases the width of the electronic bands of the surface metal atoms. Expanding the lattice of the surface atoms leads to smaller atomic orbital overlap and thus reduces the width of the d-band. If the d-band is more than half filled, charge conservation causes an upshift of the d-band centre closer to the Fermi level and increases the surface reactivity. On the contrary, a compressed lattice makes the surface atoms less reactive due to the downshift of the d-band centre away from the Fermi level. The ligand effect is due to the change in the chemical properties of the metal surface atoms by the neighbouring element and causes similar variation of the chemisorption properties. Decoupling the strain and ligand effect is hardly feasible experimentally. An elegant approach by Gsell et al. [17] consisted of producing local strain on a Ru(0001) single crystal by means of subsurface gas bubbles resulting from Ar+ ion sputtering and annealing at moderate temperature (T ~1000 K). The Ru lattice was submitted to a tensile strain close to the top part of the bubbles, while the periphery of the regions was subjected to compressive strain. Oxygen molecules were found to adsorb preferentially on local areas of the surface with expanded lattice (tensile strain) [17]. Surface strain may also be applied by using metal films grown on single crystals [15, 16, 18]. In case of small lattice mismatch between the metal surface atoms and the underlying substrate, the surface atoms are deposited pseudomorphically with a lattice constant nearly identical to the substrate material. If the lattice mismatch is large, the deposited film relaxes and grows with its own lattice parameter upon a certain film thickness [19]. Thus, by varying the film thickness or the film–substrate pair, different systems with varied amounts of tensile or compressive strain can be obtained. Using this approach, Kibler et al. [15, 16] tailored the electrocatalytic activity of Pd atoms for the electrooxidation of formic acid. Hoster et al. [20] showed that the affinity to hydrogen atoms (Hads) and hydroxyl species (OHads) of Pt atoms deposited onto Ru(0001) increases with an increase in the Pt film thickness (i.e. a released compressive strain). This was reflected in cyclic voltammograms by a positive shift of the reversible adsorption of under-potentially deposited H (Hupd) and by a negative shift of the OHads adsorption features. Using a Cu(111) single crystal modified by thin Pt films, Strasser et al. [18] demonstrated that dealloyed Pt-Cu nanoparticles feature enhanced oxygen reduction reaction kinetics because of changes in the electronic structure of Pt due to lateral strain.
In this paper, thin Pt/W(111) films with coverage of 2.2, 3.3 and 5.5 physical monolayers (PMLs) were prepared by molecular beam epitaxy (MBE) and characterized by low-energy electron diffraction (LEED) and X-ray photoelectron spectroscopy (XPS). W(111) is a body-centred cubic surface that is atomically rough and open. As shown by Fig. 1, three outermost layers are exposed to the surface: these three layers form one PML. The surface reactivity of the Pt/W(111) thin films was tested for the adsorption/desorption of under-potentially deposited hydrogen and oxygen (OH species) and for the electrooxidation of a COads monolayer.

Top and side view of W(111) surface demonstrates that atoms from the three outermost layers are exposed to the surface—these three atomic layers together form one physical monolayer (PML). The top layer is shown in white, the layer beneath is shown in light grey and the bottom layer is shown in grey
Experimental Section
The molecular beam epitaxy (MBE) system used to deposit the Pt films was composed of a characterization chamber and a deposition chamber. The characterization chamber was equipped with surface preparation (Ar+ ion bombardment) and characterization (XPS and LEED) facilities. The substrate holder was a modified Riber 1″ molyblock onto which the single crystal was tightened using tantalum wires. A furnace capable of flash annealing the single crystal at temperatures T > 2273 K was positioned at a distance of 1 mm behind the sample. The W(111) single crystal was purchased from Mateck GmbH (99, 999 at%, 0.786 cm2 geometric). It was oriented and polished to within 0.1° of the (111) crystallographic orientation. The W(111) surface was prepared by several cycles composed of Ar+ ion bombardment at an energy of 600 eV followed by rapid annealing at T ~2300 K. The annealing of the W(111) single crystal induced surface segregation of chemical impurities such as carbon and oxygen contained in the bulk of the material. These contaminants were removed by Ar+ bombardment. After each cycle, the structural order was examined by LEED until the diagram showed a hexagonal (1 × 1) pattern with extinction of only a few spots (Fig. 2).

LEED diagram of a W(111) surface. The energy of the electron beam was 222 eV
These extinctions were attributed to a slight residual contamination of the surface with oxygen (typically 0.1 monolayer) and quantified by XPS (Fig. 3).

XPS spectrum of the W(111) surface after several Ar+ ion bombardment/annealing cycles. The peak observed at 532 eV corresponds to a slight contamination with oxygen (O1s)
Pt was evaporated from an electron beam evaporator at a deposition rate of 0.1 Ǻ s−1. The Pt film thickness was measured with a quartz balance, while maintaining the substrate at T ~600 K. Three samples with Pt physical coverages of 2.2, 3.3 and 5.5 PML corresponding to a Pt film thickness of 0.6, 0.8 and 1.3 nm, respectively, were prepared. A home-made transfer chamber enabled the transfer of the Pt/W(111) surfaces from UHV to ambient pressure for electrochemical measurements and transfer back to UHV for chemical and structural analysis (see Ref. [21]). To that goal, the transfer chamber was first disconnected from the main UHV chamber by closing the gate valve and then filled with argon (>99.999 %, referred to as 5 N Ar in what follows) up to the atmospheric pressure. Subsequently, an electrochemical reaction cell made of polychlorotrifluoroethylene (PCTFE) was mounted on top of a metallic tube, moved up into the transfer chamber through an opened gate valve and brought into contact with the as-prepared surface. The model Pt/W(111) electrode was pressed onto an O-ring (Kalrez™6375, Dupont de Nemours) by a mobile rod controlled by a micrometre screw, which also provided direct electrical contact to the sample/sample holder. The O-rings used to seal the electrochemical cell were cleaned by mild boiling into concentrated H2SO4/H2O2 and thoroughly rinsed in Milli-Q water before their use. The electrochemical experiments were conducted at room temperature using an Autolab/PGSTAT12 potentiostat. The geometric area of the Pt/W(111) single crystal in contact with the electrolyte was 0.5 cm2. The counter-electrode was a Pt foil and the reference electrode was a freshly prepared reversible hydrogen electrode (RHE). Sulphuric acid (H2SO4—Suprapur, Merck) diluted with Milli-Q water to 0.1 M was used as electrolyte solution. Electrolyte purged with argon (5 N Ar) was introduced in the electrochemical cell while maintaining the electrode potential at E = 0.1 V vs. RHE. In COads stripping experiments, CO molecules were adsorbed by bubbling with CO the electrolyte contained in an Erlenmeyer tank for 20 min and circulating the saturated electrolyte with the help of a peristaltic pump while holding the potential of the working electrode at E = 0.1 V vs. RHE. Next, the electrolyte was purged with Ar for 30 min before stripping off the adsorbed CO monolayer during a positive potential sweep at a potential sweep rate of v = 0.02 V s−1. After electrochemical experiments, the working electrode was transferred back to UHV for chemical characterization by XPS.
Results and Discussion
Structural Characterization of the Pt/W(111) Surfaces
Since the Pt atoms feature larger Wigner–Seitz radius than the W atoms [12] and because they possess different structure and lattice parameters,Footnote 1 they are under strong compressive strain. For example, if the Pt atoms accommodate with W atoms along the [−2 1 1] direction, the lattice parameter of Pt is contracted by ca 6.8 %. According to the elasticity theory, part of this strain may be relaxed by introducing misfit dislocations above a critical Pt film thickness (h c). This critical thickness for the Pt/W(111) system was calculated using the Matthews formulation [22, 23]:
where b is the Burgers vector, θ and λ are two angles of the slip plane and the Burgers vector with respect to the surface, respectively, ν d is the Poisson ratio’s (0.39 for Pt), ε is the lattice misfit and r 0 is related to the cut-off radius of the dislocation and its core energy (frequently r 0 = b/2 is used).
Two possible slip systems across 3{111} planes exist for the Pt/W(111) system [24]. The first system corresponds to partial dislocations with a Burgers vector \( \overrightarrow{\mathrm{b}}=\frac{1}{6}\left[211\right] \){111}, while the second corresponds to perfect dislocations with a Burgers vector \( \overrightarrow{\mathrm{b}}=\frac{1}{2}\left[011\right] \){111}. Using Eq. 1, two critical Pt film thicknesses were determined (h c = 0.3 nm for \( \overrightarrow{b}=\frac{1}{6}\left[211\right] \){111} and h c = 0.5 nm for \( \overrightarrow{b}=\frac{1}{2}\left[011\right] \){111}). Figure 4 shows that, whatever the slip system considered, misfit dislocations are created in the deposited Pt film after the completion of the first monolayer. These partial dislocations are energetically favourable up to a Pt film thickness h = 0.9 nm above which perfect dislocations become energetically more favourable. In each case, Pt atoms supported on W(111) remain compressed by ca 4 to 6 %.

Relaxation curves obtained for a thin Pt film deposited onto a W(111) surface calculated for the two possible slip systems: \( \overrightarrow{b}=\frac{1}{6}\left[211\right] \){111} or \( \overrightarrow{b}=\frac{1}{2}\left[011\right] \){111}. The dashed lines indicate the values of the critical thickness above which misfit dislocations become thermodynamically favourable
Surface Reactivity of the Pt/W(111) Films
Figure 5a displays the cyclic voltammograms of the Pt2.2PML/W(111) surface before and after three COads stripping experiments in 0.1 M H2SO4. On the as-prepared surface, the adsorption-desorption of Hupd proceeds via a single broad peak. After the electrochemical experiments, two broad peaks (oxidation region) and a single peak centred at E = 0.20 V vs. RHE (reduction region) are observed.

Cyclic voltammograms (a, c, e) and X-ray photon electron spectra (b, d, f) of Ptx-PML/W(111) (x = 2.2, 3.3, 5.5 PML, respectively) recorded before and after three COads stripping experiments in 0.1 M H2SO4 at a potential sweep rate v = 0.05 V s−1. For the sake of comparison, the cyclic voltammogram of a Pt(111) surface prepared in ultra-high vacuum and transferred to 0.1 M H2SO4 is displayed as a black trace
These electrochemical features are very similar to those observed on Pt/WO3 surfaces with low Pt geometrical coverage (ca 10 %) by Jayaraman et al. [25] and Micoud et al. [26, 27] and suggest an incomplete coverage of the W(111) surface by Pt atoms. In these studies, the oxidation/reduction peaks were assigned to the intercalation/de-intercalation of tungsten bronzes according to the following reaction:
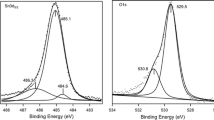
According to the Pourbaix diagram [28], exposure of the W atoms to acidic environment in the potential range 0.05 < E < 0.95 V vs. RHE results in the formation of WO3. This was confirmed by XPS spectra acquired after the electrochemical measurements (Fig. 6): the W 4f7/2 and 4f5/2 transition peaks were observed at 30.7 and 32.7 eV for the fresh Pt-2.2PML/W(111) surface and at 35.4 and 37.6 eV after the electrochemical measurements, respectively. Furthermore, the disappearance of the Pt transition peaks indicates that the Pt atoms were removed during the electrochemical measurements, most likely as a result of the oxidation of the underlying W atoms.

X-ray photoelectron spectra of the catalytic surfaces evaluated in this work
On Pt3.3-PML/W(111) and Pt5.5-PML/W(111), the Hupd adsorption/desorption features are typical of a compressed (111) surface (see, for example, the voltammogram of a Pt3Ni(111) in reference [29]), and remain stable over time (Fig. 5). The W 4f7/2 and W 4f5/2 transition peaks measured on the as-prepared Pt3.3-PML/W(111) and the Pt5.5-PML/W(111) surfaces are located at 30.7 and 32.7 eV, in agreement with the theoretical values of metallic tungsten [30]. No shift in the XPS peak position after the electrochemical experiments discards the possibility of W oxides or Pt–W alloy formation (Fig. 5d). In addition, the ratio of the intensity of the Pt and the W transition peaks remained nearly constant before/after the electrochemical experiments, therefore excluding the desorption of Pt atoms from the W(111) surface.
A 100-mV shift of the Hupd adsorption/desorption region towards negative potentials is observed with decreasing Pt film thicknesses (i.e. increasing compressive strain, Fig. 5c, e). This result agrees with the d-band theory [11, 31] and former experimental observations of Hoster et al. [20] on Pt/Ru(0001) and Van der Vliet et al. on a Pt3Ni(111)-skin surface [29]. According to Pallassana et al. [31], this shift in potential may be rationalized by considering that changes of the chemisorption energy of hydrogen atoms depend on the d-band centre of the surface metal atoms as
where V 2 is the d-band coupling matrix element for the surface metal atom and Δε = |ε d − ε a | the difference between ε d and ε a , the location of the d-band centre and the H1s orbital state, respectively.
COads Stripping Experiments
The COads monolayer electrooxidation follows a Langmuir–Hinshelwood mechanism, where OHads is formed by water splitting; COads + OHads recombine on two adjacent sites to form COOHads, which ultimately is oxidized into CO2:
where * denotes a free Pt surface site.
Figure 7a shows the COads stripping voltammograms recorded on the W(111) surfaces covered with 2.2, 3.3 or 5.5 physical monolayers and that measured on a Pt (111) single crystal for comparison. Similar to what was observed for the adsorption of Hupd, the catalytic properties of the PtxPML/W(111) are markedly different from that of the Pt(111) electrode. Figure 7 shows that both the onset and the main COads stripping peak potential shift towards positive potential with decreasing the number of deposited Pt monolayers. This suggests that the coverage with OHads species required to oxidize the adsorbed CO molecules (Eq. 4) is lowered on the thinner Pt films. The position of the COads stripping peak on Pt(111) and Pt5.5PML/W(111) is very close—E = 0.70 V vs. RHE—thereby suggesting that both surfaces have similar catalytic properties or, in other terms, that strain and ligand effects have vanished at this coverage. Interestingly, we also noticed that a pre-peak developed between E = 0.35 and 0.60 V vs. RHE (Fig. 7b) on the Pt2.2PML/W(111) and Pt5.5PML/W(111) surfaces: This pre-peak was ascribed to the oxidation of CO molecules adsorbed on surface defects (adatoms and step edges) in analogy with former observations on Pt(111) and stepped surfaces in the literature [32–35]. The increased surface reactivity of the thicker Pt films agrees with their highly defective nature in particular with the presence of strain-induced misfit dislocations as discussed in the section “Structural Characterization of the Pt/W(111) Surfaces”. The effect of the crystalline orientation, the terrace width and the density of structural defects on the COads electrooxidation kinetics has been widely discussed in the work of Lebedeva et al. [32–34]. However, it is believed to have minor influence compared to strain and ligand effects induced by the W(111) substrate.

a COads stripping voltammograms measured on a W(111) single crystal covered by 2.2, 3.3 or 5.5 physical monolayers of Pt, b zoom in the potential region 0.3 < E < 0.7 V vs. RHE and c potential of the onset and of the COads electrooxidation peak. The onset potential was defined as the potential at which 10 % of the current value at the peak potential was reached. 0.1 M H2SO4, v = 0.02 V s−1, E ads = 0.1 V vs. RHE
Finally, we stress that the COads stripping peaks become increasingly asymmetric and seem to be composed of multiple peaks when the number of Pt monolayers decreases. This phenomenon may be rationalized by considering that (i) the fraction of active sites where OH species are generated from water splitting is lowered on the most compressed Pt films, (ii) the adsorption of anions or the relaxation of CO islands during COads electrooxidation blocks the freed Pt atoms and prevents the dissociative adsorption of water molecules, and/or (iii) different multilayer structures featuring different surface reactivity are present on the thinner Pt films. In the frame of the first hypothesis, we emphasize that the potential difference between the onset and the main COads stripping peak potential varies monotonously with the number of deposited Pt monolayers (Fig. 7b), therefore suggesting that OHads formation is not limiting the reaction kinetics in the potential range investigated. The second hypothesis may also be ruled because the COads stripping peak is narrow on Pt(111). In consequence, the pronounced current tailing at high electrode potential (superior to the current maximum) was tentatively ascribed to the presence of different multilayer structures, i.e. Pt adislands featuring different thickness and morphology, in the thinner Pt/W(111) films. Although pieces of evidence of this hypothesis could not be obtained by scanning tunnelling microscopy, it is very well established that Volmer–Weber or 3D growth is favoured during heteroepitaxial metal deposition. In consequence, the surface of the thinner Pt/W(111) films is believed to be composed of Pt adislands with different sizes (number of atoms), height and thus surface reactivity. In that respect, we refer to former work of Kibler et al. [15, 16] in which the polarization curves obtained on Pt monolayers supported onto different single crystals always contained multiple peaks, which were ascribed to different reaction rates on strained and unstrained regions of the Pd films. A last but hardly provable hypothesis would be that COads molecules are stabilized on the thinnest Pt films, thereby decreasing the rate of Eq. 5. Keeping the same idea in mind, it is noteworthy that similar tailing and multiple COads electrooxidation peaks were observed by Maillard et al. [36–38] on Pt nanoparticles with different sizes and related to restricted COads surface mobility. The authors showed that the COads diffusion coefficient strongly depends on the nanoparticle size and that a transition towards fast diffusion was observed when the Pt nanoparticle size exceeded ca 3 nm.
Conclusion
In conclusion, Pt atoms were deposited on a W(111) single-crystal electrode via molecular beam epitaxy. A minimal coverage of 2.2 physical monolayers was found necessary to ensure the stability of the Pt/W(111) electrode in acidic electrolyte as shown by combined cyclic voltammetry and XPS experiments. Below this coverage, a certain number of W atoms are exposed to the acidic electrolyte, leading to the formation of WO3 and inducing the desorption of the Pt films from the W(111) single crystal. Above this coverage, the Pt films were found to be stable in the acidic electrolyte. The chemisorption energy of Hupd and OH species on PtxPML/W(111) strongly depends on the number of deposited Pt physical monolayers as reflected by cyclic voltammograms. The Hupd adsorption/desorption region is shifted to lower electrode potential as the number of Pt physical monolayers decreased, in good agreement with the d-band theory. More compressed Pt monolayers feature decreased surface reactivity for the electrochemical COads oxidation.
Notes
Bulk W crystallizes in body centred cubic (bcc) structure with a lattice parameter equal to 0.3165 nm, while Pt adopts the face centred cubic (fcc) structure with a lattice parameter equal to 0.3924 nm.
References
H.-L. Jiang, Q. Xu, J Mater Chem 21, 13705 (2011)
A.-Q. Wang, C.-M. Chang, C.-Y. Mou, J Phys Chem B 109, 18860 (2005)
R. Parsons, G. Ritzoulis, J Electroanal Chem Interfacial Electrochem 318, 1 (1991)
D.S. Strmcnik, D.V. Tripkovic, D. van der Vliet, K.C. Chang, V. Komanicky, H. You, G. Karapetrov, J. Greeley, V.R. Stamenkovic, N.M. Markovic, J Am Chem Soc 130, 15332 (2008)
W. Yu, M.A. Barteau, J.G. Chen, J Am Chem Soc 133, 20528 (2011)
W.M.H. Sachtler, Handbook of heterogeneous catalysis, vol. 3 (Wiley-VCH Verlag GmbH & Co. KGaA, Darmstadt, 2008), p. 1585
B. Hammer, Y. Morikawa, J.K. Norskov, Phys Rev Lett 76, 2141 (1996)
J.R. Kitchin, J.K. Nørskov, M.A. Barteau, J.G. Chen, J Chem Phys 120, 10240 (2004)
J.R. Kitchin, J.K. Nørskov, M.A. Barteau, J.G. Chen, Phys Rev Lett 93, 156801 (2004)
J.A. Rodriguez, D.W. Goodman, J Phys Chem 95, 4196 (1991)
B. Hammer, J.K. Nørskov, Surf Sci 343, 211 (1995)
A. Ruban, B. Hammer, P. Stoltze, H.L. Skriver, J.K. Nørskov, J Mol Catal A 115, 421 (1997)
I.E.L. Stephens, A.S. Bondarenko, F.J. Perez-Alonso, F. Calle-Vallejo, L. Bech, T.P. Johansson, A.K. Jepsen, R. Frydendal, B.P. Knudsen, J. Rossmeisl, I. Chorkendorff, J Am Chem Soc 133, 5485 (2011)
I.E.L. Stephens, A.S. Bondarenko, U. Grønbjerg, J. Rossmeisl, I. Chorkendorff, Energy Environ Sci 5, 6744 (2012)
L.A. Kibler, A.M. El-Aziz, R. Hoyer, D.M. Kolb, Angew Chem Int Ed 44, 2080 (2005)
L.A. Kibler, A.M. El-Aziz, D.M. Kolb, J Mol Catal A 199, 57 (2003)
M. Gsell, P. Jakob, D. Menzel, Science 280, 717 (1998)
P. Strasser, S. Koh, T. Anniyev, J. Greeley, K. More, C. Yu, Z. Liu, S. Kaya, D. Nordlund, H. Ogasawara, M.F. Toney, A. Nilsson, Nat Chem 2, 454 (2010)
Y. Soldo-Olivier, E. Sibert, B. Previdello, M.C. Lafouresse, F. Maillard, M. De Santis, Electrochim Acta 112, 905 (2013)
H.E. Hoster, O.B. Alves, M.T.M. Koper, Chem Phys Chem 11, 1518 (2010)
M. El-Jawad, J.-L. Chemin, B. Gilles, F. Maillard, Rev Sci Instrum 84, 064101 (2013)
J.W. Matthews, A.E. Blakeslee, J Cryst Growth 27, 118 (1974)
L.B. Freund, S. Suresh, Thin films materials: stress, defect formation and surface evolution (Cambridge University Press, Cambridge, 2006)
D. Hull, D.J. Bacon, in Introduction to dislocations, ed. by D. Hull, D.J. Bacon (Butterworth-Heinemann, Oxford, 2001), p. 82
S. Jayaraman, T.F. Jaramillo, S.H. Baeck, E.W. McFarland, J Phys Chem B 109, 22958 (2005)
F. Micoud, F. Maillard, A. Gourgaud, M. Chatenet, Electrochem Commun 11, 651 (2009)
F. Micoud, F. Maillard, A. Bonnefont, N. Job, M. Chatenet, Phys Chem Chem Phys 12, 1182 (2010)
M. Pourbaix, Atlas d’Equilibres Electrochimiques (Gauthier-Villars & Cie, Paris, 1963)
D.F. Van Der Vliet, C. Wang, D. Li, A.P. Paulikas, J. Greeley, R.B. Rankin, D. Strmcnik, D. Tripkovic, N.M. Markovic, V.R. Stamenkovic, Angew Chem Int Ed 51, 3139 (2012)
C.D. Wagner, W.M. Riggs, L.E. Davis, J.F. Moulder, Handbook of X-ray photoelectron spectroscopy (Perkin-Elmer Corp. (Physical Electronics), Eden Prairie, 1979)
V. Pallassana, M. Neurock, L.B. Hansen, B. Hammer, J.K. Norskov, Phys Rev B 60, 6146 (1999)
N.P. Lebedeva, M.T.M. Koper, E. Herrero, J.M. Feliu, R.A. van Santen, J Electroanal Chem 487, 37 (2000)
N.P. Lebedeva, A. Rodes, J.M. Feliu, M.T.M. Koper, R.A. van Santen, J Phys Chem B 106, 9863 (2002)
N.P. Lebedeva, M.T.M. Koper, J.M. Feliu, R.A. van Santen, J Phys Chem B 106, 12938 (2002)
A. López-Cudero, A. Cuesta, C. Gutiérrez, J Electroanal Chem 579, 1 (2005)
F. Maillard, M. Eikerling, O.V. Cherstiouk, S. Schreier, E. Savinova, U. Stimming, Faraday Discuss 125, 357 (2004)
F. Maillard, E.R. Savinova, P.A. Simonov, V.I. Zaikovskii, U. Stimming, J Phys Chem B 108, 17893 (2004)
F. Maillard, E.R. Savinova, U. Stimming, J Electroanal Chem 599, 221 (2007)
Compliance with Ethical Standards
Disclosure of Potential Conflict of Interest
The authors declare that they have no conflict of interest (financial or non-financial).
Funding
This study was funded by Oseo-Anvar in the framework of the H2E project.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
El Jawad, M.K., Gilles, B. & Maillard, F. Structure and Surface Reactivity of Ultra-Thin Pt/W(111) Films. Electrocatalysis 6, 398–404 (2015). https://doi.org/10.1007/s12678-015-0260-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12678-015-0260-3