
Abstract
Amyotrophic lateral sclerosis (ALS) is a progressive incurable neurodegenerative disease with selective loss of lower and upper motoneurons. Dysfunction and destruction of neuromuscular synapses leading to skeletal muscle denervation is one of the early and major events in ALS pathogenesis. Despite of the presence of studies devoted to investigation of neuromuscular transmission in ALS mouse models, no detailed information about molecular mechanisms underlying synaptic dysfunction in ALS and their temporal dynamics during ALS progression is provided. The goal of present work was to study the processes of neurotransmitter release and presynaptic vesicle recycling in neuromuscular synapses of mutated SOD1 (mSOD1) transgenic mice at different clinical stages of disease. Utilizing combination of electrophysiological recording and FM 1–43 (N-(3-triethylammoniumpropyl)-4-(4-(dibutylamino) styryl) pyridinium dibromide) fluorescent imaging, we found that mSOD1 mice at symptomatic and terminal stages of disease showed decreased baseline quantal content of end-plate potentials and prolonged synaptic vesicle recycling time comparing to wild-type mice. Despite the decrease of end-plate potential (EPP) quantal content, studied mSOD1 mice groups showed unchanged dynamics of EPP relative amplitude comparing to WT mice. We also found an increase of miniature end-plate potential amplitude in mSOD1 mice at symptomatic stage, which may reflect compensatory mechanism that alleviates reduction of EPP amplitude. Thus, we provided one of the first detailed characteristics of presynaptic dysfunction at neuromuscular junction in ALS model. Obtained data expand our understanding of the ALS pathogenesis and contribute to stage- and localization-specific description of ALS pathogenetic mechanisms.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive incurable neurodegenerative disease with selective loss of lower and upper motoneurons. There are 1–2 ALS new cases annually and 3–5 cases totally per 100,000 people in the USA and Europe, whereas cumulative lifetime risk of ALS development is about 1 in 400 [1, 2]. Disease starts with focal muscle weakness and progressively leads to atrophy and paralysis of skeletal muscles including diaphragm. In majority of clinical cases, death of patient occurs within 3–5 years after diagnosis. About 5–10% of ALS cases are familial. To date, around 20 genes are implicated in familial, sporadic, or both forms of ALS with the most common causes of ALS associated with mutations in SOD1, TARDBP, FUS, and C9orf72 genes [3, 4]. SOD1 mutations account for about 20% of familial ALS cases and were used to generate mutated SOD1 (mSOD1) transgenic mouse lines that are widely used for ALS experimental modelling [5, 6].
ALS pathogenesis reflects complex interplay between genetic and environmental factors. Heterogeneity of familial and sporadic forms of ALS suggests the convergence of cellular and molecular mechanisms leading to motor neuron degeneration. Dysfunction and destruction of neuromuscular synapses leading to skeletal muscle denervation is one of the early and major events in ALS pathogenesis [7, 8]. It was found that dramatic decline of spontaneous acetylcholine release from motor terminals occurs in mSOD1 mice at early symptomatic stage [9]. Another study showed that neuromuscular transmission in mSOD1 mice is enhanced at the pre-symptomatic phase and in part of synapses at the symptomatic phase, which is consistent with the hypothesis that skeletal muscles in ALS undergo cycles of denervation/re-innervation [10]. Despite of the presence of studies devoted to investigation of neuromuscular transmission in ALS mouse models, no detailed information about molecular mechanisms underlying synaptic dysfunction in ALS and their temporal dynamics during disease progression is provided. The contribution of complex presynaptic machinery (including synaptic vesicle recycling mechanisms) to neuromuscular dysfunction in ALS is also unclear. Data about mechanisms of synaptic dysfunction in ALS can provide a key for development of effective pathogenetic therapy.
Considering abovementioned rationale, the goal of present work was to study the processes of neurotransmitter release and presynaptic vesicle recycling in neuromuscular synapses of mSOD1 transgenic mice at different clinical stages of disease.
2 Materials and Methods
2.1 Amyotrophic Lateral Sclerosis Model
Transgenic mice B6SJL-TG (SOD1-G93A)dl1Gur/J carrying the human SOD1 gene with the G93A point mutation were purchased from Jackson Laboratory (USA), and a colony was established within Bioresource Collection of Puschino Animal Facility of the Branch of RAS Institute of Bioorganic Chemistry (Puschino, Moscow region). Two weeks prior to experiments, mice were delivered to Kazan State Medical University and housed under standard laboratory conditions. All animals were housed 4–5 animals/cage, under 12 h light/12 h dark cycle, and received food and water ad libitum. Animal experiment protocols were approved by the Kazan State Medical University Local Ethic Committee.
Two experimental groups of mSOD1 mice were formed based on age and clinical symptomatology:
-
1)
mSOD1 mice at symptomatic stage of disease: 6–7 months old, significant pareses are present, paralysis of one of hind limb is possible, but mice are able to locomote (mSOD1(6–7)).
-
2)
mSOD1 mice at terminal stage of disease: ~ 8 months old, both hind limbs are paralyzed, mice are unable to locomote (mSOD1(8)).
Wild-type (WT) mice were used as a control for mSOD1 mice groups.
2.2 Electrophysiology
Diaphragm muscle with the attached phrenic nerve was isolated, severed into two halves, and one of the hemi-diaphragms was mounted on a 5-ml Sylgard covered chamber. Preparation was perfused continuously with an aerated physiological saline solution (mM): NaCl—125; KCl—2.5; CaCl2—2; NaH2PO4—1; MgCl2—1; and glucose—11. The pH of solution was adjusted to 7.3 at 20 °C. All reagents were purchased from Sigma-Aldrich (USA), unless otherwise stated.
To prevent muscle contraction following nerve stimulation, the muscles were transected. Intracellular recording of end-plate potentials (EPPs) and miniature end-plate potentials (MEPPs) was done using Axoclamp 900A electrophysiological amplifier (Molecular Devices, CA, USA). Glass microelectrode (4–9 MΩ) filled with 3 M KCl was inserted to muscle fibers at the motor endplate (near nerve terminals), and chlorinated reference electrode was placed in the bath. Nerve terminals were visualized with an interference-polarization microscope (Biolar, Poland, × 400). The motor nerve was taken up into a suction plastic pipette and stimulated with electrical pulses of supra-threshold amplitude and 0.1–0.2 ms duration with Digitimer DS3 stimulator.
At each experiment, EPPs were recorded at low frequency stimulation of motor nerve (0.2 Hz) for 10–12 min to assess the baseline neurotransmission. To evaluate use-dependent synaptic plasticity and establish synaptic vesicle recycling parameters, few prolonged stimulation protocols were used: 500 pulses of 5, 10, 20 Hz, and 8000 pulses of 50 Hz. To evaluate spontaneous neurotransmitter release at each experiment, MEPPs were detected using amplitude thresholds set as a multiple (four to five times) of the noise standard deviation for 5–10 min; each event was also visually inspected to prevent noise disturbance of the analysis. The resting membrane potential (RMP) of the muscle fiber was monitored through the entire experiment; those experiments that showed significant drop of RMP were not analyzed.
Electrophysiological signals were digitized at 5 μs intervals by an analog-digital converter Lcard LA-2USB, stored, and processed offline with a PC and original software “Elph” (developed by Dr. Andrey Zakharov). EPPs (100–150) were analyzed to calculate amplitudes, rise times (from 20 to 80% of the amplitude), half decay times. For analysis of EPP amplitude dynamics in high-frequency stimulation trains, the first signal in train was taken for 100%. EPP quantal content (QC) was calculated based on 100–300 EPPs by amplitude dispersion analysis [11, 12]. MEPPs (100–120) were analyzed to calculate frequency, amplitude, and temporal parameters of MEPPs.
2.3 FM 1–43 Fluorescent Imaging
Fluorescent styryl dye FM 1–43 (N-(3-(triethylammoniumpropyl)-4-(4-dibutylamino) styryl) pyridinium dibromide, 6 μM) was used to estimate the rate of exo- and endocytosis of synaptic vesicles. To label nerve terminals with FM 1–43, diaphragm preparations were stimulated via motor nerves with 50 Hz frequency for up to 1 min, whereas FM 1–43 dye was applied to experimental chamber 1 min prior to beginning of stimulation and washed out from chamber in 1 min after stimulation. FM 1–43 was taken up into the vesicles during activity-induced vesicle turnover (FM 1–43 loading), which was manifested by appearance of bright fluorescent spots at the nerve terminals [13, 14]. Intensity of FM 1–43 loading reflects the intensity of endocytosis. Fifty Hertz stimulation of FM 1–43 labelled preparations resulted in destaining of nerve terminals (FM 1–43 unloading). During unloading, the imaging was performed every 60 s during 10–15 min. The speed of FM1–43 unloading reflects the intensity of exocytosis of synaptic vesicles.
The preparations were viewed with the OLYMPUS BX51 fluorescence microscope (Japan) equipped with Disc Scanning Unit (for spinning disk confocal microscopy) and 100 W Hg lamp. The objective lens used was the LUMPlanFl 60X (0.9 NA) water immersion. For FM 1–43 imaging, Olympus “U-MNB2” fluorescent cube (BP 470–490 excitation, 500 dichroic, LP 520 emission filter) was used. Only terminals on surface muscle fibers were studied. Images were captured with Hamamatsu OrcaR2 CCD camera and then stored and processed with a PC. The analysis of fluorescent images was done with use of CellP and ImageJ software. The fluorescence of the nerve terminal was calculated in relative units as an average brightness of all pixels of the part of the terminal after subtraction of background (muscle fibers) fluorescence. Background fluorescence was estimated in the neighbor part of muscle fiber. Two hundred fifty five relative units was equated to absolute white.
2.4 Synaptic Vesicles Recycling Time Estimation
An important parameter of synapse functioning is the average time of reuse of synaptic vesicles in neurotransmitter release. The rate of recycling of vesicles at presynaptic terminal directly influences level on neurotransmitter secretion and reflects functional abilities of synapse. To estimate synaptic vesicles recycling time, the juxtaposition of the cumulative amount of released neurotransmitter quanta with the dynamics of FM 1–43 dye loss by nerve terminal during FM 1–43 unloading was performed as described elsewhere [15]. For this analysis, cumulative curve of released neurotransmitter quanta was imposed on the FM 1–43 dye loss curve scaling them so that the initial parts of the curves coincide. At the beginning of the graph, the loss of dye corresponds to the cumulative curve of quantal release; however, from a certain point of time, the curves diverge, which relates to release of neurotransmitter from the vesicles that have already passed one round of exocytosis and do not contain a dye. The moment of curves divergence allows approximate estimation of recycling time.
2.5 Statistical Analysis of Experimental Results
All data are presented as mean ± SEM (n = number of synapses), with statistical significance assessed by Student’s t test. A P value of less than 0.05 was considered to indicate a statistically significant difference.
3 Results
3.1 Electrophysiological Changes of Neuromuscular Synaptic Transmission in mSOD1 Mice
At control conditions, WT mice MEPP frequency was equal to 1.04 ± 0.15 s−1, and MEPP amplitude, rise time, and half-decay time were 1.01 ± 0.11 mV, 0.45 ± 0.05 ms, and 1.89 ± 0.2 ms, correspondingly (n = 8). EPP rise time, half-decay time, and quantal content were 0.54 ± 0.03 ms, 2.42 ± 0.14 ms, and 175.8 ± 10.5, correspondingly (n = 6). In natural in vivo conditions, neuromuscular synapses conduct high-frequency trains of synaptic signals which accompanied by changes in EPP amplitude due to processes of short-term synaptic plasticity [16, 17]. High-frequency stimulation (5, 10, 20 Hz) of WT neuromuscular preparations was accompanied by typical biphasic dynamics of EPP amplitude decrease (Fig. 1). Initial (within few seconds) sharp decline of EPP amplitude down to 70–80% of baseline values was followed by relative stabilization and slow decay. So, under 5 Hz stimulation, EPP amplitude decreased at 25th signal down to 78.8 ± 1.1% and then аt 500th signal to 72.3 ± 1.9% of initial values (n = 12) (Fig. 1a). Under 10 Hz stimulation, EPP amplitude dropped down to 78.3 ± 1.3% at 10th signal and then reached 74.1 ± 1.3% of initial values at 500th signal (n = 14) (Fig. 1b). Within 20 Hz stimulation, EPP amplitude decreased down to 78.2 ± 1.0% and then to 72.1 ± 1.4% at 10th and 500th signals, respectively (n = 14) (Fig. 1c). For further analysis of presynaptic vesicle cycle, we also studied EPP amplitude dynamics during prolonged (160 s) 50 Hz stimulation. Under 50 Hz stimulation, the exponential decline of EPP amplitude was found. EPP amplitudes were equal to 84.7 ± 4.5%, 27.9 ± 6.0%, and 13.4 ± 5.0% of baseline level at 10th, 3000th (60 s), and 8000th (160 s) signals , respectively (n = 15) (Fig. 1d).

Dynamics of amplitudes of end-plate potentials in mSOD1 transgenic mice. The figure shows the dynamics of average EPP amplitudes at rhythmic stimulation with frequencies of 5 Hz (a), 10 Hz (b), 20 Hz (c), and 50 Hz (d) in WT (white squares), mSOD1(6–7) (black triangles), and mSOD1(8) (black circles) mice. Significant differences from WT mice (asterisk)
There were no significant changes found in abovementioned electrophysiological parameters of neuromuscular synaptic transmission between 3–4, 6–7, and 8 months old WT mice, which is in line with previous findings [18].
In mSOD1(6–7) mice, MEPP amplitude was 1.5 ± 0.15 mV, being significantly higher comparing to WT mice (n = 7, P < 0.05); MEPP temporal parameters and frequency did not differ from those in WT mice (n = 7). EPP quantal content in mSOD1(6–7) mice (109 ± 14, n = 7) was significantly lower comparing to WT mice (P < 0.05), whereas EPP rise time and half-decay time did not differ from WT mice. There were no significant changes in EPP amplitude dynamics under 5, 20, and 50 Hz stimulations in mSOD1(6–7) mice comparing to WT mice (Fig. 1a, c, d), whereas at 10 Hz stimulation, the depression of EPP amplitude at 10–50 s of stimulation was more expressed comparing to WT mice (n = 8) (Fig. 1b).
There were no significant changes found in MEPP parameters between mSOD1(8) mice and WT mice (n = 7, P > 0.05). EPP quantal content in mSOD1(8) mice was 130.5 ± 7.8 mV, which is significantly lower comparing to WT mice (n = 7, P < 0.05); EPP temporal parameters did not differ from WT mice. Dynamics of EPP amplitude in mSOD1(8) mice under 5, 10, 20, and 50 Hz stimulation also did not differ from those in WT mice (n = 7) (Fig. 1).
Thus, although the parameters of neuromuscular synaptic transmission were slightly different in studied age groups of transgenic mice, both mSOD1 mice groups were characterized by marked reduction in EPP quantal content and mostly unchanged dynamics of EPP amplitude during high-frequency trains comparing to WT mice.
3.2 Cumulative Neurotransmitter Quantal Release in mSOD1 and WT Mice Under High-Frequency Stimulation
To compare the intensity of FM 1–43 loading into nerve terminals of different experimental groups of mice, the stimulation protocols that elicit the release of same cumulative amounts of neurotransmitter quanta (and possess similar stimulating effect on the endocytosis of synaptic vesicles) in different groups were adjusted by varying the duration of motor nerve stimulation.
According to the cumulative curves of EPP quantal release, 1 min 50 Hz stimulation of mSOD1(6–7) neuromuscular synapse induces release of 93,988.3 ± 16,464.3 neurotransmitter quanta (n = 6), which is more than two times lower comparing to WT mice under the same stimulation protocol (222,573.0 ± 37,239.0 quanta, n = 8); the revealed discrepancy is explained mainly by different levels of baseline EPP quantal content in two experimental groups. We were able to empirically find that 1 min 50 Hz stimulation of mSOD1 (6–7) neuromuscular synapse induces release of same amount of neurotransmitter quanta as 20 s 50 Hz stimulation of WT neuromuscular synapse (96,136.8 ± 13,967.8 quanta, n = 6). 1 min 50 Hz stimulation of mSOD1(8) neuromuscular synapse induces release of 170,327.6 ± 23,713.7 neurotransmitter quanta (n = 6), which is equal to cumulative quanta release elicited by 38 s 50 Hz stimulation at WT synapse.
Thus, we were able to empirically find high-frequency stimulation protocols that induce release of equal amount of neurotransmitter quanta in synapses of different mice experimental groups. Obtained data provide important tool for further experiments with FM 1–43 imaging of endo- and exocytosis of synaptic vesicles.
3.3 FM 1–43 Imaging of Synaptic Vesicles Endo- and Exocytosis in mSOD1 Mice
High-frequency stimulation of neuromuscular preparation in presence of FM 1–43 dye in experimental chamber resulted in increase of fluorescence intensity of nerve terminals and appearance of bright fluorescent spots representing clusters of synaptic vesicles that have undergone cycle of exo- and endocytosis and contain dye (Fig. 2a). FM 1–43 stained synapses of mouse diaphragm were visualized as compact, oval shape branchings of axon terminals with fluorescence noticeable almost throughout the area of nerve terminals. Consequent 50 Hz stimulation of FM 1–43 loaded synapses resulted in exponential decay of terminal fluorescence intensity. At 7–9 min of stimulation, the unloading curve reached the plateau at the level of 35–40% of initial values (Fig. 3a).

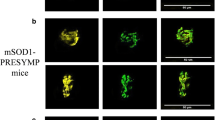
Imaging of FM 1–43 loaded nerve terminals of mSOD1 transgenic mice. Fluorescent images represent FM 1–43 loading in WT mice (20 s 50 Hz stimulation; a), mSOD1(6–7) (1 min 50 Hz stimulation; b), and mSOD1(8) (1 min 50 Hz stimulation; c) mice synapses

Recycling of synaptic vesicles in neuromuscular synapses of mSOD1 transgenic mice. a Curves representing decrease of average fluorescence intensity of FM 1–43 pre-loaded nerve terminals during prolonged 50 Hz stimulation (FM 1–43 unloading) in WT (white squares), mSOD1(6–7) (black triangles), and mSOD1(8) (black circles) mice. Significant differences from WT mice (asterisk). b–d Evaluation of vesicle recycle time by juxtaposing the cumulative curve of neurotransmitter secretion (Σm) and the curve of FM 1–43 loss (ΔF) under the 50 Hz stimulation of FM 1–43 pre-loaded nerve endings in WT mice (b), mSOD1(6–7) (c), and mSOD1(8) (d) mice
The fluorescence intensity of FM 1–43 loaded (50 Hz 1 min stimulation) terminals at mSOD1(6–7) mice was 26.9 ± 0.9 (n = 99), which is significantly higher comparing to WT (10.7 ± 0.6 (n = 90) after 50 Hz 20 s stimulation) (Fig. 2b). Prolonged 50 Hz stimulation of pre-stained mSOD1 (6–7) mice synapses resulted in a more rapid decrease in fluorescence intensity compared to WT mice in the first minutes of stimulation. On the fourth and subsequent minutes of stimulation, the fluorescence intensity in mSOD1(6–7) mice did not differ significantly from WT mice (Fig. 3a).
mSOD1(8) mice were also characterized by increased fluorescence intensity of FM 1–43 stained (50 Hz 1 min stimulation) terminals comparing to WT mice (50 Hz 38 s stimulation): 41.2 ± 2.1 (n = 69) vs. 16.0 ± 1.1 (n = 52), correspondingly (significant difference) (Fig. 2c). High-frequency stimulation of pre-stained preparations of mSOD1(8) mice led to accelerated release of the dye by exocytosis processes and a drop of fluorescence intensity in comparison with WT mice: at the 5th minute of stimulation, the intensity of fluorescence reached 25.4 ± 4.1% (n = 6) of the initial level vs. 42.9 ± 4.5% in WT mice (n = 7).
Obtained data certify that neuromuscular synapses of studied mSOD1 mice are characterized by enhanced FM 1–43 loading and accelerated FM 1–43 unloading comparing to WT mice; both phenomena are more pronounced in mSOD1(8) than in mSOD1(6–7) mice.
3.4 Evaluation of Synaptic Vesicles Recycling Time in mSOD1 Mice
We found that cumulative curve of released quanta of neurotransmitter and dynamics of FM 1–43 loss by nerve terminal in WT mice starts to diverge 60 s after beginning of stimulation (Fig. 3a). Thus, we estimated synaptic vesicles recycling time in WT mice as approximately 60 s, which is in line with previous findings [12]. Both in mSOD1(6–7) and mSOD1(8) mice, no signs of divergence of superimposed cumulative quantal release curve and FM 1–43 loss dynamics were revealed during at least 140 s (Fig. 3b, c). We concluded that synaptic vesicles recycling times in both tested mSOD1 mice groups are longer than 140 s which is significantly more comparing to WT mice.
4 Discussion
Over recent years, a lot of experimental data have been obtained in favor of the “dying-back hypothesis” of ALS pathogenesis, suggesting that pathological process begins in the skeletal muscle and/or neuromuscular synapse [7], which promotes the development of degeneration of the lower and then upper motor neurons [19]. In accordance with this theory, ALS is a distal axonopathy with impairments that occur in the neuromuscular synapse and extend proximally in the direction of the body of the motor neuron. By the way, the presence of signs of distal axonopathy was found in Alzheimer’s and Parkinson’s diseases, while axonal defects were observed before cell death, and the loss of axonal function correlated with the onset of functional disorders [20,21,22]. In general, dysfunction and destruction of neuromuscular synapses leading to skeletal muscle denervation is recognized as one of the earliest and most important events in ALS pathogenesis [7, 23].
Key pathogenetic mechanisms underlying neurotoxicity and motoneuron death in ALS are well described and include oxidative stress, abnormal protein aggregation, mitochondrial dysfunction, glutamate excitotoxicity, impairment of axonal transport, synaptic dysfunction, and glial pathology [1, 24, 25]. However, performed studies have not allowed to develop a single hypothesis that combines different pathogenetic mechanisms; that fact makes it impossible to develop an effective neuroprotective therapy, because the targeted correction of one potential pathogenetic process does not take into account other factors involved in the development of the disease. The situation is complicated by the fact that there is no specific information about the timing of the development of various dysfunctions during the course of the disease.
In present study, we found that at symptomatic and terminal stages of disease mSOD1 mice showed decreased baseline EPP quantal content and prolonged synaptic vesicle recycling time. Despite the decrease of EPP quantal content, studied mSOD1 mice groups showed unchanged dynamics of EPP relative amplitude comparing to WT mice, which may certify reduction of functional abilities of synapse, in particular, due to the increase of synaptic vesicles recycling time. Fluorescent experiments showed that mSOD1 mice are characterized by increased FM 1–43 loading and accelerated FM 1–43 unloading comparing to WT mice; both phenomena are more pronounced in mSOD1(8) than in mSOD1(6–7) mice. Revealed prolongation of synaptic vesicles recycling time may explain enhanced FM 1–43 loading in mSOD1 mice: during FM 1–43 loading, larger (comparing to WT) portion of synaptic vesicles that captured FM 1–43 does not release dye, which results in increased FM 1–43 staining. Generally, in the current work, we provided one of the first experimental evidences for impairment of synaptic vesicle recycling at neuromuscular synapses in ALS model. Impairment of synaptic vesicle reuse mechanisms that we found in mSOD1 mice may be associated with dysregulation of molecular mechanism(s) underlying the functioning of presynaptic vesicle pools.
Significant decrease of EPP quantal content revealed in mSOD1 mice should result in reduction of safety factor of neuromuscular transmission [26] with most pronounced manifestation under high-frequency activity due to EPP depression. Interestingly, we found an increase of MEPP amplitude in mSOD1(6–7) mice, which may reflect compensatory mechanism that alleviates reduction of EPP amplitude and safety factor of neuromuscular transmission at symptomatic stage of modelled disease.
Since ALS leads to the death of patients from respiratory failure, the study of the plasticity of respiratory motor neurons (including neuromuscular synapses) is of great importance for the development of ways to improve the quality and life expectancy of such patients. In mSOD1 ALS animal model, major loss of phrenic motor neurons is observed at disease end-stage; however, reduction of phrenic nerve activity is less pronounced than loss of motor neurons, representing amplification of activity in spared motor neurons [27, 28]. The contribution of possible changes of safety factor of neuromuscular transmission on the respiratory muscle activity in ALS should be also considered.
Neuromuscular synaptic dysfunction in mSOD1 model of ALS may result from impaired axon transport, mitochondrial dysfunction, oxidative stress, and/or other factors of mSOD1-mediated toxicity. Schwann cells and skeletal muscle fibers also contribute to neurotoxicity [7, 29]. In turn, presynaptic dysfunction can lead to dysregulatory and degenerative processes in skeletal muscle. “Vicious circle” of ALS pathogenesis consisting of presynaptic nerve terminals, skeletal muscle, and Schwann cells can be formed at the level of neuromuscular junction, in which the dysfunction of each element worsens the state of the entire system.
Thus, in present study, we found that mSOD1 mice at symptomatic and terminal stages of disease showed decreased baseline EPP quantal content and prolonged synaptic vesicle recycling time comparing to WT mice, providing one of the first detailed characteristics of presynaptic dysfunction at neuromuscular junction in ALS model. Obtained data expand our understanding of the ALS pathogenesis and contribute to stage- and localization-specific description of ALS pathogenetic mechanisms.
References
Brown, R. H., & Al-Chalabi, A. (2017). Amyotrophic lateral sclerosis. The New England Journal of Medicine, 377(2), 162–172. https://doi.org/10.1056/NEJMra1603471.
Chio, A., Logroscino, G., Traynor, B. J., Collins, J., Simeone, J. C., Goldstein, L. A., & White, L. A. (2013). Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology, 41(2), 118–130. https://doi.org/10.1159/000351153.
Therrien, M., Dion, P. A., & Rouleau, G. A. (2016). ALS: recent developments from genetics studies. Current Neurology and Neuroscience Reports, 16(6), 59. https://doi.org/10.1007/s11910-016-0658-1.
Mukhamedyarov, M. A., Petrov, A. M., Grigoryev, P. N., Giniatullin, A. R., Petukhova, E. O., & Zefirov, A. L. (2018). Amyotrophic lateral sclerosis: modern views on the pathogenesis and experimental models. Zhurnal Vyssheĭ Nervnoĭ Deiatelnosti Imeni I P Pavlova, 68(5), 551–566.
Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A., Donaldson, D., Goto, J., O’Regan, J. P., Deng, H. X., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature, 362(6415), 59–62. https://doi.org/10.1038/362059a0.
Van Damme, P., Robberecht, W., & Van Den Bosch, L. (2017). Modelling amyotrophic lateral sclerosis: progress and possibilities. Disease Models & Mechanisms, 10(5), 537–549. https://doi.org/10.1242/dmm.029058.
Dupuis, L., & Loeffler, J. P. (2009). Neuromuscular junction destruction during amyotrophic lateral sclerosis: insights from transgenic models. Current Opinion in Pharmacology, 9(3), 341–346. https://doi.org/10.1016/j.coph.2009.03.007.
Moloney, E. B., de Winter, F., & Verhaagen, J. (2014). ALS as a distal axonopathy: molecular mechanisms affecting neuromuscular junction stability in the presymptomatic stages of the disease. Frontiers in Neuroscience, 8, 252. https://doi.org/10.3389/fnins.2014.00252.
Naumenko, N., Pollari, E., Kurronen, A., Giniatullina, R., Shakirzyanova, A., Magga, J., Koistinaho, J., & Giniatullin, R. (2011). Gender-specific mechanism of synaptic impairment and its prevention by GCSF in a mouse model of ALS. Frontiers in Cellular Neuroscience, 5, 26. https://doi.org/10.3389/fncel.2011.00026.
Rocha, M. C., Pousinha, P. A., Correia, A. M., Sebastiao, A. M., & Ribeiro, J. A. (2013). Early changes of neuromuscular transmission in the SOD1(G93A) mice model of ALS start long before motor symptoms onset. PLoS One, 8(9), e73846. https://doi.org/10.1371/journal.pone.0073846.
Elmqvist, D., & Quastel, D. M. (1965). A quantitative study of end-plate potentials in isolated human muscle. The Journal of Physiology, 178(3), 505–529.
Zefirov, A. L., Zakharov, A. V., Mukhametzianov, R. D., Petrov, A. M., & Sitdikova, G. F. (2008). Vesicle cycle in mouse diaphragm motor nerve terminals. Rossiiskii fiziologicheskii zhurnal imeni IM Sechenova, 94(2), 129–141.
Betz, W. J., & Bewick, G. S. (1992). Optical analysis of synaptic vesicle recycling at the frog neuromuscular junction. Science, 255(5041), 200–203.
Betz, W. J., Mao, F., & Bewick, G. S. (1992). Activity-dependent fluorescent staining and destaining of living vertebrate motor nerve terminals. The Journal of neuroscience : the official journal of the Society for Neuroscience, 12(2), 363–375.
Zefirov, A. L., Zakharov, A. V., Mukhametzyanov, R. D., Petrov, A. M., & Sitdikova, G. F. (2009). The vesicle cycle in motor nerve endings of the mouse diaphragm. Neuroscience and Behavioral Physiology, 39(3), 245–252. https://doi.org/10.1007/s11055-009-9122-x.
Zucker, R. S., & Regehr, W. G. (2002). Short-term synaptic plasticity. Annual Review of Physiology, 64, 355–405. https://doi.org/10.1146/annurev.physiol.64.092501.114547.
Zefirov, A. L., & Mukhamed’iarov, M. A. (2004). The mechanisms of short-term forms of synaptic plasticity. Rossiiskii fiziologicheskii zhurnal imeni IM Sechenova, 90(8), 1041–1059.
Banker, B. Q., Kelly, S. S., & Robbins, N. (1983). Neuromuscular transmission and correlative morphology in young and old mice. The Journal of Physiology, 339, 355–377.
Dadon-Nachum, M., Melamed, E., & Offen, D. (2011). The “dying-back” phenomenon of motor neurons in ALS. Journal of molecular neuroscience : MN, 43(3), 470–477. https://doi.org/10.1007/s12031-010-9467-1.
Selkoe, D. J. (2002). Alzheimer’s disease is a synaptic failure. Science, 298(5594), 789–791. https://doi.org/10.1126/science.1074069.
Dauer, W., & Przedborski, S. (2003). Parkinson’s disease: mechanisms and models. Neuron, 39(6), 889–909.
Arendt, T. (2009). Synaptic degeneration in Alzheimer’s disease. Acta Neuropathologica, 118(1), 167–179. https://doi.org/10.1007/s00401-009-0536-x.
Cappello, V., & Francolini, M. (2017). Neuromuscular junction dismantling in amyotrophic lateral sclerosis. International Journal of Molecular Sciences, 18(10). https://doi.org/10.3390/ijms18102092.
Rothstein, J. D. (2009). Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Annals of Neurology, 65 Suppl 1, S3–S9. https://doi.org/10.1002/ana.21543.
Robberecht, W., & Philips, T. (2013). The changing scene of amyotrophic lateral sclerosis. Nature Reviews Neuroscience, 14(4), 248–264. https://doi.org/10.1038/nrn3430.
Wood, S. J., & Slater, C. R. (2001). Safety factor at the neuromuscular junction. Progress in Neurobiology, 64(4), 393–429.
Nichols, N. L., Gowing, G., Satriotomo, I., Nashold, L. J., Dale, E. A., Suzuki, M., Avalos, P., Mulcrone, P. L., McHugh, J., Svendsen, C. N., & Mitchell, G. S. (2013). Intermittent hypoxia and stem cell implants preserve breathing capacity in a rodent model of amyotrophic lateral sclerosis. American Journal of Respiratory and Critical Care Medicine, 187(5), 535–542. https://doi.org/10.1164/rccm.201206-1072OC.
Nichols, N. L., & Mitchell, G. S. (2016). Quantitative assessment of integrated phrenic nerve activity. Respiratory Physiology & Neurobiology, 226, 81–86. https://doi.org/10.1016/j.resp.2015.12.005.
Lasiene, J., & Yamanaka, K. (2011). Glial cells in amyotrophic lateral sclerosis. Neurology Research International, 2011, 718987. https://doi.org/10.1155/2011/718987.
Funding
The work was supported by Russian Science Foundation (grant #14-15-00847-П). Fluorescent studies within the work were supported by Grant of President of Russian Federation for young doctors of sciences (MD-6877.2018.4).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Animal experiment protocols were approved by the Kazan State Medical University Local Ethic Committee.
Rights and permissions
About this article

Cite this article
Mukhamedyarov, M.A., Grigoryev, P.N., Khisamieva, G.A. et al. Dysfunction of Neuromuscular Synaptic Transmission and Synaptic Vesicle Recycling in Motor Nerve Terminals of mSOD1 Transgenic Mice with Model of Amyotrophic Lateral Sclerosis. BioNanoSci. 9, 66–73 (2019). https://doi.org/10.1007/s12668-018-0590-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12668-018-0590-8