
Abstract
Amyotrophic lateral sclerosis (ALS) is a fatal neurological disorder characterized by progressive degeneration of motor neurons leading to skeletal muscle denervation. Earlier studies have shown that motor neuron degeneration begins in motor cortex and descends to the neuromuscular junction (NMJ) in a dying forward fashion. However, accumulating evidences support that ALS is a distal axonopathy where early pathological changes occur at the NMJ, prior to onset of clinical symptoms and propagates towards the motor neuron cell body supporting “dying back” hypothesis. Despite several evidences, series of events triggering NMJ disassembly in ALS are still obscure. Neuromuscular junction is a specialized tripartite chemical synapse which involves a well-coordinated communication among the presynaptic motor neuron, postsynaptic skeletal muscle, and terminal Schwann cells. This review provides comprehensive insight into the role of NMJ in ALS pathogenesis. We have emphasized the molecular alterations in cellular components of NMJ leading to loss of effective neuromuscular transmission in ALS. Further, we provide a preview into research involved in exploring NMJ as potential target for designing effective therapies for ALS.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal progressive disorder with degeneration of corticospinal/corticobulbar (upper) motor neurons (UMN) in cerebral cortex and bulbar/spinal (lower) motor neurons (LMN) in brain stem and spinal cord. Major clinical symptoms are weakness and atrophy of limb muscles, difficulty in swallowing and speaking, and finally death due to respiratory failure. The clinical profile of ALS is heterogeneous with mean survival period of 3 to 5 years, but based on age, gender, and site of onset of symptoms, the duration of survival is variable [1,2,3,4]. While familial ALS constitutes ~ 5 to 10% in the global context, in India, however, it is much less at 1 to 2% [5]. Genetic mutations in more than 100 genes have been reported in familial ALS. Among these, mutations in SOD1, C9ORF72, TDP-43, and FUS genes are the most frequent. Many of these genes have also been implicated in sporadic ALS [6].
UMN makes direct (monosynaptic) or indirect (via interneurons) connections with LMN which innervates effector muscles in the periphery and triggers their contraction (Fig. 1a). Loss of motor neurons (MN) causes rapid weakness of voluntary muscles due to denervation and corresponding changes of neuromuscular junction (NMJ) structure. Fatigue or easy tiredness is an important symptom of ALS, and this feature was the basis for evaluation of neuromuscular transmission in the clinical setting. Neuromuscular transmission defect can be demonstrated by neurophysiology technique of repetitive nerve stimulation (RNS). After the first report by Mulder et al. [7], several studies observed that ALS patients with muscle fatigue showed a decremental response during RNS which indicates instability of neuromuscular transmission [8,9,10]. Further, decrease in motor unit number estimation (MUNE) in ALS patients indicated progressive loss of motor axons which affects neuron-muscle communication [11,12,13].

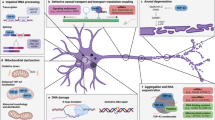
Structural and functional organization of healthy motor neurons and neuromuscular junction. a Upper motor neurons projects from motor cortex and descends to brainstem/spinal cord via corticospinal tracts where it synapses with lower motor neurons which innervate skeletal muscles. b Interaction of tripartite components of neuromuscular junction: presynaptic motor neuron, postsynaptic skeletal muscle, terminal Schwann cells and capping kranocytes. Motor neuron and skeletal muscles are separated via synaptic cleft filled with basal lamina consisting of different laminins isoforms. Action potential arriving at nerve terminal triggers clustered voltage-gated Ca2+ channels at the active zones. This leads to (i) influx of Ca2+ ions, (ii) docking, (iii) fusion, and release of synaptic vesicles filled with neurotransmitter ACh into synaptic cleft. Binding of ACh to postsynaptic clustered nAChRs generates localized endplate potential (EPP). EPP triggers (iv) voltage-gated Na+ channels and generates muscle action potential which propagates along the muscle fiber and activates dihydropyridine receptors (DHPR) located in T-Tubules. DHPR-mediated ryanodine receptor (RyR) activation causes (v) release of Ca2+ from sarcoplasmic reticulum which eventually causes contraction of muscle fiber via the actin myosin contractile units. Inset 1 Organization of synaptic vesicles at active zone. Synaptotagmin, synaptobrevin, SNAP-25, and syntaxin are crucial for docking, fusion, and release of ACh into the synaptic cleft. Inset 2 Organization and signaling of postsynaptic Lrp4/MuSK/nAChRs complex
Despite advances in the field of ALS, site of disease origin still remains a key unresolved question. Initiation of neurodegeneration in ALS can be explained by the dying forward and dying backward hypotheses [14]. The dying forward hypothesis proposes that UMN drives anterograde degeneration of LMN via glutamate excitotoxicity which eventually descends to NMJ [15,16,17]. This hypothesis was proposed by Eisen and team in 1992 [18], and they demonstrated inverse correlation between cortical threshold and motor evoked potentials (MEP)/ compound muscle action potential (CMAP) ratio indicating cortical hyperexcitability [19]. Later, several studies supported early cortical dysfunction in ALS patients which probably arises due to altered cortical excitatory/inhibitory circuitry [15, 20,21,22,23,24,25]. Even transgenic ALS mice displayed increased intrinsic excitability and bioenergetics defects in UMN [26, 27]. Further, exposure of LMN to excitotoxin in mice caused motor neuron degeneration and NMJ abnormalities in an anterograde manner [17].
These studies raised an important question, whether excitotoxicity mediated neurodegeneration arises due to intrinsic hyperexcitability of MN? Several research groups have reported variable excitability of LMN during the course of ALS progression. At early stage, LMN from mutant SOD1 mice displayed hyperexcitability which arises due to increased persistent Na ( +) current in large LMN [28,29,30,31,32]. However, some studies have also observed unaltered or decreased excitability of LMNs [33,34,35]. At later stage, most LMN displayed normal excitability despite increase in input conductance. These MN were able to achieve homeostasis for excitability by upregulating depolarizing current, whereas some MN exhibited hypoexcitability [36, 37]. The inability of LMN to fire repetitively probably arises due to homeostatic deregulation of excitability [38]. Altogether, these reports indicate that alterations in electrical properties are not caused by intrinsic excitability of MN but rather involves extrinsic factors such as synaptic activity. Some of the crucial findings supporting dying forward phenomenon have been outlined in Fig. 2a. For better understanding, readers can refer to excellent recent reviews by Brunet and Eisen [39, 40] on early cortical dysfunction.

Timeline of major findings supporting a dying forward and b dying backward hypothesis in ALS. UMN, upper motor neuron; LMN, lower motor neuron; VEGF, vascular endothelial growth factor; GDNF, glial cell line-derived neurotrophic factor; TSC, terminal Schwann cells
Contrary to dying forward hypothesis, the dying back hypothesis propounds that MN degeneration begins distally at the nerve terminal/neuromuscular junction and progresses towards the cell body in a retrograde fashion [41, 42]. Below, we provide a comprehensive overview of cellular and molecular perturbations at neuromuscular junction in ALS and thereby focusing on evidencing supporting the dying back hypothesis.
Neuromuscular Junction
Neuromuscular junction is a specialized chemical synapse where information in the form of electrical impulses is transmitted from motor neurons to skeletal muscles to initiate muscle contraction and execute voluntary motor functions. The NMJ is a tripartite synapse comprising of three major components, viz., presynaptic motor neurons, postsynaptic muscles, and terminal Schwann cells (Fig. 1b). Kranocytes, the fibroblast-like cells identified as the fourth component of NMJ, are responsible for capping TSCs and entire endplate area [43]. Although their role in synapse formation and functioning is not known, they appear to affect synaptic regeneration [44].
The well-coordinated reciprocal interactions among these components are essential for regulating formation and maturation of NMJ. Structural organization of pre- and postsynaptic apparatus, such as active zones on nerve terminals, clustering of nicotinic acetylcholine receptors (nAChRs) on the postsynaptic membrane, is indispensable for proper functioning of NMJ as illustrated in Fig. 1b. Active zones are electron dense multiprotein complex responsible for exocytosis of neurotransmitter. Active zones contain voltage-gated calcium channels (VGCC), soluble N-ethylmaleimide-sensitive factor attachment receptor (SNARE) proteins including syntaxin-1 and SNAP-25, and cytoskeletal matrix at the active zone (Fig. 1b inset 1). VGCCs are responsible for rapid influx of Ca2+ which helps in interaction of SNARE proteins and leads to docking, fusion, and release of neurotransmitter acetylcholine (ACh) into the synaptic cleft (Fig. 1b).
Postsynaptic muscle membrane contains densely clustered nAChR that directly oppose presynaptic active zones. During NMJ development, agrin released by motor nerve terminal binds to postsynaptic lipoprotein receptor-related protein 4 (Lrp4) which activates muscle-specific receptor tyrosine kinase (MuSK) domain and leading to its self-phosphorylation [45]. Activated MuSK phosphorylates Dok-7, a muscle cytoplasmic adaptor protein, which in turn phosphorylates and further activates MuSK. Dok-7 interacts with MuSK through its phosphotyrosine-binding domain and controls its activity and responsiveness to agrin [46]. MuSK acts as a master regulator of synaptic differentiation by activating a series of downstream molecules which eventually induces dense clustering of nAChR at the postsynaptic membrane. MuSK activates ETS-related molecule (ERM) which regulates expression of various postsynaptic genes including nAChR subunits genes [47]. MuSK recruits rapsyn, an intracellular scaffolding protein which strongly interacts with nAChRs, leading to recruitment and clustering of nAChRs at the NMJ (Fig. 1b inset 2) [48]. Clustered nAChRs are anchored into the cell membrane by actin cytoskeleton and dystrophin-glycoprotein complex. Detailed cellular pathway involved in NMJ formation is reviewed in Li et al. [49].
The space between the presynaptic nerve terminal and postsynaptic muscle membrane is called synaptic cleft. Synaptic cleft spanning ~ 50 nm contains basal lamina, made up of extracellular matrix (ECM), which surrounds the muscle fibers [50]. Basal lamina contains various ECM proteins such as laminins, collagens, perlecan, and fibronectins which play a pivotal role in NMJ assembly and functioning [50, 51]. Laminins play a pivotal role in organizing pre- and postsynaptic components and thereby maintain structural integrity of NMJ. Laminin β2, secreted by muscles, interacts with presynaptic VGCC which leads to clustering of VGCC and recruitment of other presynaptic components at the active zones [52]. Laminin β2-deficient mice displayed decreased calcium sensitivity and co-localization of active zone proteins and led to alteration in motor endplate maturation and thereby suggested its role in synapse stabilization [53]. Laminin α4 and α5 interaction with dystroglycan helps in maturation of nAChR clusters which promotes postsynaptic differentiation [54]. Loss of laminin α4 disrupted alignment and stabilization of pre- and postsynaptic components at the NMJ and resulted in altered neuromuscular transmission [55].
This spatiotemporal organization of NMJ components is cardinal for efficient neuromuscular transmission which involves a series of highly complex and dynamic processes. Any alterations in organization and/or signal transmission can impair NMJ functionality leading to denervation, muscle weakness, or paralysis.
Evidences of Neuromuscular Junction Disruption as Primary Event in ALS
Most of the initial research encompassing ALS majorly revolved around MN degeneration as it was believed that ALS primarily affects MN and alterations in skeletal muscle arise as a consequence of loss of neurons. The hypothesis of distal involvement stemmed from early observations that LMN loss was much more evident than UMN [56] and further loss of axonal integrity led to dysfunctional NMJ [57]. Importantly, preserving MN by deletion of Bax, a key regulator of apoptosis, in SOD1G93A ALS mice successfully rescued MN from mutant SOD1-mediated toxicity but failed to prevent NMJ denervation [58]. Similarly, inhibition of p38 MAPK, a stress activated protein kinase involved in initiating cell death, exerted positive effects on MN survival but failed to rescue NMJ denervation in ALS mice [59]. These reports have highlighted crucial role of NMJ disruption in disease mechanism.
To understand the mechanism underlying NMJ dysfunction in ALS, various cellular models based on mutations in SOD1, FUS, TARDBP, and C9ORF72 genes have been exploited. Studies have reported that transgenic SOD1 ALS mice showed significant NMJ denervation followed by degeneration of ventral root axons and eventually loss of MN cell bodies indicating early loss of NMJ circuitry [60,61,62]. Apart from mutant SOD1, transgenic mice expressing mutant FUS experienced early structural and functional alterations at the NMJ followed by MN degeneration [63, 64]. Transgenic mice with inducible expression of mutant TDP-43 (∆NLS, loss of nuclear localization signal) displayed significant denervation with no changes in number of LMN in spinal cord. Suppression of mutant TDP-43 in postsymptomatic stages prevented ongoing MN loss and increased NMJ re-innervation which helped in functional recovery from mutant TDP-43-mediated toxicity [65]. Similarly, inducible expression of hexanucleotide (G4C2) C9ORF72 repeats in mice showed dipeptide repeats expression which led to structural NMJ abnormalities and rapid muscle dystrophy with no change in number of LMN in spinal cord. Interestingly, early suppression of C9ORF72 expression was able to prevent but not reverse muscle dystrophy [66]. Mutant FUS-ALS drosophila displayed significantly impaired synaptic neuromuscular transmission with normal electrical excitability of MN cell bodies indicating early neuromuscular dysfunction [67]. Similarly, SOD1G93A zebrafish displayed early loss of intact NMJs including decrease in postsynaptic volume followed by loss of MN cell bodies [68, 69].
It is important to mention that there is a temporal loss of different motor units during disease progression. ALS pathology involves presymptomatic loss of NMJs of fast fatigable (FF) motor units followed by loss of fast fatigue-resistant (FFR) motor units at symptomatic stage and eventual loss of slow (S) motor units at end-stage disease [60, 70, 71] as illustrated in Fig. 3b. High metabolic demand and large size of FF MN makes them more vulnerable to degeneration whereas small size of type S MN makes them resistant. It has recently been highlighted that reduced neurotransmitter release in NMJs of FF motor units precedes NMJ denervation affecting pre- and postsynaptic elements in SOD1G37R mice [72]. However, the initial denervation triggers a compensatory mechanism where the nearby neurons increase their axonal arbors to form new synaptic connection to mask ongoing NMJ dismantling [73,74,75]. Repeated cycles of denervation and reinnervation results into an intermittent conduction block in newly formed nerve endings, indicating that immature sprouts and unstable conduction of NMJ are a result of degenerating axons in ALS.

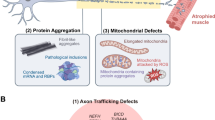
Neuromuscular junction disassembly in amyotrophic lateral sclerosis. a Structural and functional alterations in NMJ in ALS. Left panel shows the interaction of presynaptic and postsynaptic membranes in humans and rodents in healthy and ALS condition. Presynaptic membrane is in purple for healthy and gray for ALS, whereas postsynaptic is in red for healthy and light pink for ALS. Arrow heads denote orphaned nAChRs. Right panel defines the altered functional parameters of NMJ in ALS. b Specificity and temporal loss of motor units during ALS progression. Arrow heads denote retraction of presynaptic nerve endings from skeletal muscle. c Cellular and molecular alterations in the presynaptic neuron, postsynaptic muscle, and terminal Schwann cells resulting in NMJ denervation in ALS. ALS-associated mutant proteins differentially affect the components of NMJ which trigger and/or amplify malfunctioning of NMJ in ALS
In humans, very few studies have identified early changes at NMJ as it is difficult to obtain presymptomatic samples. Therefore, it is challenging to validate the findings from animal model in human patients at presymptomatic disease stages. In 1993, Maselli et al. [76] reported altered synaptic transmission in muscle biopsies of ALS patients. Histological examination of NMJ in these patients showed absence of axon terminal indicating denervation and corresponding decrease in amplitudes of miniature endplate potentials as measured by electrophysiological techniques. In this study, normal postsynaptic folds were observed in limited number of denervated muscle samples analyzed. In contrast, a recent study revealed significantly altered postsynaptic apparatus and NMJ denervation with fragmented synaptic gutters in muscle samples of early and long-term ALS patients. They also observed flattened and fragmented postsynaptic apparatus being partially innervated by axon terminal indicating reinnervation and remodeling of NMJ as observed in animal models. [77]. A case report also showed severe denervation with no changes in LMN in autopsy of a 58-year-old male, who died in a minor surgery, after 6 months of onset of ALS symptoms [61]. These studies on ALS patients indicate altered NMJ integrity as an early event in disease course. It is important to note that there are considerable differences in cellular architecture of NMJ of different species. In humans, NMJs are smaller and have “nummular” morphology, whereas mouse NMJs are larger and “pretzel”-shaped. Despite these differences, evidences from human and animal models clearly supports that NMJ is compromised in early ALS stages (Fig. 3a).
Altogether, such studies corroborate that malfunctioning of NMJ may be the primary pathogenic event in ALS and MN loss proceeds in a dying back fashion. Evidences from animal and human studies supporting early role of NMJ in ALS have been summarized in Fig. 2b. In the sections below, role played by presynaptic motor neurons, postsynaptic skeletal muscle cells, and terminal Schwann cells in triggering NMJ disassembly is described to delineate the series of events culminating in neurodegeneration.
Role of Motor Neuron in NMJ Disruption in ALS
Lower motor neuron communicates with skeletal muscles and executes voluntary actions by means of neuromuscular transmission. LMN cell bodies lie within the central nervous system, i.e., brainstem and anterior horns of spinal cord from where they extend long processes to reach target muscle and form an indispensable link between CNS and skeletal muscles. Due to long axons, communication between cell body and nerve terminal is majorly dependent upon anterograde and retrograde axonal transport system. Maintaining homeostasis in cell body and distal nerve terminal is necessary for healthy functioning of NMJ. ALS-associated mutant proteins may lead to deregulation of protein homeostasis, malfunctioning of axonal transportation, and cholinergic dysfunction and impairs mitochondrial dynamics as illustrated in Fig. 3c.
Mutant Protein and Deregulated Protein Homeostasis
ALS-associated mutant proteins can cause structural and functional changes in the presynaptic nerve terminal which may trigger NMJ degeneration. Motor neuron of SOD1G93A mice showed the presence of enlarged and vacuolated mitochondria in axons and presynaptic nerve terminal. There was also decrease in soma size and accumulation of small, empty vacuoles in axons which results in axonal loss and muscle denervation [78]. AAV vector-mediated silencing of SOD1 in motor neurons of SOD1G93A mice led to delayed disease onset, preserved NMJ functionality, and prolonged survival [79]. In addition, interneurons play a key role in modulating synaptic activity. Transgenic SOD1 ALS zebrafish showed the earliest sign of neuronal stress in inhibitory glycinergic interneurons at embryonic stage. This resulted in loss of inhibitory input which generated neuronal stress in LMN. In adult life, these stressed LMN showed retracted nerve terminal resulting in NMJ abnormalities whereas non-stressed neurons have normal NMJs [80, 81]. Systematic and structural screening of NMJs in mutant FUS and TDP-43 ALS mice showed early structural changes in presynaptic nerve terminal but no corresponding changes at the motor endplate. These changes preceded denervation which led to reduced interaction between pre- and postsynaptic membrane suggesting that cues for NMJ degeneration may arise from MN terminal [82]. Overexpression of mutant FUS in motor neurons of transgenic flies resulted in reduction in number and area of presynaptic bouton which led to significant locomotive impairment [83].
The large size of LMN makes them more vulnerable to proteome imbalance which causes ER stress, mitochondrial impairment, altered excitability, and synapse dysfunction [84]. Prolonged deregulation of protein homeostasis leads to aberrant accumulation of misfolded mutant proteins such as SOD1, TDP-43, and FUS which have been implicated in ALS. Most of the mutations in SOD1 enzymes have a propensity to form high molecular weight aggregates. These mutant SOD1 aggregates have been reported in ALS patients as well as mice models [85, 86]. Ubiquitin proteasome system (UPS) was found to be impaired with increased accumulation of ubiquitin and phosphorylated neurofilaments in motor neurons of SOD1G93A mice [87]. Even, we have observed effect of modulation of protein homeostasis pathways on high molecular-weight aggregates of SOD1L84F [5] in motor neuronal cell line (unpublished work).
Expression of mutant FUS at physiological level in mice showed cytoplasmic mislocalization of mutant protein which accumulates at rough endoplasmic reticulum and alters expression of gene associated with ribosomes, mitochondria, and proteasome suggesting altered proteostasis as a potential pathomechanism of mutant FUS [88]. Mutant FUS decreases synaptic activity by downregulating expression of transporters and ion channels required for synaptic functioning in ALS mice. Moreover, accumulation of mutant FUS generates stress response and inhibits local, intra-axonal protein synthesis [89]. Mutant TDP-43 mice exhibited alterations in gene splicing, and downregulation of genes is involved in ubiquitin proteasome pathway along with ubiquitin‐positive inclusions [90]. ALS-associated polydipeptide repeat expansion in C9ORF72 gene causes ribosome-mediated distress and hampers protein translation in ALS mice [91].
Degradation of improperly folded protein is mediated by ubiquitin proteasome system and autophagy. A recent study showed that cranial MN exhibit high proteasome activity making them more resistant to proteostasis stress as compared to spinal MN indicating time-dependent death of spinal and cranial MN in ALS disease course [92]. Autophagy plays a key role in maintaining cellular homeostasis in distal axons by balancing synthesis and degradation of proteins [93]. Activation and upregulation of autophagy has been observed in cytoplasm of MN in sALS patients [94]. Mutations in several genes associated with autophagy including SQSTM1, OPTN, UBQLN2, and TBK1 have been associated with ALS [95,96,97,98]. NMJs of SOD1G93A mice showed increased accumulation of autophagosomes in nerve terminals of MN along with reduced expression of SQSTM1 suggesting altered autophagy [99]. Spinal MN restricted expression of mutant UBQLN2P497H in transgenic rats recapitulated ALS-like phenotype such as MN degeneration, denervated NMJs, and abnormal protein accumulation [100]. Autophagy inhibition by Atg7 or TBK1 deletion in MN of SOD1G93A mice accelerated NMJ denervation accompanied by significant decrease in endplate current amplitude in tibialis anterior muscle [101, 102]. Moreover, deletion of TBK1 in TDP-43G298S mice aggravated NMJ integrity [103]. These studies provide a vital link between mutant proteins, alteration in protein degradation pathways, and NMJ stability in ALS pathology.
Disturbed Axonal Transport
Axonal transportation involves neuronal cytoskeleton (microtubules, actin, and intermediate filaments) and motor proteins such as dyneins, kinesins, and myosins. Anterograde transportation of various organelles and cargos (containing mRNA, proteins, and lipids) from cell body to axon terminals as well as retrograde transportation of neurotrophic factors, signaling molecules, and damaged organelles from distal nerve terminal to cell body are crucial for functioning and survival of MNs [104]. Most of kinesin motor family proteins are involved in unidirectional anterograde transportation, whereas dynein is responsible for retrograde transportation (Fig. 1b).
Dynein is a dimeric multisubunit complex and requires an essential cofactor dynactin to mediate axonal transportation. Dynactin is a large protein complex made up of several subunits and acts as a processivity factor for dynein. Mutations in largest subunit, p150 (Glued), of dynactin complex have been reported in ALS patients [105,106,107]. Mutant dynactin p150 (Glued) mice displayed pathological features of motor neuron disease characterized by impaired vesicular transport, denervation, axonal swelling, and axon-terminal degeneration [108]. Moreover, decreased mRNA and protein levels of dynactin1 are observed in spinal MN of sALS patients [109, 110]. Depletion of dynactin 1 in C. elegans and zebrafish displayed axonal degeneration accompanied by severe motor defects, abnormal innervation of fast twitch muscle fibers, NMJ instability, and locomotor defects [111, 112].
Aberrant neurofilament aggregation is a pathological hallmark of ALS. Mutant SOD1-mediated toxicity is responsible for abnormal accumulation of neurofilaments which disrupts axonal transport and leads to axonal degeneration and profound reduction in retrograde uptake in early stages of disease [113,114,115,116,117]. These defects in axonal transportation has been attributed to activated p38 MAP kinase. Inhibition of p38 MAPK rescued proximal axons from SOD1-mediated toxicity and improved survival of SOD1 ALS mice [59, 118]. In addition, mutant SOD1 inhibits anterograde fast axonal transport by activating p38 MAP kinase [119]. Mutant TDP-43 impairs axonal trafficking in drosophila and MN derived from ALS patients [120]. Recently, a genome-wide association study identified mutation in C-terminal of kinesin family member 5A (KIF5A) gene in ALS patients [121, 122]. These studies highlight the role of axonal transport in maintaining distal synapse, and defects in transportation machinery may strongly impact NMJ stability.
Cholinergic Dysfunction and Altered Neurotransmission
Efficient cholinergic transmission is crucial for maintaining structural and functional integrity of NMJ. Cholinergic dysfunction is a common feature of various neurodegenerative disorders including ALS. Choline acetyltransferase (ChAT) is responsible for synthesis of ACh, patterning, and formation of neuromuscular synapses [123]. Decreased ChAT activity has been reported in spinal MN of ALS patients [124, 125]. Even SOD1G93A ALS mice displayed early reduced ChAT content in soma and synaptic terminals of MNs indicating that presymptomatic cholinergic dysfunction could be the earliest event in ALS [126]. Previously, SOD1 aggregates also impaired transportation of ChAT by sequestering kinesin-associated protein 3 and thereby disrupt cholinergic system in SOD1 ALS mice [127].
Vesicular ACh transporter (VAChT) is involved in packing of ACh in synaptic vesicles at nerve terminals and serves as a marker for cholinergic synapses. Immunohistochemical staining of spinal MN of ALS patients displayed decreased VAChT but no decrease in activity of synaptophysin, a marker for synapse integrity, indicating loss of cholinergic input prior to MN degeneration [128]. Interestingly, increasing synaptic ACh by overexpressing VAChT diminished motor functions and accelerated NMJ degeneration and death of SOD1G93A mice [129]. These observations suggest that increased or decreased levels of VAChT have detrimental effect on cholinergic transmission.
Efficient neurotransmission is quantified by measuring localized postsynaptic depolarization (endplate potential (EPP)) caused by release of synaptic vesicles. These changes in postsynaptic membrane in turn reflects the functioning of presynaptic neuron. Muscle biopsies from ALS patients revealed decreased amplitude of miniature EPP (MEPP) along with decreased mean quantal stores and EPP quantal content suggesting impaired neuron and muscle communication [76]. Prior to morphological NMJ alterations, SOD1G37R ALS mice showed decreased quantal content indicating presymptomatic functional changes at NMJ [72]. Similarly, mutant TDP-43 Q331K mice displayed increased MEPP amplitude, decreased MEPP frequency, and quantal content at the NMJs prior to motor neuron loss [130]. These results indicate early molecular alterations in fusion and release of synaptic vesicles from the presynaptic axon terminal.
Transgenic zebrafish harboring mutant TDP-43 or mutant FUS also showed aberrant synaptic fidelity with reduced frequency of miniature endplate currents (MEPCs) and quantal neurotransmission [131,132,133]. Mutant FUS ALS mice showed marked depletion in synaptic vesicles in axon terminal and reduced motor response amplitude in tibialis anterior muscles suggesting altered neurotransmission [63]. Mutant FUS transgenic flies developed presynaptic structural and function defects prior to degeneration of MN cell body and axons. Mutant flies displayed disorganized active zone protein and reduced quantal content resulting in marked decrease in postsynaptic evoked current [67]. Overexpression of mutant FUS in C. elegans and in motor neurons of drosophila resulted in impaired synaptic vesicle docking and reduced quantal size and quantal content demonstrating reduced neuromuscular transmission [134, 135]. In addition, silencing of C9ORF72 (C9-miR) in zebrafish resulted in NMJ abnormalities accompanied with reduced frequency of MEPC suggesting impaired presynaptic functionality. C9ORF72 has been shown to interact with SV2a, a key component of presynaptic active zones. C9-miR causes decrease in release and uptake of neurotransmitter suggesting loss of C9ORF72 affects exocytosis [136]. These studies suggest that derangements in neurotransmission (synthesis, packaging, and release of neurotransmitter) are early events in ALS pathology which may contribute to distal degeneration.
Mitochondrial Dysfunction
MNs are large, polarized cells that rely heavily on mitochondrial ATP for their high energy demands. Nerve terminals at the NMJ are packed with mitochondria which are transported from cell body to synaptic terminals by means of fast axonal transport. Synaptic mitochondria are crucially involved in ATP production, calcium handling, synthesis, and release of neurotransmitter and thereby play a critical role in neuronal transmission [137].
Deficits in mitochondrial dynamics and their axonal transport have been observed in ALS [138, 139]. Mutant SOD1 and TDP-43 transgenic mice exhibited abnormal mitochondrial morphology and transport in distal part of motor axons of phrenic or sciatic nerve before disease onset indicating that altered mitochondria may trigger cascade of events leading to MN degeneration [140, 141]. Even, FUS ALS transgenic mice showed early mitochondrial abnormalities in presynaptic MN along with decreased synaptic vesicles and NMJ area before onset of clinical symptoms [142]. Similarly, transgenic mice with regulatable mutant TDP-43 (∆NLS) expression also displayed altered mitochondrial activity. Accumulation of mutant TDP-43 in axons and nerve terminals leads to formation of RNP granules and affects localized protein translation which in turn decreases level of key nuclear-encoded mitochondrial proteins (Altman et al. unpublished work, https://doi.org/10.21203/rs.3.rs-87662/v1).
Mutant SOD1 induces degradation of mitochondrial Rho GTPase 1 (Miro1) which inhibits mitochondrial axonal transport [143]. Miro1 is responsible for attachment of mitochondria to kinesin-1, and its activity depends on cytosolic Ca2+ levels. Mutations in another ALS causing gene, VAPB, increase cytosolic Ca2+ which effects Miro1/kinesin-1 interaction and hampers mitochondrial axonal movement [144]. NMJs of SOD1G93A mice showed increased number of degenerating mitochondria with large vacuoles and irregular cristae spacing in presynaptic nerve terminals. Moreover, there was decreased expression of mitophagy-related proteins SQSTM1, Pink1, Parkin, and Bnip3 indicating impaired mitophagy [99]. Double knockout mice with ablation of Pink1 and Parkin resulted in increased accumulation of mitochondria in presynaptic terminal, axon swelling, and NMJ fragmentation which highlights the key role of impaired mitophagy in NMJ degeneration in ALS [99].
Impaired mitochondrial axonal transport and mitophagy causes decrease in healthy mitochondrial pool and improper turnover of damaged mitochondria in synaptic terminals which could significantly alter NMJ dynamics. Therefore, disruption of mitochondrial integrity alters NMJ dynamics and may lead to neurodegeneration.
Role of the Skeletal Muscle in NMJ Disruption in ALS
Skeletal muscle comprises the postsynaptic component of NMJ. Multiple mechanisms, such as alterations in postsynaptic apparatus, acetylcholine esterase (AChE) abnormalities, misfolding, and clearance of mutant proteins and mitochondrial dysfunction, have been implicated for its degeneration in ALS (Fig. 3c). The findings from various studies provide insight into the role of muscles in disease initiation and have been discussed below in-depth.
Alterations in Postsynaptic Apparatus
Structural and functional integrity of postsynaptic apparatus including clustered nicotinic acetylcholine receptors (nAChRs), muscle-specific kinase (MuSK), and low-density lipoprotein receptor-related protein 4 (Lrp4) present on skeletal muscle is crucial for stability and functioning of NMJ (Fig. 1b inset 2). Earlier, muscles of ALS patients showed denervated endplates with various degrees of postsynaptic structural changes including flattening of primary cleft but well-preserved nAChRs [145, 146]. Similar findings were also observed in ALS transgenic mice [147]. However, elegant ex vivo experiments by Palma and team showed decreased ACh affinity and abnormal expression of AChR in skeletal muscle of ALS patients [148, 149]. Recently, muscle-specific expression of SOD1G93A in ALS mice resulted in smaller and fragmented NMJ with dispersed endplates, decreased ramification of primary cleft, and high turnover, and fragmentation of AChRs indicating mutant SOD1 affects AChR clustering and NMJ stability [150]. In addition, FUS ALS mice showed reduced endplate area in newborn mutants. Even impaired endplate maturation was observed in iPSC-derived myotubes from FUS-ALS patients [64]. siRNA-mediated knockdown of FUS in C2C12 cell line decreased gene expression of nAChR subunits. Similar results were obtained in FUSΔNLS/ΔNLS and FUSΔNLS/+ mice [64]. Moreover, knockdown of TDP-43 in zebrafish resulted in denervation and fragmentation of nAChR cluster [151].
These studies show that AChRs can be targeted for ALS pharmacological therapy. In fact, riluzole, the first FDA-approved drug for ALS, has been shown to block postsynaptic AChRs and may affect its function in denervated muscle fibers of ALS patients, but its biological significance still remains elusive [152, 153].
In addition to AChRs, alterations in other key postsynaptic proteins have also been documented in ALS. Lrp4 protein is essential for structural and functional integrity of NMJ. Loss of Lrp4 led to fragmented AChR clusters, reduced synaptic vesicles and junctional folds, and impaired neuromuscular transmission [154]. SOD1G93A transgenic mice also showed loss of postsynaptic structural proteins such as Lrp4, nestin, rapsyn, and dystrophin from NMJ before complete disassembly of AChRs [62]. Furthermore, neonatal rat muscle injected with cerebrospinal fluid (CSF) from ALS patients displayed structural damage and fragmentation of NMJ along with decreased rapsyn and increased calpain expression. However, no change in levels of AChRs protein was observed [155].
MuSK is another crucial postsynaptic protein involved in efficient functioning of NMJ. Increasing MuSK expression or stimulating MuSK via agonist antibody has been shown to reduce muscle denervation, delay in disease onset, and improve motor function in ALS mice [156, 157]. In contrast, another study showed that MuSK agonist antibody is not sufficient for preservation of motor neuron function and survival of ALS mice [158]. These opposing findings raise concern regarding MuSK activation as therapeutic for ALS.
Several studies have targeted Dok-7, a muscle cytoplasmic protein, for enhanced activation of MuSK. Muscle-specific overexpression of Dok-7 in mice increased pre- and postsynaptic area and enhanced neuromuscular transmission in diaphragm muscle [159] as previously observed for neuromuscular disorders [160]. Recently, Dok-7 gene therapy in SOD1G93A mice protected NMJ from degeneration, suppressed muscle atrophy, improved locomotor activity, and prolonged life span [161].
Acetylcholine Esterase Activity in ALS
One of the primary roles of AChE is termination of neuromuscular transmission by degradation of ACh at the NMJ. AChE is also believed to play non-enzymatic roles such as regulating fate of AChRs, NMJ formation, and survival. AChE knockout mice (AChE−/−) exhibited reduced AChR cluster density, shallow and irregular junctional folds, and fragmented nerve terminals [162]. Initial studies have identified decreased AChE activity in muscle biopsies and increased AChE activity in plasma and serum of ALS patients [163,164,165]. Muscle biopsies of ALS patients also revealed alterations in various isoforms of AChE which resulted in decreased AChE activity in motor endplates [146, 166]. Loss of function of TDP-43 in zebrafish resulted in decreased expression of AChE leading to its decreased activity in skeletal muscles [151]. Earlier clinical trials with AChE inhibitors displayed no therapeutic effects in ALS patients whereas downregulating AChE expression using anti-sense nucleotide in presymptomatic ALS mice exerted beneficial effects by reducing MN loss and increasing its survival [167, 168]. Abnormalities in AChE seem to play an important role in ALS but more studies are needed to elucidate its pathomechanism.
Mutant Protein Toxicity
Studies have demonstrated that MN-specific expression of mutant SOD1 in transgenic mice is not sufficient to produce ALS phenotype [169, 170] whereas muscle-specific expression of mutant SOD1 is sufficient to develop ALS phenotype [171,172,173]. Transgenic mice with muscle restricted expression of SOD1G93A caused accumulation of reactive oxygen species (ROS) leading to structural and functional aberration in muscles [171]. Later on, expression of mutant SOD in skeletal muscles was shown to cause NMJ abnormalities, distal axonopathy with swollen or vacuolated axon terminals, and loss of MN in spinal cord [172]. NMJ abnormalities included loss of presynaptic synaptophysin and postsynaptic nAChRs and rapsyn [173]. NMJ disruption and retrograde degeneration of MN may involve target deprivation of neurotrophic factors. Altogether, these studies strongly proposed the crucial involvement of skeletal muscles in ALS pathogenesis and challenge the well-established dogma that muscle atrophy occurs as a consequence of MN degeneration and supports dying back phenomenon.
A longitudinal MRI analysis of SOD1G93A mice showed significant reduction in muscle volume followed by progressive muscle atrophy prior to onset of clinical symptoms indicating skeletal muscle could be the primary target of mutant SOD1-mediated toxicity [174]. Mutant SOD1 causes early changes in muscle metabolic properties by altering glucose metabolism and fiber type composition and increasing lipid catabolism [175].
Turner et al. [176] reported the presence of intracellular aggregates of mutant SOD1 in skeletal muscles of ALS mice. On the other hand, Onesto and colleagues showed that enhanced proteasome activity and autophagy in murine myoblast cell line (C2C12) efficiently removed mutant SOD1 aggregates whereas these aggregates impaired proteasome activity in motor neuronal cell line (NSC-34) indicating mutant SOD1 differentially affects MN and skeletal muscles [177]. Even in ALS mice, mutant SOD1 did not form aggregates in skeletal muscles throughout the disease course, possibly due to enhanced proteasomal activity [178]. These recent studies highlight that muscle cells are more efficient in managing mutant SOD1 aggregates as compared to MN, and it has been suggested that mutant SOD1-mediated toxicity in muscle may involve a different mechanism [179].
Interestingly, Wei et al. [180] showed that despite the absence of insoluble mutant SOD1 aggregates in muscles, skeletal muscle proteins are sensitive to misfolding similar to spinal cord proteins. Moreover, they observed lower steady-state levels of heat shock proteins in skeletal muscles as compared to spinal cord in SOD1G93A mice and postulated that differential expression of molecular chaperones makes skeletal muscles proteins more vulnerable to misfolding, which probably explains early degeneration of muscles in ALS pathogenesis [180]. In addition, the two major protein degradation pathways showed a time-dependent activation in skeletal muscles during disease progression: ubiquitin–proteasome degradation system (UPS) is activated during early symptomatic stages, whereas autophagy upregulation is observed in presymptomatic and end stages of disease [181].
Another important observation showed that mutant SOD1-associated toxicity in muscles can be rescued by decreasing mRNA and protein levels of SOD1 using antisense oligonucleotides. The findings clearly demonstrated that lowering the expression of mutant SOD1 can maintain neuromuscular innervation, preserve compound muscle action potential, restore neuronal dysfunction, and prolong the life of ALS transgenic mice [182].
Similar to SOD1, hyper-phosphorylated TDP-43 aggregates also plays a pathogenic role in central nervous system in ALS, but their role in skeletal muscles is unclear. Previous study reported the absence of phosphorylated TDP-43 (pTDP-43) aggregates in quadriceps muscles [183], whereas recent studies have identified dense filamentous aggregates of pTDP-43 in diaphragm and axial muscles of ALS patients [184, 185]. These authors suggest that the opposing findings could arise due to muscle group-specific difference in muscle pathology. Further studies are warranted to better understand TDP-43 pathology in skeletal muscles.
Mitochondrial Dysfunction
Mitochondrial dysfunction in muscle plays a pivotal role in ALS pathogenesis and contributes to rapid disease progression [186, 187]. Numerous studies revealed abnormalities in morphology, quantity, membrane potential, and disposition of skeletal muscle mitochondria in ALS [150, 171, 188,189,190,191,192]. Mitochondrial dysfunction is also responsible for hypermetabolism, which is a common feature of ALS [193, 194]. Mitochondrial abnormalities are often accompanied by defective mitochondrial respiratory chain complex and increased oxidative stress. Mice with mutation in CHCHD10, a mitochondrial protein, resulted in severe mitochondrial defects in skeletal muscles which led to NMJ abnormalities including hyper-fragmentation of motor endplate. Loss of MN in spinal cord was evident in end disease stages indicating abnormalities in muscle mitochondria can trigger MN degeneration in a dying back fashion [195]. SOD1G93A mice showed tremendous increase in muscle mitochondrial ROS production accompanied by muscle atrophy [196, 197]. Recently, it has been shown that administration of cerebrospinal fluid from sALS patients into muscles of neonatal rats caused mitochondrial abnormalities and increased oxidative stress in skeletal muscles [155]. An in vitro model system developed by differentiating iPSCs from C9ORF72 ALS patients into skeletal myocytes also showed altered mitochondrial gene expression and oxidative stress [192].
Muscle mitochondria are crucially involved in maintaining NMJ integrity. Overexpression of mitochondrial uncoupling protein 1(UCP1) in muscles significantly affected NMJ stability and led to motor neuron degeneration. Moreover, heterozygous (mutant SOD1/UCP1) transgenic ALS mice exhibited shortened progression from onset and survival as compared to homozygous mutant SOD1 ALS mice [198]. Overexpression of mutant SOD1 in skeletal muscle of normal mice also led to defective mitochondrial dynamics by forming aggregates in muscle mitochondria without causing MN degeneration, indicating that ALS-associated muscle pathology is an early event in disease course [199].
Interestingly, mitochondrial functionality can be rescued by reducing oxidative-mediated damage. Treating SOD1G93A mice with Trolox, a potent antioxidant, inhibited ROS and rescued mitochondrial function which in turn stabilized NMJ turnover and complexity [88]. In addition, increased mitochondrial biogenesis by overexpression of peroxisome proliferator-activated receptor gamma (PPARγ) coactivator-1α (PGC-1α), in skeletal muscle of SOD1G37R mice, maintained mitochondrial activity throughout disease course which improved muscle endurance, delayed muscle atrophy without improving lifespan [200]. Taken together, the above studies highlight the involvement of muscle mitochondrial abnormalities in ALS.
Autoantibodies Against Postsynaptic Proteins in ALS
Interestingly, autoantibodies against various postsynaptic proteins have been reported in ALS. The presence of anti-acetylcholine receptor antibodies, a hallmark feature of myasthenia gravis, has been observed in ALS patients with no history of myasthenia gravis or exposure to snake’s venom [201, 202]. Similarly, antibodies against Lrp4 have also been reported in peripheral blood of 9.8% American, ~ 15% Israeli, ~ 23% in Greek and Italian, and 5.4% in Chinese ALS patients [203,204,205,206]. Some studies have also reported the presence of autoantibodies against AChE in serum of ALS patients whereas others failed to observe the same [207, 208]. Although these are rare events but it raises an important question, whether autoimmune component is involved in ALS? These autoantibodies may be involved in denervation process and appear to be an important aspect to explore their role in disease mechanism.
Role of Terminal Schwann Cells in NMJ Disruption in ALS
Unlike well-characterized nerve-muscle interaction, terminal Schwann cells (TSCs) are important but overlooked component of tripartite synapse. TSCs play an imperative role in NMJ formation, maturation, maintenance, and regeneration [209, 210]. TSCs can sense and modulate synaptic activity to ensure appropriate neuromuscular transmission. Due to the close association, TSCs have a bidirectional relationship with motor nerve terminals in regulating synaptic output. Release of neurotransmitter from nerve terminal activates TSC’s muscarinic AChRs (mAChRs) which causes intracellular release of Ca2+ from internal stores of TSCs [211, 212]. In response, TSCs release gliotransmitter such as ATP which acts on presynaptic A1 (inhibitory) and A2A (excitatory) adenosine receptors and fine-tunes acetylcholine release from the nerve terminal [213]. It has been demonstrated that degenerating nerve terminals release signaling molecules which causes TSC to acquire a macrophage like behavior. The acquired phagocytic activity helps TSCs to engulf degenerating nerve terminal, assist nerve regeneration [214], and induces new AChR clustering and helps in neuromuscular junction remodeling [215].
Studies based on ALS transgenic mice showed involvement of TSCs abnormalities in ALS (Fig. 3c). Altered Ca2+ signaling accompanied by higher mAChR-mediated TSC activation led to major synaptic alterations [216]. NMJs of fast medial gastrocnemius muscle lacked TSC cell bodies followed by an increased denervation as compared to NMJs of slow soleus muscle [217]. Previously, TSCs showed increased expression of Sema3A, an axon repellent molecule, in NMJs of FF type IIb/x muscle fibers which highlights the early loss of FF motor units observed in ALS pathology [218]. Another study demonstrated increased level of adenosine at the synaptic cleft during symptomatic phase suggesting impaired release of TSC gliotransmitter [219].
Importantly, muscle samples from ALS patients also exhibited altered TSC morphology with extensive cytoplasmic processes. It was observed that in certain NMJs, TSC processes invaded synaptic cleft leaving limited accessible space for synaptic neuromuscular transmission [77]. Owing to the dynamic role played by TSC in regulating NMJ homeostasis, its altered function may exacerbate ALS phenotype.
Is Targeting NMJ a Possible Treatment for ALS?
Currently, riluzole and edaravone are the only two FDA-approved drugs for ALS treatment. Edaravone, a potent antioxidant, scavenges oxygen radicals, whereas riluzole exerts inhibitory effects on glutamate release [220]. Both drugs display modest benefits in prolonged survival and/or function by alleviating symptoms and slowing disease progression [221].
As several evidences discussed above support that NMJ impairment is an early event in ALS pathology, therapeutic treatments aimed at preserving NMJs may play an imperative role in fighting against ALS. Globally, the major focus of research has been to investigate role of neurotrophic factors, microRNAs, and small molecules to preserve NMJ integrity (Fig. 4).

Rescuing potential of neurotrophic factors and miRNAs in preserving NMJ integrity in ALS. a Intramuscular transplantation of neurotrophic factors helps preserve NMJ integrity and improves life span of ALS mice. b Manipulating muscle-specific miRNA expression rescues MN degeneration and NMJ disruption
Neurotrophic Factors
Neurotrophic factors are endogenous soluble signaling proteins which promote growth and survival of neurons. Skeletal muscle is a rich source of neurotrophic factors such as glial-derived neurotrophic factor (GDNF), transforming growth factor-beta (TGF-β), vascular endothelial growth factor (VEGF), insulin-like growth factor-1 (IGF-1), and brain-derived neurotrophic factor (BDNF) which are crucial for NMJ stability, MN survival, and axonal growth. Neurotrophic factors are internalized by axon terminals at NMJ and retrogradely transported to cell bodies of MN. Alterations in expression of muscle-derived neurotrophic factors influence NMJ denervation and spinal MN degeneration.
-
a)
Glial Derived Neurotrophic Factor
GDNF is a potential neuronal growth factor which plays pivotal role in presynaptic branching and hyperinnervation [222]. Exogenous GDNF treatment in healthy mice resulted in enhanced axonal branching and arborization at the NMJ in early post-natal days suggesting GNDF plays a key role in synaptic maintenance and remodeling [223]. Therefore, it is important to evaluate its role during NMJ denervation in ALS. Increased GDNF mRNA levels were observed in muscle biopsies of alive ALS patients, whereas muscle biopsies from post mortem patient samples showed decreased expression of GDNF [224, 225]. These observations indicate GDNF expression varies drastically with disease progression.
Three independent studies observed that increasing expression of GDNF in skeletal muscle of SOD1G93A mice resulted in significant delay in disease onset, improved locomotor performance, and prolonged survival [226,227,228]. Interestingly, delivery of GDNF secreting neural progenitor cells (NPCs) in spinal cords remarkably preserved MN degeneration but failed to preserve NMJ denervation resulting in no beneficial effect on limb function [229]. However, when GDNF secreting mesenchymal stem cells (MSCs) were injected in skeletal muscles, an apparent increase in NMJ connections, survival of spinal MNs delayed disease progression [230]. These findings indicate that survival of motor neurons and innervation of muscle fibers are independent events. GDNF, secreted by muscle and terminal Schwann cells, binds to Ret tyrosine kinase receptor localized at the nerve terminals of presynaptic region and promotes synapse maturation [231]. On the other hand, it has been suggested that binding of GDNF to GPI-linked coreceptors and Ret tyrosine kinase receptors on motor neurons increases its survival by activating PI3-kinase signaling pathway [232, 233]. Therefore, combined therapy aimed at targeting MN cell perikarya and NMJ would help reduce disease progressing and alleviate ALS symptoms.
-
b)
Transforming Growth Factor-Beta
TGF-β, a motoneuron survival factor, is involved in the development and maintenance of NMJ. TGF-β is localized at the synaptic region of muscle fibers, and its receptors are distributed along the length of motor axons and nerve terminals [234]. Schwann cells also express TGF-β and promote synapse formation at NMJ. TGF-βs belong to a small family of multi-functional cytokines, consisting of three isoforms: TGF-β1, β2, and β3. A significant increase in all three isoforms of TGF-β has been observed in human and mouse ALS muscle [235]. Circulating levels (serum, plasma, and CSF) of TGF-β1 have been found to be significantly enhanced in ALS patients [236,237,238,239]. TGF-β1 activation has also been shown in both spinal cord and muscle of ALS mice [240]. In skeletal muscle of symptomatic SOD1G93A mice, TGF-β-mediated signaling is enhanced as compared to presymptomatic mice [241]. It has been hypothesized that reduced TGF-β signaling at early stage induces glutamate excitotoxicity and prevents its neuroprotective effect. And at the later stage, increased TGF-β levels activate microglia and hamper NMJ [242]. Another study showed increased levels of TGF-β1 at NMJs of even presymptomatic SOD1 ALS mice which repressed FGFBP1 expression and modulated NMJ innervation [243].
Interestingly, intraperitoneal injection of TGF-β2 in SOD1 mice with resulted in enhancement of motor performance without preventing motor neuronal degeneration [244]. Though the mechanistic action was not defined, but TGF-β2 has been shown to locally regulate neurotransmission by increasing the quantal content (presynaptic vesicles) [245]. All these evidences suggest that TGF-β pathway plays a role in ALS pathogenesis, but detailed analysis to understand the fine balance between levels of different TGF-β isoforms and downstream targets is important to explore its role in diagnostics and therapeutics.
-
iii)
Vascular Endothelial Growth Factor
VEGF exerts its protective effects by decreasing astrogliosis and increasing NMJ formation in ALS mouse [246]. Lentiviral expression of VEGF in skeletal muscles of ALS mice protected MN degeneration, slowed disease onset and progression, and enhanced life expectancy by 30% [247]. Intramuscular transplantation of MSCs expressing GDNF and VEFG improved lifespan, NMJ stability, and decreased MN loss in SOD1G93A transgenic rats [248].
-
iv)
Insulin-Like Growth Factor-1
IGF-1 plays a key role in neuronal survival and maintenance of neuromuscular connections. Significantly reduced serum levels of IGF-1 were observed in ALS patients [249]. Even skeletal muscle samples from ALS patients showed decreased levels of IGF-1 and IGF binding proteins [250]. However, a recent study reported increased serum IGF-1 levels in ALS patients with longer survival indicating higher IGF-1 levels may be involved in delaying disease progression [251].
Muscle-specific expression of IGF-1 in ALS mice attenuated muscle atrophy and displayed well-preserved AChR clusters and stabilized innervation of muscle fibers. Moreover, increased IGF-1 led to prolonged MN function and life span [252, 253]. IGF-1 injected in skeletal muscle of ALS mice was retrogradely internalized by MN which protected mitochondria from autophagy by reducing cytochrome C release and inhibited apoptosis [254].
-
e)
Brain-Derived Neurotrophic Factor
BDNF is involved in maintaining structural integrity and synaptic function in adult muscular synapses by binding to tropomyosin-related kinase B receptor (TrkB) [255]. Nerve-induced muscle contraction regulates BDNF/TrkB signaling which in turn affects release of ACh by modulating presynaptic protein kinases PKC and PKA [256]. Earlier studies have reported alterations in BDNF expression and TrkB signaling in spinal cord and muscle tissues of ALS patients [257,258,259]. A recent study identified C270T polymorphism in BDNF as susceptibility locus in Han Chinese ALS patients [260].
Impaired BDNF/TrkB signaling was observed in NMJs of plantaris muscle in symptomatic SOD1G93A mice [261]. Exercise regime modulated BDNF/TrkB signaling resulting in reduced MN death [262]. Interestingly, in vitro incubation with ALS-CSF downregulated BDNF expression and induced neurodegeneration in NSC-34 motor neuronal cell line [263, 264] and exogenous supplementation of BDNF-ameliorated ALS-CSF-induced neurodegeneration [264]. Even intrathecal administration of ALS-CSF led to significant downregulation of BDNF mRNA levels in spinal cord and upregulation in extensor digitorum longus muscle of neonatal rats [155, 265]. The increased muscle BDNF expression could be a compensatory mechanism to restore homeostasis, but due to increased NMJ denervation, MNs are not able to harness the enhanced neurotrophic levels.
MicroRNAs
MicroRNAs (miRNAs) are non-coding, single-stranded RNA molecules involved in post-transcriptional regulation of gene expression. miRNAs typically bind to 3′ untranslated region (3′UTR) of messenger RNAs (mRNAs) and lead to translational repression or mRNA degradation. Recent evidences suggest that deregulated expression of miRNAs can contribute to motor neuron disorders [266, 267]. miRNAs are key mediators for development of skeletal muscle and nervous system. They have also been implicated in maturation, maintenance, and repair of NMJ. Altered levels of miRNA expression have been reported in motor neurons and skeletal muscles of ALS patients [268, 269]. Moreover, mutations in FUS, TDP-43, and SOD1 have been shown to reduce miRNA biogenesis in MN by inhibiting DICER catalytic activity [269]. Thus, it is important to evaluate the role of these non-coding RNAs in ALS pathology.
-
a)
miR-1 and miR-133
miR-1, a conserved muscle-specific miRNA, has been shown to contribute to NMJ stability. miR-1 and miR-133 mediate differentiation and proliferation of skeletal muscle by regulating histone deacetylase 4 (HDAC4, a transcriptional repressor of muscle gene expression) and serum response factor, respectively [270, 271].
Lower levels of miR-1, miR-133a, miR-133b, and miR-206 were observed in ALS patients undergoing physical rehabilitation which shows that miRNA expression is altered during skeletal muscle recovery in ALS patients [272].
Muscle-specific expression of SOD1G93A has been shown to affect spinal cord miRNA. The levels of miR-1, miR-9, miR-133, and miR-330 were reduced in ALS transgenic mice. These miRNAs are involved in activation of genes linked with myelination events in spinal cord and upregulate expression of peripheral myelin protein 22 and myelin protein zero [273]. In contrast, miR-133b was found to be upregulated following denervation in ALS mice [274].
-
b)
miR-126-5p
miR-126-5p has been shown to regulate ALS-associated genes, like VEGF-A, agrin, and C9orf72. In presymptomatic ALS mice model, lower levels of miR-126-5p induced overexpression of Sema3A and other destabilizing factors in skeletal muscles and its coreceptor Neuropilin 1 in axons. This triggered NMJ disruption and axon degeneration [275]. At the same time, overexpressed miR-126-5p transiently rescued axonal degeneration and NMJ disruption.
-
iii)
miR-206
miR-206, a skeletal muscle-specific microRNA, has been shown to promote maintenance and repair of NMJ [274]. In SOD1G93A mice, knocking out miR-206 enhanced disease progression and accelerated skeletal muscle atrophy. Elevated expression of miR-206 in SOD1G93A mice after nerve damage promoted reinnervation at NMJ by regulating HDAC4 and fibroblast growth factor pathway, thereby delaying disease progression [276]. Further, levels of miR-206 were found to be upregulated in plasma and muscles of SOD1G93A mice and serum of ALS patients suggesting its possible role in ALS pathogenesis [277].
-
iv)
Other miRs
In skeletal muscle of ALS patients, miR-23a, miR-29b, miR-206, miR-455, and miR-31 were found to be significantly upregulated. It was demonstrated that miR-23a binds 3′UTR of PGC-1α mRNA, a regulator of mitochondrial biogenesis, and suppresses protein translation, thereby impairing skeletal muscle mitochondrial function [268]. On the contrary, miR‐9 and miR‐124 were downregulated in MN of ALS patients [269].
Taken together, these evidences suggest that miRNAs are key elements responsible for regeneration and repair of NMJ, proliferation, and differentiation of skeletal muscles. However, the role of miRNAs in disruption of NMJ in ALS etiology remains to be elucidated.
Small Molecules
Various therapeutic compounds including neuroleptics are being explored for stabilizing NMJ in ALS. Pimozide, T-type Ca2+ channel blocker, has been shown to stabilize synaptic transmission in C. elegans, zebrafish, and mice model of ALS. Further, pimozide displayed its beneficial effects on NMJ function in a 6-week long phase II randomized controlled trial on ALS patients [278]. On the contrary, Pozzi et al. [279] reported chronic administration of pimozide exerted no beneficial effect on muscle strength performance but rather resulted in diminished motor performance and exacerbated NMJ loss in SOD1G93A and TDP-43A315T ALS mice. Pimozide is undergoing a long-term clinical trial to determine its effectiveness in slowing ALS progression (ClinicalTrials.gov Identifier: NCT03272503). Recently, TRVA242, a derivative of pimozide, substantially rescued locomotor deficits, NMJ structural abnormalities, and restored NMJ transmission in C. elegans, zebrafish, and mice model of ALS [280]. Although TRVA242 showed promising results, further studies evaluating pharmacodynamics, pharmacokinetics, behavioral effects, and other measures are required.
Conclusion
ALS, primarily considered as a motor neuron disorder, is now being viewed as a non-cell autonomous disease which involves interplay of neuronal and non-neuronal cell populations which act together to exacerbate the disease [281]. Non-neuronal cell populations such as glial cells, skeletal muscles, and peripheral blood mononuclear cells play a key role in ALS pathophysiology [171, 282,283,284]. Despite leaps of advancement in the field of ALS, one of the key unresolved issues is site of disease initiation. Considering the complex etiology of ALS, it is postulated that MN degeneration involves multifactorial origin and can progress in either a “dying forward” or a “dying backward” manner. There is an ongoing debate regarding cortical hyperexcitability or neuromuscular junction as the site of ALS onset. Evidences supporting both the hypothesis have been reviewed, but we have focused more on the dying back hypothesis.
Research in the past 2 decades have identified neuromuscular junction alterations in early disease course. This review presents a plethora of clinical and experimental evidences describing cellular and molecular alterations in motor neurons, skeletal muscles, and terminal Schwann cells in ALS. MN in ALS display disturbed anterograde and retrograde axonal transport leading to insufficient maintenance of distal terminals affecting cholinergic transmission and mitochondrial dynamics as well as poor uptake of neurotrophic factors. Impaired protein homeostasis and proteins aggregation have been shown to trigger NMJ denervation. Similar to MNs, skeletal muscles are also involved in NMJ malfunctioning. Muscle-specific expression of ALS-associated protein is able to generate ALS phenotype indicating causal role of skeletal muscle in disease pathogenesis [171, 172]. Alterations in postsynaptic structures such as AChRs, Lrp4, and AChE lead to endplate dispersion, fragmentation of AChRs, decreased ramification of junctional folds, and impaired neuromuscular transmission in ALS. Interestingly, autoantibodies against these postsynaptic structures have been reported in ALS, but their pathological significance is still elusive. Unlike MNs, muscle cells have a robust proteasome machinery due to which ALS-associated mutant proteins fail to accumulate in muscles. These deleterious proteins cause mitochondrial dysfunction and alters muscle metabolic properties which leads to hypermetabolism in ALS. Decreasing mutant SOD1 expression or reducing oxidative stress have been shown to rescue mitochondrial function, restore neuronal dysfunction, and stabilize NMJ complexity [150, 182]. Terminal Schwann cells, a less explored component of NMJ, plays a pivotal role during NMJ reinnervation. TSCs abnormalities such as altered Ca2+ signaling, morphology, and irregular release of gliotransmitter, axon repellant molecule can impair NMJ maintenance and regenerative capabilities of TSCs. Apart from these tripartite components, basal lamina also plays a critical role in structural and functional organization of NMJ. Limb muscle samples from ALS patients showed absence of laminins α4 from NMJs [285].
The current therapies targeting motor neurons have not been very effective against ALS, and therefore, new targets for developing effective therapies for ALS are being explored. Experimental strategies aimed at preserving NMJ by increasing muscle expression of neurotrophic factors have shown to exert beneficial effects such as preserving innervation, enhancing MN survival, delaying disease onset, and improving lifespan. Similarly modulating miRNAs expression rescues NMJ denervation and aids in alleviating ALS symptoms.
Although recent studies have highlighted early NMJ pathomechanism in ALS, information regarding the series of events leading to NMJ denervation are scattered. It is still unclear whether NMJ denervation is a MN or muscle induced event? In order to find answer to such questions, extensive research is required to closely observe early pathological events involved in disease initiation. In vivo studies focusing on monitoring various aspects of NMJ during presymptomatic stages will help in better understanding of cellular and molecular changes occurring in early disease stages. Similarly, in vitro studies exploring co-culture of motor neuron, skeletal muscles, and TSCs could provide deep insights into NMJ formation, functioning, and maintenance in ALS. Furthermore, time-dependent studies comparing NMJs of vulnerable (limb muscles) and resistant (extraocular muscles) motor units in ALS will enhance our understanding of altered synaptic functionality in ALS. Altogether, such studies will provide a better understanding of early pathological changes at NMJ and help in connecting the missing dots that will unravel the mystery behind ALS.
Data Availability
Not applicable.
Code Availability
Not applicable.
References
Nalini A, Thennarasu K, Gourie-Devi M, Shenoy S, Kulshreshtha D (2008) Clinical characteristics and survival pattern of 1153 patients with amyotrophic lateral sclerosis: experience over 30 years from India. J Neurol Sci 272(1–2):60–70. https://doi.org/10.1016/j.jns.2008.04.034
Pupillo E, Messina P, Logroscino G, Beghi E, SLALOM Group (2014) Long-term survival in amyotrophic lateral sclerosis: a population-based study. Ann Neurol 75(2):287–297. https://doi.org/10.1002/ana.24096
Moura MC, Novaes MRCG, Eduardo EJ, Zago YSSP, Freitas RDNB, Casulari LA (2015) Prognostic factors in amyotrophic lateral sclerosis: a population-based study. PLoS One 10(10):e0141500. https://doi.org/10.1371/journal.pone.0141500
Knibb JA, Keren N, Kulka A, Leigh PN, Martin S, Shaw CE, Tsuda M, Al-Chalabi A (2016) A clinical tool for predicting survival in ALS. J Neurol Neurosurg Psychiatry 87(12):1361–1367. https://doi.org/10.1136/jnnp-2015-312908
Vats A, Gourie-Devi M, Verma M, Ramachandran S, Taneja B, Kukreti R, Taneja V (2016) Identification of L84F mutation with a novel nucleotide change c.255G > T in the superoxide dismutase gene in a North Indian Family with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener 17(3–4):253–259. https://doi.org/10.3109/21678421.2015.1111906
Renton AE, Chiò A, Traynor BJ (2014) State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci 17(1):17–23. https://doi.org/10.1038/nn.3584
Mulder DW, Lambert EH, Eaton LM (1959) Myasthenic syndrome in patients with amyotrophic lateral sclerosis. Neurology 9:627–631. https://doi.org/10.1212/wnl.9.10.627
Killian JM, Wilfong AA, Burnett L, Appel SH, Boland D (1994) Decremental motor responses to repetitive nerve stimulation in ALS. Muscle Nerve 17(7):747–754. https://doi.org/10.1002/mus.880170708
Hu F, Jin J, Kang L, Jia R, Qin X, Liu X, Liu X, Liu C et al (2018) Decremental responses to repetitive nerve stimulation in amyotrophic lateral sclerosis. Eur Neurol 80(3–4):151–156. https://doi.org/10.1159/000494670
Shang L, Chu H, Lu Z (2020) Can the Large-Scale Decrement in repetitive nerve stimulation be used as an exclusion criterion for amyotrophic lateral sclerosis? Front Neurol 11:101. https://doi.org/10.3389/fneur.2020.00101
Yuen EC, Olney RK (1997) Longitudinal study of fiber density and motor unit number estimate in patients with amyotrophic lateral sclerosis. Neurology 49(2):573–578. https://doi.org/10.1212/wnl.49.2.573
van Dijk JP, Schelhaas HJ, Van Schaik IN, Janssen HMHA, Stegeman DF, Zwarts MJ (2010) Monitoring disease progression using high-density motor unit number estimation in amyotrophic lateral sclerosis. Muscle Nerve 42(2):239–244. https://doi.org/10.1002/mus.21680
Shefner JM, Watson ML, Simionescu L, Caress JB, Burns TM, Maragakis NJ, Benatar M, David WS et al (2011) Multipoint incremental motor unit number estimation as an outcome measure in ALS. Neurology 77(3):235–241. https://doi.org/10.1212/WNL.0b013e318225aabf
Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, Burrell JR, Zoing MC (2011) Amyotrophic lateral sclerosis. Lancet 377(9769):942–955. https://doi.org/10.1016/S0140-6736(10)61156-7
Vucic S, Nicholson GA, Kiernan MC (2008) Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain 131(Pt6):1540–1550. https://doi.org/10.1093/brain/awn071
Menon P, Kiernan MC, Vucic S (2015) Cortical hyperexcitability precedes lower motor neuron dysfunction in ALS. Clin Neurophysiol 126(4):803–809. https://doi.org/10.1016/j.clinph.2014.04.023
Blizzard CA, Southam KA, Dawkins E, Lewis KE, King AE, Clark JA, Dickson TC (2015) Identifying the primary site of pathogenesis in amyotrophic lateral sclerosis - vulnerability of lower motor neurons to proximal excitotoxicity. Dis Model Mech 8(3):215–224. https://doi.org/10.1242/dmm.018606
Eisen A, Kim S, Pant B (1992) Amyotrophic Lateral Sclerosis (ALS): A Phylogenetic disease of the corticomotoneuron? Muscle Nerve 15(2):219–224. https://doi.org/10.1002/mus.880150215
Eisen A, Pant B, Stewart H (1993) Cortical excitability in amyotrophic lateral sclerosis: a clue to pathogenesis. Can J Neurol Sci 20(1):11–16. https://doi.org/10.1017/s031716710004734x
Mills KR, Nithi KA (1997) Corticomotor threshold is reduced in early sporadic amyotrophic lateral sclerosis. Muscle Nerve 20(9):1137–1141. https://doi.org/10.1002/(sici)1097-4598(199709)20:9<3c1137::aid-mus7>3e3.0.co;2-9
Eisen A, Nakajima M, Weber M (1998) Corticomotorneuronal hyper-excitability in amyotrophic lateral sclerosis. J Neurol Sci 160(Suppl 1):S64-68. https://doi.org/10.1016/s0022-510x(98)00200-7
Weber M, Eisen A, Nakajima M (2000) Corticomotoneuronal activity in ALS: changes in the peristimulus time histogram over time. Clin Neurophysiol 111(1):169–177. https://doi.org/10.1016/s1388-2457(99)00190-x
Zanette G, Tamburin S, Manganotti P, Refatti N, Forgione A, Rizzuto N (2002) Different mechanisms contribute to motor cortex hyperexcitability in amyotrophic lateral sclerosis. Clin Neurophysiol 113(11):1688–1697. https://doi.org/10.1016/s1388-2457(02)00288-2
Geevasinga N, Menon P, Nicholson GA, Ng K, Howells J, Kril JJ, Yiannikas C, Kiernan MC et al (2015) Cortical function in asymptomatic carriers and patients with C9orf72 amyotrophic lateral sclerosis. JAMA Neurol 72(11):1268–1274. https://doi.org/10.1001/jamaneurol.2015.1872
Van den Bos MAJ, Higashihara M, Geevasinga N, Menon P, Kiernan MC, Vucic S (2018) Imbalance of cortical facilitatory and inhibitory circuits underlies hyperexcitability in ALS. Neurology 91(18):e1669–e1676. https://doi.org/10.1212/WNL.0000000000006438
Browne SE, Yang L, DiMauro JP, Fuller SW, Licata SC, Beal MF (2006) Bioenergetic abnormalities in discrete cerebral motor pathways presage spinal cord pathology in the G93A SOD1 mouse model of ALS. Neurobiol Dis 22(3):599–610. https://doi.org/10.1016/j.nbd.2006.01.001
Kim J, Hughes EG, Shetty AS, Arlotta P, Goff LA, Bergles DE, Brown SP (2017) Changes in the excitability of neocortical neurons in a mouse model of amyotrophic lateral sclerosis are not specific to corticospinal neurons and are modulated by advancing disease. J Neurosci 37(37):9037–9053. https://doi.org/10.1523/JNEUROSCI.0811-17.2017
Kuo JJ, Schonewille M, Siddique T, Schults ANA, Fu R, Bär PR, Anelli R, Heckman CJ et al (2004) Hyperexcitability of cultured spinal motoneurons from presymptomatic ALS mice. J Neurophysiol 91(1):571–575. https://doi.org/10.1152/jn.00665.2003
Kuo JJ, Siddique T, Fu R, Heckman CJ (2005) Increased persistent Na (+) current and its effect on excitability in motoneurones cultured from mutant SOD1 mice. J Physiol 563(Pt 3):843–854. https://doi.org/10.1113/jphysiol.2004.074138
van Zundert B, Peuscher MH, Hynynen M, Chen A, Neve RL, Brown RH, Constantine-Paton M, Bellingham MC (2008) Neonatal neuronal circuitry shows hyperexcitable disturbance in a mouse model of the adult-onset neurodegenerative disease amyotrophic lateral sclerosis. J Neurosci 28(43):10864–10874. https://doi.org/10.1523/JNEUROSCI.1340-08.2008
Elbasiouny SM, Amendola J, Durand J, Heckman CJ (2010) Evidence from computer simulations for alterations in the membrane biophysical properties and dendritic processing of synaptic inputs in mutant superoxide dismutase-1 motoneurons. J Neurosci 30(16):5544–5558. https://doi.org/10.1523/JNEUROSCI.0434-10.2010
Martin E, Cazenave W, Cattaert D, Branchereau P (2013) Embryonic alteration of motoneuronal morphology induces hyperexcitability in the mouse model of amyotrophic lateral sclerosis. Neurobiol Dis 54:116–126. https://doi.org/10.1016/j.nbd.2013.02.011
Pambo-Pambo A, Durand J, Gueritaud JP (2009) Early excitability changes in lumbar motoneurons of transgenic SOD1G85R and SOD1G(93A-Low) mice. J Neurophysiol 102(6):3627–3642. https://doi.org/10.1152/jn.00482.2009
Quinlan KA, Schuster JE, Fu R, Siddique T, Heckman CJ (2011) Altered postnatal maturation of electrical properties in spinal motoneurons in a mouse model of amyotrophic lateral sclerosis. J Physiol 589(Pt 9):2245–2260. https://doi.org/10.1113/jphysiol.2010.200659
Bories C, Amendola J, Lamotte d’Incamps B, Durand J (2007) Early electrophysiological abnormalities in lumbar motoneurons in a transgenic mouse model of amyotrophic lateral sclerosis. Eur J Neurosci 25(2):451–459. https://doi.org/10.1111/j.1460-9568.2007.05306.x
Delestrée N, Manuel M, Iglesias C, Elbasiouny SM, Heckman CJ, Zytnicki D (2014) Adult spinal motoneurones are not hyperexcitable in a mouse model of inherited amyotrophic lateral sclerosis. J Physiol 592(7):1687–1703. https://doi.org/10.1113/jphysiol.2013.265843
de Martínez-Silva ML, Imhoff-Manuel RD, Sharma A, Heckman CJ, Shneider NA, Roselli F, Zytnicki D, Manuel M (2018) Hypoexcitability precedes denervation in the large fast-contracting motor units in two unrelated mouse models of ALS. Elife 7:e30955. https://doi.org/10.7554/eLife.30955
Kuo SW, Binder MD, Heckman CJ (2020) Excessive homeostatic gain in spinal motoneurons in a mouse model of amyotrophic lateral sclerosis. Sci Rep 10(1):9049. https://doi.org/10.1038/s41598-020-65685-8
Brunet A, Stuart-Lopez G, Burg T, Scekic-Zahirovic J, Rouaux C (2020) Cortical circuit dysfunction as a potential driver of amyotrophic lateral sclerosis. Front Neurosci 14:363. https://doi.org/10.3389/fnins.2020.00363
Eisen A (2021) The dying forward hypothesis of ALS: tracing its history. Brain Sci 11(3):300. https://doi.org/10.3390/brainsci11030300
Dadon-Nachum M, Melamed E, Offen D (2011) The “dying-back” phenomenon of motor neurons in ALS. J Mol Neurosci 43(3):470–477. https://doi.org/10.1007/s12031-010-9467-1
Moloney EB, de Winter F, Verhaagen J (2014) ALS as a distal axonopathy: molecular mechanisms affecting neuromuscular junction stability in the presymptomatic stages of the disease. Front Neurosci 8:252. https://doi.org/10.3389/fnins.2014.00252
Court FA, Gillingwater TH, Melrose S, Sherman DL, Greenshields KN, Morton AJ, Harris JB, Willison HJ et al (2008) Identity, developmental restriction and reactivity of extralaminar cells capping mammalian neuromuscular junctions. J Cell Sci 121(Pt 23):3901–3911. https://doi.org/10.1242/jcs.031047
Sugiura Y, Lin W (2011) Neuron-glia interactions: the roles of schwann cells in neuromuscular synapse formation and function. Biosci Rep 31(5):295–302. https://doi.org/10.1042/BSR20100107
Kim N, Stiegler AL, Cameron TO, Hallock PT, Gomez AM, Huang JH, Hubbard SR, Dustin ML et al (2008) Lrp4 is a receptor for agrin and forms a complex with MuSK. Cell 135(2):334–342. https://doi.org/10.1016/j.cell.2008.10.002
Inoue A, Setoguchi K, Matsubara Y, Okada K, Sato N, Iwakura Y, Higuchi O, Yamanashi Y (2009) Dok-7 activates the muscle receptor kinase MuSK and shapes synapse formation. Sci Signal 2(59):ra7. https://doi.org/10.1126/scisignal.2000113
Hippenmeyer S, Huber RM, Ladle DR, Murphy K, Arber S (2007) ETS transcription factor Erm controls subsynaptic gene expression in skeletal muscles. Neuron 55(5):726–740. https://doi.org/10.1016/j.neuron.2007.07.028
Lee Y, Rudell J, Ferns M (2009) Rapsyn interacts with the muscle acetylcholine receptor via alpha-helical domains in the alpha beta and epsilon subunit intracellular Loops. Neuroscience 163(1):222–232. https://doi.org/10.1016/j.neuroscience.2009.05.057
Li L, Xiong WC, Mei L (2018) Neuromuscular junction formation aging and disorders. Annu Rev Physiol 80:159–188. https://doi.org/10.1146/annurev-physiol-022516-034255
Sanes JR (2003) The basement membrane/basal lamina of skeletal muscle. J Biol Chem 278(15):12601–12604. https://doi.org/10.1074/jbc.R200027200
Patton BL (2003) Basal lamina and the organization of neuromuscular synapses. J Neurocytol 32(5–8):883–903. https://doi.org/10.1023/B:NEUR.0000020630.74955.19
Nishimune H, Sanes JR, Carlson SS (2004) A synaptic laminin-calcium channel interaction organizes active zones in motor nerve terminals. Nature 432(7017):580–587. https://doi.org/10.1038/nature03112
Chand KK, Lee KM, Schenning MP, Lavidis NA, Noakes PG (2015) Loss of β2-laminin alters calcium sensitivity and voltage-gated calcium channel maturation of neurotransmission at the neuromuscular junction. J Physiol 593(1):245–265. https://doi.org/10.1113/jphysiol.2014.284133
Nishimune H, Valdez G, Jarad G, Moulson CL, Müller U, Miner JH, Sanes JR (2008) Laminins promote postsynaptic maturation by an autocrine mechanism at the neuromuscular junction. J Cell Biol 182(6):1201–1215. https://doi.org/10.1083/jcb.200805095
Chand KK, Lee KM, Lavidis NA, Noakes PG (2017) Loss of laminin-α4 results in pre- and postsynaptic modifications at the neuromuscular junction. FASEB J 31(4):1323–1336. https://doi.org/10.1096/fj.201600899R
Chou SM, Norris FH (1993) Amyotrophic lateral sclerosis: lower motor neuron disease spreading to upper motor neurons. Muscle Nerve 16(8):864–869. https://doi.org/10.1002/mus.880160810
Kennel PF, Finiels F, Revah F, Mallet J (1996) Neuromuscular function impairment is not caused by motor neuron loss in FALS Mice: An Electromyographic Study. NeuroReport 7(8):1427–1431. https://doi.org/10.1097/00001756-199605310-00021
Gould TW, Buss RR, Vinsant S, Prevette D, Sun W, Knudson CM, Milligan CE, Oppenheim RW (2006) Complete dissociation of motor neuron death from motor dysfunction by Bax deletion in a mouse model of ALS. J Neurosci 26(34):8774–8786. https://doi.org/10.1523/JNEUROSCI.2315-06.2006
Dewil M, Dela Cruz VF, Van Den Bosch L, Robberecht W (2007) Inhibition of P38 mitogen activated protein kinase activation and mutant SOD1(G93A)-induced motor neuron death. Neurobiol Dis 26(2):332–341. https://doi.org/10.1016/j.nbd.2006.12.023
Frey D, Schneider C, Xu L, Borg J, Spooren W, Caroni P (2000) Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J Neurosci 20(7):2534–2542. https://doi.org/10.1523/JNEUROSCI.20-07-02534.2000
Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, Khan J, Polak MA et al (2004) Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol 185(2):232–240. https://doi.org/10.1016/j.expneurol.2003.10.004
Clark JA, Southam KA, Blizzard CA, King AE, Dickson TC (2016) Axonal degeneration, distal collateral branching and neuromuscular junction architecture alterations occur prior to symptom onset in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. J Chem Neuroanat 76(Pt A):35–47. https://doi.org/10.1016/j.jchemneu.2016.03.003
Sharma A, Lyashchenko AK, Lu L, Nasrabady SE, Elmaleh M, Mendelsohn M, Nemes A, Tapia JC et al (2016) ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat Commun 7:10465. https://doi.org/10.1038/ncomms10465
Picchiarelli G, Demestre M, Zuko A, Been M, Higelin J, Dieterlé S, Goy MA, Mallik M et al (2019) FUS-mediated regulation of acetylcholine receptor transcription at neuromuscular junctions is compromised in amyotrophic lateral sclerosis. Nat Neurosci 22(11):1793–1805. https://doi.org/10.1038/s41593-019-0498-9
Walker AK, Spiller KJ, Ge G, Zheng A, Xu Y, Zhou M, Tripathy K, Kwong LK (2015) Functional recovery in new mouse models of ALS/FTLD after clearance of pathological cytoplasmic TDP-43. Acta Neuropathol 130(5):643–660. https://doi.org/10.1007/s00401-015-1460-x
Riemslagh FW, van der Toorn EC, Verhagen RFM, Maas A, Bosman LWJ, Hukema RK, Willemsen R (2021) Inducible expression of human C9ORF72 36x G4C2 hexanucleotide repeats is sufficient to cause RAN translation and rapid muscular atrophy in mice. Dis Model Mech dmm 044842. https://doi.org/10.1242/dmm.044842.
Shahidullah M, Le Marchand SJ, Fei H, Zhang J, Pandey UB, Dalva MB, Pasinelli P, Levitan IB (2013) Defects in synapse structure and function precede motor neuron degeneration in drosophila models of FUS-related ALS. J Neurosci 33(50):19590–19598. https://doi.org/10.1523/JNEUROSCI.3396-13.2013
Sakowski SA, Lunn JS, Busta AS, Oh SS, Zamora-Berridi G, Palmer M, Rosenberg AA, Philip SG (2012) Neuromuscular effects of G93A-SOD1 expression in zebrafish. Mol Neurodegener 7:44. https://doi.org/10.1186/1750-1326-7-44
Ramesh T, Lyon AN, Pineda RH, Wang C, Janssen PML, Canan BD, Burghes AHM, Beattie CE (2010) A genetic model of amyotrophic lateral sclerosis in zebrafish displays phenotypic hallmarks of motoneuron disease. Dis Model Mech 3(9–10):652–662. https://doi.org/10.1242/dmm.005538
Pun S, Santos AF, Saxena S, Xu L, Caroni P (2006) Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat Neurosci 9(3):408–419. https://doi.org/10.1038/nn1653
Hegedus J, Putman CT, Gordon T (2007) Time course of preferential motor unit loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neurobiol Dis 28(2):154–164. https://doi.org/10.1016/j.nbd.2007.07.003
Tremblay E, Martineau É, Robitaille R (2017) Opposite synaptic alterations at the neuromuscular junction in an ALS mouse model: when motor units matter. J Neurosci 37(37):8901–8918. https://doi.org/10.1523/JNEUROSCI.3090-16.2017
Schaefer AM, Sanes JR, Lichtman JWA (2005) Compensatory subpopulation of motor neurons in a mouse model of amyotrophic lateral sclerosis. J Comp Neurol 490(3):209–219. https://doi.org/10.1002/cne.20620
Hegedus J, Putman CT, Tyreman N, Gordon T (2008) Preferential motor unit loss in the SOD1 G93A transgenic mouse model of amyotrophic lateral sclerosis. J Physiol 586(14):3337–3351. https://doi.org/10.1113/jphysiol.2007.149286
Martineau É, Di Polo A, Vande Velde C, Robitaille R 2018 Dynamic neuromuscular remodeling precedes motor-unit loss in a mouse model of ALS Elife 7https://doi.org/10.7554/eLife.41973
Maselli RA, Wollman RL, Leung C, Distad B, Palombi S, Richman DP, Salazar-Grueso EF, Roos RP (1993) Neuromuscular transmission in amyotrophic lateral sclerosis. Muscle Nerve 16(11):1193–1203. https://doi.org/10.1002/mus.880161109
Bruneteau G, Bauché S, Gonzalez de Aguilar JL, Brochier G, Mandjee N, Tanguy ML, Hussain G, Behin A et al (2015) Endplate denervation correlates with Nogo-A muscle expression in amyotrophic lateral sclerosis patients. Ann Clin Transl Neurol 2(4):362–372. https://doi.org/10.1002/acn3.179
Vinsant S, Mansfield C, Jimenez-Moreno R, Del Gaizo MV, Yoshikawa M, Hampton TG, Prevette D, Caress J (2013) Characterization of early pathogenesis in the SOD1(G93A) mouse model of ALS: Part II Results and Discussion. Brain Behav 3(4):431–457. https://doi.org/10.1002/brb3.142
Dirren E, Aebischer J, Rochat C, Towne C, Schneider BL, Aebischer P (2015) SOD1 Silencing in motoneurons or glia rescues neuromuscular function in ALS mice. Ann Clin Transl Neurol 2:167–184. https://doi.org/10.1002/acn3.162
McGown A, McDearmid JR, Panagiotaki N, Tong H, Al Mashhadi S, Redhead N, Lyon AN, Beattie CE et al (2013) Early interneuron dysfunction in ALS: insights from a mutant Sod1 zebrafish model. Ann Neurol 73(2):246–258. https://doi.org/10.1002/ana.23780
Ramesh TM, Shaw PJ, McDearmid J (2014) A zebrafish model exemplifies the long preclinical period of motor neuron disease. J Neurol Neurosurg Psychiatry 85(11):1288–1289. https://doi.org/10.1136/jnnp-2014-308288
Mejia Maza A, Jarvis S, Lee WC, Cunningham TJ, Schiavo G, Secrier M, Fratta P, Sleigh JN et al (2021) NMJ-analyser identifies subtle early changes in mouse models of neuromuscular disease. Sci Rep 11(1):12251. https://doi.org/10.1038/s41598-021-91094-6
Xia R, Liu Y, Yang L, Gal J, Zhu H, Jia J (2012) Motor neuron apoptosis and neuromuscular junction perturbation are prominent features in a drosophila model of Fus-mediated ALS. Mol Neurodegener 7:10. https://doi.org/10.1186/1750-1326-7-10
Yerbury JJ, Farrawell NE, McAlary L (2020) Proteome homeostasis dysfunction: a unifying principle in ALS pathogenesis. Trends Neurosci 43(5):274–284. https://doi.org/10.1016/j.tins.2020.03.002
Watanabe M, Dykes-Hoberg M, Culotta VC, Price DL, Wong PC, Rothstein JD (2001) Histological evidence of protein aggregation in mutant SOD1 transgenic mice and in amyotrophic lateral sclerosis neural tissues. Neurobiol Dis 8(6):933–941. https://doi.org/10.1006/nbdi.2001.0443
Jonsson PA, Ernhill K, Andersen PM, Bergemalm D, Brännström T, Gredal O, Nilsson P, Marklund SL (2004) Minute quantities of misfolded mutant superoxide dismutase-1 cause amyotrophic lateral sclerosis. Brain 127(Pt1):73–88. https://doi.org/10.1093/brain/awh005
Cheroni C, Marino M, Tortarolo M, Veglianese P, De Biasi S, Fontana E, Zuccarello LV, Maynard C (2009) Functional alterations of the ubiquitin-proteasome system in motor neurons of a mouse model of familial amyotrophic lateral sclerosis. Hum Mol Genet 18(1):82–96. https://doi.org/10.1093/hmg/ddn319
Devoy A, Kalmar B, Stewart M, Park H, Burke B, Noy SJ, Redhead Y, Humphrey J et al (2017) Humanized mutant FUS drives progressive motor neuron degeneration without aggregation in “FUSDelta14” Knockin Mice. Brain 140(11):2797–2805. https://doi.org/10.1093/brain/awx248
López-Erauskin J, Tadokoro T, Baughn MW, Myers B, McAlonis-Downes M, Chillon-Marinas C, Asiaban JN, Artates J et al (2018) ALS/FTD-linked mutation in FUS suppresses intra-axonal protein synthesis and drives disease without nuclear loss-of-function of FUS. Neuron 100(4):816-830.e7. https://doi.org/10.1016/j.neuron.2018.09.044
Fratta P, Sivakumar P, Humphrey J, Lo K, Ricketts T, Oliveira H, Brito-Armas JM, Kalmar B et al (2018) Mice with endogenous TDP-43 mutations exhibit gain of splicing function and characteristics of amyotrophic lateral sclerosis. EMBO J 37(11):e98684. https://doi.org/10.15252/embj.201798684
Zhang YJ, Gendron TF, Ebbert MTW, O’Raw AD, Yue M, Jansen-West K, Zhang X, Prudencio M et al (2018) Poly (GR) Impairs protein translation and stress granule dynamics in C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis. Nat Med 24(8):1136–1142. https://doi.org/10.1038/s41591-018-0071-1
An D, Fujiki R, Iannitelli DE, Smerdon JW, Maity S, Rose MF, Gelber A, Wanaselja EK et al 2019 Stem cell-derived cranial and spinal motor neurons reveal proteostatic differences between ALS resistant and sensitive motor neurons Elife 8https://doi.org/10.7554/eLife.44423
Maday S, Holzbaur ELF (2016) Compartment-specific regulation of autophagy in primary neurons. J Neurosci 36(22):5933–5945. https://doi.org/10.1523/JNEUROSCI.4401-15.2016
Sasaki S (2011) Autophagy in spinal cord motor neurons in sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 70(5):349–359. https://doi.org/10.1097/NEN.0b013e3182160690
Fecto F, Yan J, Vemula SP, Liu E, Yang Y, Chen W, Zheng JG, Shi Y et al (2011) SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol 68(11):1440–1446. https://doi.org/10.1001/archneurol.2011.250
Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y, Kinoshita Y, Kamada M et al (2010) Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465(7295):223–226. https://doi.org/10.1038/nature08971
Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N, Yang Y, Fecto F et al (2011) Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 477(7363):211–215. https://doi.org/10.1038/nature10353
Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, Couthouis J, Lu YF (2015) Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347(6229):1436–1441. https://doi.org/10.1126/science.aaa3650
Rogers RS, Tungtur S, Tanaka T, Nadeau LL, Badawi Y, Wang H, Ni HM, Ding WX et al (2017) Impaired mitophagy plays a role in denervation of neuromuscular junctions in ALS mice. Front Neurosci 11:473. https://doi.org/10.3389/fnins.2017.00473
Chen T, Huang B, Shi X, Gao L, Huang C (2018) Mutant UBQLN2P497H in Motor Neurons Leads to ALS-like Phenotypes and Defective Autophagy in Rats. Acta Neuropathol Commun 6(1):122. https://doi.org/10.1186/s40478-018-0627-9
Rudnick ND, Griffey CJ, Guarnieri P, Gerbino V, Wang X, Piersaint JA, Tapia JC, Rich MM et al (2017) Distinct roles for motor neuron autophagy early and late in the SOD1G93A mouse model of ALS. Proc Natl Acad Sci U S A 114(39):E8294–E8303. https://doi.org/10.1073/pnas.1704294114
Gerbino V, Kaunga E, Ye J, Canzio D, O’Keeffe S, Rudnick ND, Guarnieri P, Lutz CM et al (2020) The loss of TBK1 kinase activity in motor neurons or in all cell types differentially impacts ALS disease progression in SOD1 mice. Neuron 106(5):789-805.e5. https://doi.org/10.1016/j.neuron.2020.03.005
Sieverding K, Ulmer J, Bruno C, Satoh T, Tsao W, Freischmidt A, Akira S, Wong PC et al (2021) Hemizygous deletion of Tbk1 worsens neuromuscular junction pathology in TDP-43G298S transgenic mice. Exp Neurol 335:113496. https://doi.org/10.1016/j.expneurol.2020.113496
Chevalier-Larsen E, Holzbaur ELF (2006) Axonal transport and neurodegenerative disease. Biochim Biophys Acta 1762(11–12):1094–1108. https://doi.org/10.1016/j.bbadis.2006.04.002
Münch C, Sedlmeier R, Meyer T, Homberg V, Sperfeld AD, Kurt A, Prudlo J, Peraus G et al (2004) Point mutations of the P150 subunit of dynactin (DCTN1) gene in ALS. Neurology 63(4):724–726. https://doi.org/10.1212/01.wnl.0000134608.83927.b1
Cooper-Knock J, Robins H, Niedermoser I, Wyles M, Heath PR, Higginbottom A, Walsh T, Kazoka M et al (2017) Targeted genetic screen in amyotrophic lateral sclerosis reveals novel genetic variants with synergistic effect on clinical phenotype. Front Mol Neurosci 10:370. https://doi.org/10.3389/fnmol.2017.00370
Liu X, Yang L, Tang L, Chen L, Liu X, Fan D (2017) DCTN1 gene analysis in Chinese patients with sporadic amyotrophic lateral sclerosis. PLoS ONE 12(8):e0182572. https://doi.org/10.1371/journal.pone.0182572
Laird FM, Farah MH, Ackerley S, Hoke A, Maragakis N, Rothstein JD, Griffin J, Price DL et al (2008) Motor neuron disease occurring in a mutant dynactin mouse model is characterized by defects in vesicular trafficking. J Neurosci 28(9):1997–2005. https://doi.org/10.1523/JNEUROSCI.4231-07.2008
Tanaka F, Niwa JI, Ishigaki S, Katsuno M, Waza M, Yamamoto M, Doyu M, Sobue G (2006) Gene expression profiling toward understanding of ALS pathogenesis. Ann N Y Acad Sci 1086:1–10. https://doi.org/10.1196/annals.1377.011
Kuźma-Kozakiewicz M, Chudy A, Kaźmierczak B, Dziewulska D, Usarek E, Barańczyk-Kuźma A (2013) Dynactin deficiency in the CNS of humans with sporadic ALS and mice with genetically determined motor neuron degeneration. Neurochem Res. https://doi.org/10.1007/s11064-013-1160-7
Ikenaka K, Kawai K, Katsuno M, Huang Z, Jiang YM, Iguchi Y, Kobayashi K, Kimata T et al (2013) Dnc-1/Dynactin 1 knockdown disrupts transport of autophagosomes and induces motor neuron degeneration. PLoS ONE 8(2):e54511. https://doi.org/10.1371/journal.pone.0054511
Bercier V, Hubbard JM, Fidelin K, Duroure K, Auer TO, Revenu C, Wyart C, Del Bene F (2019) Dynactin1 depletion leads to neuromuscular synapse instability and functional abnormalities. Mol Neurodegener 14(1):27. https://doi.org/10.1186/s13024-019-0327-3
Williamson TL, Cleveland DW (1999) Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat Neurosci 2(1):50–56. https://doi.org/10.1038/4553
Rouleau GA, Clark AW, Rooke K, Pramatarova A, Krizus A, Suchowersky O, Julien JP, Figlewicz D (1996) SOD1 mutation is associated with accumulation of neurofilaments in amyotrophic lateral sclerosis. Ann Neurol 39(1):128–131. https://doi.org/10.1002/ana.410390119
Parkhouse WS, Cunningham L, McFee I, Miller JML, Whitney D, Pelech SL, Krieger C (2008) Neuromuscular dysfunction in the mutant superoxide dismutase mouse model of amyotrophic lateral sclerosis. Amyotroph Lateral Scler 9(1):24–34. https://doi.org/10.1080/17482960701725646
Bilsland LG, Sahai E, Kelly G, Golding M, Greensmith L, Schiavo G (2010) Deficits in axonal transport precede ALS symptoms in vivo. Proc Natl Acad Sci USA 107(47):20523–20528. https://doi.org/10.1073/pnas.1006869107
Chen H, Qian K, Du Z, Cao J, Petersen A, Liu H, Blackbourn LW, Huang CL et al (2014) Modeling ALS with IPSCs reveals that mutant SOD1 misregulates neurofilament balance in motor neurons. Cell Stem Cell 14(6):796–809. https://doi.org/10.1016/j.stem.2014.02.004
Gibbs KL, Kalmar B, Rhymes ER, Fellows AD, Ahmed M, Whiting P, Davies CH, Greensmith L et al (2018) Inhibiting P38 MAPK alpha rescues axonal retrograde transport defects in a mouse model of ALS. Cell Death Dis 9(6):596. https://doi.org/10.1038/s41419-018-0624-8
Morfini GA, Bosco DA, Brown H, Gatto R, Kaminska A, Song Y, Molla L, Baker L et al (2013) Inhibition of fast axonal transport by pathogenic SOD1 involves activation of P38 MAP kinase. PLoS One 8(6):e65235. https://doi.org/10.1371/journal.pone.0065235
Alami NH, Smith RB, Carrasco MA, Williams LA, Winborn CS, Han SSW, Kiskinis E, Winborn B et al (2014) Axonal transport of TDP-43 MRNA granules is impaired by ALS-causing mutations. Neuron 81(3):536–543. https://doi.org/10.1016/j.neuron.2013.12.018
Nicolas A, Kenna KP, Renton AE, Ticozzi N, Faghri F, Chia R, Dominov JA, Kenna BJ et al (2018) Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron 97(6):1268-1283.e6. https://doi.org/10.1016/j.neuron.2018.02.027
Brenner D, Yilmaz R, Müller K, Grehl T, Petri S, Meyer T, Grosskreutz J, Weydt P et al (2018) German ALS network MND-NET. Hot-spot KIF5A mutations cause familial ALS. Brain 141(3):688–697. https://doi.org/10.1093/brain/awx370
Brandon EP, Lin W, D’Amour KA, Pizzo DP, Dominguez B, Sugiura Y, Thode S, Ko CP (2003) Aberrant patterning of neuromuscular synapses in choline acetyltransferase-deficient mice. J Neurosci 23(2):539–549. https://doi.org/10.1523/JNEUROSCI.23-02-00539.2003
Kato T (1989) Choline acetyltransferase activities in single spinal motor neurons from patients with amyotrophic lateral sclerosis. J Neurochem 52(2):636–640. https://doi.org/10.1111/j.1471-4159.1989.tb09167.x
Oda Y, Imai S, Nakanishi I, Ichikawa T, Deguchi T (1995) Immunohistochemical study on choline acetyltransferase in the spinal cord of patients with amyotrophic lateral sclerosis. Pathol Int 45(12):933–939. https://doi.org/10.1111/j.1440-1827.1995.tb03418.x
Casas C, Herrando-Grabulosa M, Manzano R, Mancuso R, Osta R, Navarro X (2013) Early presymptomatic cholinergic dysfunction in a murine model of amyotrophic lateral sclerosis. Brain Behav 3(2):145–158. https://doi.org/10.1002/brb3.104
Tateno M, Kato S, Sakurai T, Nukina N, Takahashi R, Araki T (2009) Mutant SOD1 impairs axonal transport of choline acetyltransferase and acetylcholine release by sequestering KAP3. Hum Mol Genet 18(5):942–955. https://doi.org/10.1093/hmg/ddn422
Nagao M, Misawa H, Kato S, Hirai S (1998) Loss of cholinergic synapses on the spinal motor neurons of amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 57(4):329–333. https://doi.org/10.1097/00005072-199804000-00004
Sugita S, Fleming LL, Wood C, Vaughan SK, Gomes MPSM, Camargo W, Naves LA, Prado VF et al (2016) VAChT overexpression increases acetylcholine at the synaptic cleft and accelerates aging of neuromuscular junctions. Skelet Muscle 6:31. https://doi.org/10.1186/s13395-016-0105-7
Chand KK, Lee KM, Lee JD, Qiu H, Willis EF, Lavidis NA, Hilliard MA, Noakes PG (2018) Defects in synaptic transmission at the neuromuscular junction precede motor deficits in a TDP-43Q331K transgenic mouse model of amyotrophic lateral sclerosis. FASEB J 32(5):2676–2689. https://doi.org/10.1096/fj.201700835R
Armstrong GAB, Drapeau P (2013) Calcium channel agonists protect against neuromuscular dysfunction in a genetic model of TDP-43 mutation in ALS. J Neurosci 33(4):1741–1752. https://doi.org/10.1523/JNEUROSCI.4003-12.2013
Armstrong GAB, Drapeau P (2013) Loss and Gain of FUS Function impair neuromuscular synaptic transmission in a genetic model of ALS. Hum Mol Genet 22(21):4282–4292. https://doi.org/10.1093/hmg/ddt278
Bose P, Armstrong GAB, Drapeau P (2019) Neuromuscular junction abnormalities in a zebrafish loss-of-function model of TDP-43. J Neurophysiol 121(1):285–297. https://doi.org/10.1152/jn.00265.2018
Markert SM, Skoruppa M, Yu B, Mulcahy B, Zhen M, Gao S, Sendtner M, Stigloher C (2020) Overexpression of an ALS-associated FUS mutation in C. elegans disrupts NMJ morphology and leads to defective neuromuscular transmission. Biol Open 9(12):bio055129. https://doi.org/10.1242/bio.055129
Machamer JB, Collins SE, Lloyd TE (2014) The ALS gene FUS regulates synaptic transmission at the drosophila neuromuscular junction. Hum Mol Genet 23(14):3810–3822. https://doi.org/10.1093/hmg/ddu094
Butti Z, Pan YE, Giacomotto J, Patten SA (2021) Reduced C9orf72 function leads to defective synaptic vesicle release and neuromuscular dysfunction in zebrafish. Commun Biol 4(1):792. https://doi.org/10.1038/s42003-021-02302-y
Vos M, Lauwers E, Verstreken P (2010) Synaptic mitochondria in synaptic transmission and organization of vesicle pools in health and disease. Front Synaptic Neurosci 2:139. https://doi.org/10.3389/fnsyn.2010.00139
De Vos KJ, Chapman AL, Tennant ME, Manser C, Tudor EL, Lau KF, Brownlees J, Ackerley S (2007) Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum Mol Genet 16(22):2720–2728. https://doi.org/10.1093/hmg/ddm226
Farrawell NE, Lambert-Smith I, Mitchell K, McKenna J, McAlary L, Ciryam P, Vine KL, Saunders DN et al (2018) SOD1A4V aggregation alters ubiquitin homeostasis in a cell model of ALS. J Cell Sci 131(11). https://doi.org/10.1242/jcs.209122.
Bendotti C, Calvaresi N, Chiveri L, Prelle A, Moggio M, Braga M, Silani V, De Biasi S (2001) Early Vacuolization and mitochondrial damage in motor neurons of FALS Mice are not associated with apoptosis or with changes in cytochrome oxidase histochemical reactivity. J Neurol Sci 191(1–2):25–33. https://doi.org/10.1016/s0022-510x(01)00627-x
Magrané J, Cortez C, Gan WB, Manfredi G (2014) Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum Mol Genet 23(6):1413–1424. https://doi.org/10.1093/hmg/ddt528
So E, Mitchell JC, Memmi C, Chennell G, Vizcay-Barrena G, Allison L, Shaw CE, Vance C (2018) Mitochondrial Abnormalities and Disruption of the Neuromuscular Junction precede the clinical phenotype and motor neuron loss in HFUSWT transgenic mice. Hum Mol Genet 27(3):463–474. https://doi.org/10.1093/hmg/ddx415
Moller A, Bauer CS, Cohen RN, Webster CP, De Vos KJ (2017) Amyotrophic lateral sclerosis-associated mutant SOD1 inhibits anterograde axonal transport of mitochondria by reducing Miro1 levels. Hum Mol Genet 26(23):4668–4679. https://doi.org/10.1093/hmg/ddx348
Mórotz GM, De Vos KJ, Vagnoni A, Ackerley S, Shaw CE, Miller CCJ (2012) Amyotrophic lateral sclerosis-associated mutant VAPBP56S perturbs calcium homeostasis to disrupt axonal transport of mitochondria. Hum Mol Genet 21(9):1979–1988. https://doi.org/10.1093/hmg/dds011
Tsujihata M, Hazama R, Yoshimura T, Satoh A, Mori M, Nagataki S (1984) The motor end-plate fine structure and ultrastructural localization of acetylcholine receptors in amyotrophic lateral sclerosis. Muscle Nerve 7(3):243–249. https://doi.org/10.1002/mus.880070310
Yoshihara T, Ishii T, Iwata M, Nomoto M (1998) Ultrastructural and histochemical study of the motor end plates of the intrinsic laryngeal muscles in amyotrophic lateral sclerosis. Ultrastruct Pathol 22(2):121–126. https://doi.org/10.3109/01913129809032266
Narai H, Manabe Y, Nagai M, Nagano I, Ohta Y, Murakami T, Takehisa Y, Kamiya T et al (2009) Early detachment of neuromuscular junction proteins in ALS Mice with SODG93A mutation. Neurol Int 1(1):e16. https://doi.org/10.4081/ni.2009.e16
Palma E, Inghilleri M, Conti L, Deflorio C, Frasca V, Manteca A, Pichiorri F, Roseti C et al (2011) Physiological characterization of human muscle acetylcholine receptors from ALS patients. Proc Natl Acad Sci U S A 108(50):20184–20188. https://doi.org/10.1073/pnas.1117975108
Palma E, Reyes-Ruiz JM, Lopergolo D, Roseti C, Bertollini C, Ruffolo G, Cifelli P, Onesti E et al (2016) Acetylcholine receptors from human muscle as pharmacological targets for ALS therapy. Proc Natl Acad Sci U S A 113(11):3060–3065. https://doi.org/10.1073/pnas.1600251113
Dobrowolny G, Martini M, Scicchitano BM, Romanello V, Boncompagni S, Nicoletti C, Pietrangelo L, De Panfilis S et al (2018) Muscle expression of SOD1G93A triggers the dismantlement of neuromuscular junction via PKC-Theta. Antioxid Redox Signal 28(12):1105–1119. https://doi.org/10.1089/ars.2017.7054
Campanari ML, Marian A, Ciura S, Kabashi E (2021) TDP-43 Regulation of AChE expression can mediate ALS-like phenotype in zebrafish. Cells 10(2):221. https://doi.org/10.3390/cells10020221
Bellingham MC (2011) A review of the neural mechanisms of action and clinical efficiency of riluzole in treating amyotrophic lateral sclerosis: what have we learned in the last decade? CNS Neurosci Ther 17(1):4–31. https://doi.org/10.1111/j.1755-5949.2009.00116.x
Deflorio C, Palma E, Conti L, Roseti C, Manteca A, Giacomelli E, Catalano M, Limatola C et al (2012) Riluzole blocks human muscle acetylcholine receptors. J Physiol 590(10):2519–2528. https://doi.org/10.1113/jphysiol.2012.230201
Barik A, Lu Y, Sathyamurthy A, Bowman A, Shen C, Li L, Xiong W, Mei L (2014) LRP4 is critical for neuromuscular junction maintenance. J Neurosci 34(42):13892–13905. https://doi.org/10.1523/JNEUROSCI.1733-14.2014
Shanmukha S, Narayanappa G, Nalini A, Alladi PA, Raju TR (2018) Sporadic amyotrophic lateral sclerosis (SALS) - skeletal muscle response to cerebrospinal fluid from SALS patients in a rat model. Dis Model Mech 11:(4). https://doi.org/10.1242/dmm.031997.
Pérez-García MJ, Burden SJ (2012) Increasing MuSK activity delays denervation and improves motor function in ALS mice. Cell Rep 2(3):497–502. https://doi.org/10.1016/j.celrep.2012.08.004
Cantor S, Zhang W, Delestrée N, Remédio L, Mentis GZ, Burden SJ 2018 Preserving neuromuscular synapses in ALS by stimulating MuSK with a therapeutic agonist antibody Elife 7https://doi.org/10.7554/eLife.34375
Sengupta-Ghosh A, Dominguez SL, Xie L, Barck KH, Jiang Z, Earr T, Imperio J, Phu L et al (2019) Muscle specific kinase (MuSK) activation preserves neuromuscular junctions in the diaphragm but is not sufficient to provide a functional benefit in the SOD1G93A mouse model of ALS. Neurobiol Dis 124:340–352. https://doi.org/10.1016/j.nbd.2018.12.002
Eguchi T, Tezuka T, Fukudome T, Watanabe Y, Sagara H, Yamanashi Y (2020) Overexpression of Dok-7 in skeletal muscle enhances neuromuscular transmission with structural alterations of neuromuscular junctions: implications in robustness of neuromuscular transmission. Biochem Biophys Res Commun 523(1):214–219. https://doi.org/10.1016/j.bbrc.2019.12.011
Arimura S, Okada T, Tezuka T, Chiyo T, Kasahara Y, Yoshimura T, Motomura M, Yoshida N et al (2014) Neuromuscular disease. DOK7 gene therapy benefits mouse models of diseases characterized by defects in the neuromuscular junction. Science 345(6203):1505–1508. https://doi.org/10.1126/science.1250744
Miyoshi S, Tezuka T, Arimura S, Tomono T, Okada T, Yamanashi Y (2017) DOK7 gene therapy enhances motor activity and life span in ALS model mice. EMBO Mol Med 9(7):880–889. https://doi.org/10.15252/emmm.201607298
Adler M, Manley HA, Purcell AL, Deshpande SS, Hamilton TA, Kan RK, Oyler G, Lockridge O et al (2004) Reduced acetylcholine receptor density, morphological remodeling, and butyrylcholinesterase activity can sustain muscle function in acetylcholinesterase knockout mice. Muscle Nerve 30(3):317–327. https://doi.org/10.1002/mus.20099
Rasool CG, Bradley WG, Connolly B, Baruah JK (1983) Acetylcholinesterase and ATPases in motor neuron degenerative diseases. Muscle Nerve 6(6):430–435. https://doi.org/10.1002/mus.880060606
Festoff BW, Fernandez HL (1981) Plasma and red blood cell acetylcholinesterase in amyotrophic lateral sclerosis. Muscle Nerve 4(1):41–47. https://doi.org/10.1002/mus.880040108
Niebrój-Dobosz I, Domitrz I, Mickielewicz A (1999) Cytotoxic activity of serum and cerebrospinal fluid of amyotrophic lateral sclerosis (ALS) patients against acetylcholinesterase. Folia Neuropathol 37(2):107–112
Fernandez HL, Stiles JR, Donoso JA (1986) Skeletal muscle acetylcholinesterase molecular forms in amyotrophic lateral sclerosis. Muscle Nerve 9(5):399–406. https://doi.org/10.1002/mus.880090504
Aquilonius SM, Askmark H, Eckernäs SA, Gillberg PG, Hilton-Brown P, Rydin E, Stålberg E (1986) Cholinesterase inhibitors lack therapeutic effect in amyotrophic lateral sclerosis. A controlled study of physostigmine versus neostigmine. Acta Neurol Scand 73(6):628–632. https://doi.org/10.1111/j.1600-0404.1986.tb04610.x
Gotkine M, Marc G, Rozenstein L, Leah R, Einstein O, Ofira E, Abramsky O, Oded A et al (2013) Presymptomatic treatment with acetylcholinesterase antisense oligonucleotides prolongs survival in ALS (G93A-SOD1) mice. Biomed Res Int 2013:845345. https://doi.org/10.1155/2013/845345
Pramatarova A, Laganière J, Roussel J, Brisebois K, Rouleau GA (2001) Neuron-specific expression of mutant superoxide dismutase 1 in transgenic mice does not lead to motor impairment. J Neurosci 21(10):3369–3374. https://doi.org/10.1523/JNEUROSCI.21-10-03369.2001
Lino MM, Schneider C, Caroni P (2002) Accumulation of SOD1 mutants in postnatal motoneurons does not cause motoneuron pathology or motoneuron disease. J Neurosci 22(12):4825–4832. https://doi.org/10.1523/JNEUROSCI.22-12-04825.2002
Dobrowolny G, Aucello M, Rizzuto E, Beccafico S, Mammucari C, Boncompagni S, Bonconpagni S, Belia S et al (2008) Skeletal muscle is a primary target of SOD1G93A-mediated toxicity. Cell Metab 8(5):425–436. https://doi.org/10.1016/j.cmet.2008.09.002
Wong M, Martin LJ (2010) Skeletal muscle-restricted expression of human SOD1 causes motor neuron degeneration in transgenic mice. Hum Mol Genet 19(11):2284–2302. https://doi.org/10.1093/hmg/ddq106
Martin LJ, Wong M (2020) Skeletal muscle-restricted expression of human SOD1 in transgenic mice causes a fatal ALS-like syndrome. Front Neurol 11:592851. https://doi.org/10.3389/fneur.2020.592851
Marcuzzo S, Zucca I, Mastropietro A, de Rosbo NK, Cavalcante P, Tartari S, Bonanno S, Preite L et al (2011) Hind limb muscle atrophy precedes cerebral neuronal degeneration in G93A-SOD1 mouse model of amyotrophic lateral sclerosis: a longitudinal MRI study. Exp Neurol 231(1):30–37. https://doi.org/10.1016/j.expneurol.2011.05.007
Dobrowolny G, Lepore E, Martini M, Barberi L, Nunn A, Scicchitano BM, Musarò A (2018) Metabolic changes associated with muscle expression of SOD1G93A. Front Physiol 9:831. https://doi.org/10.3389/fphys.2018.00831
Turner BJ, Lopes EC, Cheema SS (2003) Neuromuscular accumulation of mutant superoxide dismutase 1 aggregates in a transgenic mouse model of familial amyotrophic lateral sclerosis. Neurosci Lett 350(2):132–136. https://doi.org/10.1016/s0304-3940(03)00893-0
Onesto E, Rusmini P, Crippa V, Ferri N, Zito A, Galbiati M, Poletti A (2011) Muscle cells and motoneurons differentially remove mutant SOD1 causing familial amyotrophic lateral sclerosis. J Neurochem 118(2):266–280. https://doi.org/10.1111/j.1471-4159.2011.07298.x
Wei R, Bhattacharya A, Chintalaramulu N, Jernigan AL, Liu Y, Van Remmen H, Chaudhuri AR (2012) Protein misfolding mitochondrial dysfunction and muscle loss are not directly dependent on soluble and aggregation state of MSOD1 protein in skeletal muscle of ALS. Biochem Biophys Res Commun 417(4):1275–1279. https://doi.org/10.1016/j.bbrc.2011.12.126
Crippa V, Galbiati M, Boncoraglio A, Rusmini P, Onesto E, Giorgetti E, Cristofani R, Zito A et al (2013) Motoneuronal and muscle-selective removal of ALS-related misfolded proteins. Biochem Soc Trans 41(6):1598–1604. https://doi.org/10.1042/BST20130118
Wei R, Bhattacharya A, Hamilton RT, Jernigan AL, Chaudhuri AR (2013) Differential effects of mutant SOD1 on protein structure of skeletal muscle and spinal cord of familial amyotrophic lateral sclerosis: role of chaperone network. Biochem Biophys Res Commun 438(1):218–223. https://doi.org/10.1016/j.bbrc.2013.07.060
Oliván S, Calvo AC, Gasco S, Muñoz MJ, Zaragoza P, Osta R (2015) Time-point dependent activation of autophagy and the UPS in SOD1G93A mice skeletal muscle. PLoS One 10(8):e0134830. https://doi.org/10.1371/journal.pone.0134830
McCampbell A, Cole T, Wegener AJ, Tomassy GS, Setnicka A, Farley BJ, Schoch KM, Hoye ML et al (2018) Antisense oligonucleotides extend survival and reverse decrement in muscle response in ALS models. J Clin Invest 128(8):3558–3567. https://doi.org/10.1172/JCI99081
Sorarú G, Orsetti V, Buratti E, Baralle F, Cima V, Volpe M, D’ascenzo C, Palmieri A et al (2010) TDP-43 in skeletal muscle of patients affected with amyotrophic lateral sclerosis. Amyotroph Lateral Scler 11(1–2):240–243. https://doi.org/10.3109/17482960902810890
Cykowski MD, Powell SZ, Appel JW, Arumanayagam AS, Rivera AL, Appel SH (2018) Phosphorylated TDP-43 (PTDP-43) Aggregates in the axial skeletal muscle of patients with sporadic and familial amyotrophic lateral sclerosis. Acta Neuropathol Commun 6(1):28. https://doi.org/10.1186/s40478-018-0528-y
Mori F, Tada M, Kon T, Miki Y, Tanji K, Kurotaki H, Tomiyama M, Ishihara T et al (2019) Phosphorylated TDP-43 Aggregates in skeletal and cardiac muscle are a marker of myogenic degeneration in amyotrophic lateral sclerosis and various conditions. Acta Neuropathol Commun 7(1):165. https://doi.org/10.1186/s40478-019-0824-1
Echaniz-Laguna A, Zoll J, Ponsot E, N’guessan B, Tranchant C, Loeffler JP, Lampert E, (2006) Muscular mitochondrial function in amyotrophic lateral sclerosis is progressively altered as the disease develops: a temporal study in man. Exp Neurol 198(1):25–30. https://doi.org/10.1016/j.expneurol.2005.07.020
Muyderman H, Chen T (2014) Mitochondrial dysfunction in amyotrophic lateral sclerosis - a valid pharmacological target? Br J Pharmacol 171(8):2191–2205. https://doi.org/10.1111/bph.12476
Vielhaber S, Winkler K, Kirches E, Kunz D, Büchner M, Feistner H, Elger CE, Ludolph AC (1999) Visualization of defective mitochondrial function in skeletal muscle fibers of patients with sporadic amyotrophic lateral sclerosis. J Neurol Sci 169(1–2):133–139. https://doi.org/10.1016/s0022-510x(99)00236-1
Chung MJ, Suh YL (2002) Ultrastructural Changes of mitochondria in the skeletal muscle of patients with amyotrophic lateral sclerosis. Ultrastruct Pathol 26(1):3–7. https://doi.org/10.1080/01913120252934260
Dupuis L, di Scala F, Rene F, de Tapia M, Oudart H, Pradat PF, Meininger V, Loeffler JP (2003) Up-regulation of mitochondrial uncoupling protein 3 reveals an early muscular metabolic defect in amyotrophic lateral sclerosis. FASEB J 17(14):2091–2093. https://doi.org/10.1096/fj.02-1182fje
Dupuis L, Oudart H, René F, Gonzalez de Aguilar JL, Loeffler JP (2004) Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: benefit of a high-energy diet in a transgenic mouse model. Proc Natl Acad Sci U S A 101(30):11159–11164. https://doi.org/10.1073/pnas.0402026101
Lynch E, Semrad T, Belsito VS, FitzGibbons C, Reilly M, Hayakawa K, Suzuki M (2019) C9ORF72-related cellular pathology in skeletal myocytes derived from ALS-patient induced pluripotent stem cells. Dis Model Mech 12(8). https://doi.org/10.1242/dmm.039552.
Bouteloup C, Desport JC, Clavelou P, Guy N, Derumeaux-Burel H, Ferrier A, Couratier P (2009) Hypermetabolism in ALS patients: an early and persistent phenomenon. J Neurol 256(8):1236–1242. https://doi.org/10.1007/s00415-009-5100-z
Funalot B, Desport JC, Sturtz F, Camu W, Couratier P (2009) High metabolic level in patients with familial amyotrophic lateral sclerosis. Amyotroph Lateral Scler 10(2):113–117. https://doi.org/10.1080/17482960802295192
Genin EC, Madji Hounoum B, Bannwarth S, Fragaki K, Lacas-Gervais S, Mauri-Crouzet A, Lespinasse F, Neveu J et al (2019) Mitochondrial defect in muscle precedes neuromuscular junction degeneration and motor neuron death in CHCHD10S59L/+ Mouse. Acta Neuropathol 138(1):123–145. https://doi.org/10.1007/s00401-019-01988-z
Muller FL, Song W, Jang YC, Liu Y, Sabia M, Richardson A, Van Remmen H (2007) Denervation-induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am J Physiol Regul Integr Comp Physiol 293(3):R1159-1168. https://doi.org/10.1152/ajpregu.00767.2006
Xiao Y, Karam C, Yi J, Zhang L, Li X, Yoon D, Wang H, Dhakal K et al (2018) ROS-related mitochondrial dysfunction in skeletal muscle of an ALS mouse model during the disease progression. Pharmacol Res 138:25–36. https://doi.org/10.1016/j.phrs.2018.09.008
Dupuis L, Gonzalez de Aguilar JL, Echaniz-Laguna A, Eschbach J, Rene F, Oudart H, Halter B, Huze C et al (2009) Muscle mitochondrial uncoupling dismantles neuromuscular junction and triggers distal degeneration of motor neurons. PLoS One 4(4):e5390. https://doi.org/10.1371/journal.pone.0005390
Luo G, Yi J, Ma C, Xiao Y, Yi F, Yu T, Zhou J (2013) Defective mitochondrial dynamics is an early event in skeletal muscle of an amyotrophic lateral sclerosis mouse model. PLoS One 8(12):e82112. https://doi.org/10.1371/journal.pone.0082112
Da Cruz S, Parone PA, Lopes VS, Lillo C, McAlonis-Downes M, Lee SK, Vetto AP, Petrosyan S (2012) Elevated PGC-1α activity sustains mitochondrial biogenesis and muscle function without extending survival in a mouse model of inherited ALS. Cell Metab 5:778–786. https://doi.org/10.1016/j.cmet.2012.03.019
Okuyama Y, Mizuno T, Inoue H, Kimoto K (1997) Amyotrophic lateral sclerosis with anti-acetylcholine receptor antibody. Intern Med 36(4):312–315. https://doi.org/10.2169/internalmedicine.36.312
Mehanna R, Patton EL, Phan CL, Harati Y (2012) Amyotrophic lateral sclerosis with positive anti-acetylcholine receptor antibodies. Case report and review of the literature. J Clin Neuromuscul Dis 14(2):82–85. https://doi.org/10.1097/CND.0b013e31824db163
Rivner MH, Liu S, Quarles B, Fleenor B, Shen C, Pan J, Mei L (2017) Agrin and low-density lipoprotein-related receptor protein 4 antibodies in amyotrophic lateral sclerosis patients. Muscle Nerve 55(3):430–432. https://doi.org/10.1002/mus.25438
Tzartos JS, Zisimopoulou P, Rentzos M, Karandreas N, Zouvelou V, Evangelakou P, Tsonis A, Thomaidis T et al (2014) LRP4 antibodies in serum and CSF from amyotrophic lateral sclerosis patients. Ann Clin Transl Neurol 1(2):80–87. https://doi.org/10.1002/acn3.26
Tzartos J, Zisimopoulou P, Tsonis A, Evangelakou P, Rentzos M, Karandreas N, Zouvelou V, Thomaidis T et al (2015) LRP4 antibodies are frequent in serum and CSF from amyotrophic lateral sclerosis patients (S34.004). Neurology 84(14 Supplement). https://doi.org/10.1002/acn3.26.
Lei L, Shen XM, Wang SY, Lu Y, Wang SB, Chen H, Liu Z, Ouyang YS et al (2019) Presence of antibodies against low-density lipoprotein receptor-related protein 4 and impairment of neuromuscular junction in a Chinese cohort of amyotrophic lateral sclerosis. Chin Med J (Engl) 132(12):1487–1489. https://doi.org/10.1097/CM9.0000000000000284
Conradi S, Ronnevi LO (1994) Further studies on the occurrence of serum autoantibodies against a membrane bound AChE fraction in ALS/MND patients and controls. J Neurol Sci 124(Suppl):67–69. https://doi.org/10.1016/0022-510x(94)90182-1
Häggström B, Andersen PM, Hjalmarsson K, Binzer M, Forsgren L (1997) Autoimmunity and ALS: studies on antibodies to acetylcholinesterase in sera. Acta Neurol Scand 95(2):111–114. https://doi.org/10.1111/j.1600-0404.1997.tb00079.x
Feng Z, Ko CP (2008) The role of glial cells in the formation and maintenance of the neuromuscular junction. Ann N Y Acad Sci 1132:19–28. https://doi.org/10.1196/annals.1405.016
Barik A, Li L, Sathyamurthy A, Xiong WC, Mei L (2016) Schwann cells in neuromuscular junction formation and maintenance. J Neurosci 36(38):9770–9781. https://doi.org/10.1523/JNEUROSCI.0174-16.2016
Jahromi BS, Robitaille R, Charlton MP (1992) Transmitter release increases intracellular calcium in perisynaptic Schwann cells in situ. Neuron 8(6):1069–1077. https://doi.org/10.1016/0896-6273(92)90128-z
Rochon D, Rousse I, Robitaille R (2001) Synapse-glia interactions at the mammalian neuromuscular junction. J Neurosci 21(11):3819–3829. https://doi.org/10.1523/JNEUROSCI.21-11-03819.2001
Todd KJ, Darabid H, Robitaille R (2010) Perisynaptic glia discriminate patterns of motor nerve activity and influence plasticity at the neuromuscular junction. J Neurosci 30(35):11870–11882. https://doi.org/10.1523/JNEUROSCI.3165-10.2010
Duregotti E, Negro S, Scorzeto M, Zornetta I, Dickinson BC, Chang CJ, Montecucco C, Rigoni M (2015) Mitochondrial alarmins released by degenerating motor axon terminals activate perisynaptic Schwann cells. Proc Natl Acad Sci U S A 112(5):E497-505. https://doi.org/10.1073/pnas.1417108112
Kang H, Tian L, Mikesh M, Lichtman JW, Thompson WJ (2014) Terminal Schwann cells participate in neuromuscular synapse remodeling during reinnervation following nerve injury. J Neurosci 34(18):6323–6333. https://doi.org/10.1523/JNEUROSCI.4673-13.2014
Arbour D, Tremblay E, Martineau É, Julien JP, Robitaille R (2015) Early and persistent abnormal decoding by glial cells at the neuromuscular junction in an ALS model. J Neurosci 35(2):688–706. https://doi.org/10.1523/JNEUROSCI.1379-14.2015
Carrasco DI, Seburn KL, Pinter MJ (2016) Altered terminal Schwann cell morphology precedes denervation in SOD1 mice. Exp Neurol 275(Pt 1):172–181. https://doi.org/10.1016/j.expneurol.2015.09.014
De Winter F, Vo T, Stam FJ, Wisman LAB, Bär PR, Niclou SP, van Muiswinkel FL, Verhaagen J (2006) The expression of the chemorepellent semaphorin 3A is selectively induced in terminal schwann cells of a subset of neuromuscular synapses that display limited anatomical plasticity and enhanced vulnerability in motor neuron disease. Mol Cell Neurosci 32(1–2):102–117. https://doi.org/10.1016/j.mcn.2006.03.002
Nascimento F, Sebastião AM, Ribeiro JA (2015) Presymptomatic and symptomatic ALS SOD1(G93A) mice differ in adenosine A1 and A2A receptor-mediated tonic modulation of neuromuscular transmission. Purinergic Signal 11(4):471–480. https://doi.org/10.1007/s11302-015-9465-4
Petrov D, Mansfield C, Moussy A, Hermine O (2017) ALS clinical trials review: 20 years of failure. are we any closer to registering a new treatment? Front Aging Neurosci 9:68. https://doi.org/10.3389/fnagi.2017.00068
Chen JJ (2020) Overview of current and emerging therapies for amytrophic lateral sclerosis. Am J Manag Care 26(9 Suppl):S191–S197. https://doi.org/10.37765/ajmc.2020.88483
Nguyen QT, Parsadanian AS, Snider WD, Lichtman JW (1998) Hyperinnervation of neuromuscular junctions caused by GDNF overexpression in muscle. Science 279(5357):1725–1729. https://doi.org/10.1126/science.279.5357.1725
Keller-Peck CR, Feng G, Sanes JR, Yan Q, Lichtman JW, Snider WD (2001) Glial cell line-derived neurotrophic factor administration in postnatal life results in motor unit enlargement and continuous synaptic remodeling at the neuromuscular junction. J Neurosci 21(16):6136–6146. https://doi.org/10.1523/JNEUROSCI.21-16-06136.2001
Grundström E, Askmark H, Lindeberg J, Nygren I, Ebendal T, Aquilonius SM (1999) Increased expression of glial cell line-derived neurotrophic factor MRNA in muscle biopsies from patients with amyotrophic lateral sclerosis. J Neurol Sci 162(2):169–173. https://doi.org/10.1016/s0022-510x(98)00333-5
Yamamoto M, Sobue G, Yamamoto K, Terao S, Mitsuma T (1996) Expression of glial cell line-derived growth factor MRNA in the spinal cord and muscle in amyotrophic lateral sclerosis. Neurosci Lett 204(1–2):117–120. https://doi.org/10.1016/0304-3940(96)12342-9
Acsadi G, Anguelov RA, Yang H, Toth G, Thomas R, Jani A, Wang Y, Ianakova E et al (2002) Increased survival and function of SOD1 mice after glial cell-derived neurotrophic factor gene therapy. Hum Gene Ther 13(9):1047–1059. https://doi.org/10.1089/104303402753812458
Li W, Brakefield D, Pan Y, Hunter D, Myckatyn TM, Parsadanian A (2007) Muscle-derived but not centrally derived transgene GDNF is neuroprotective in G93A-SOD1 mouse model of ALS. Exp Neurol 203(2):457–471. https://doi.org/10.1016/j.expneurol.2006.08.028
Wang LJ, Lu YY, Muramatsu S, Ikeguchi K, Fujimoto K, Okada T, Mizukami H, Matsushita T (2002) Neuroprotective effects of glial cell line-derived neurotrophic factor mediated by an adeno-associated virus vector in a transgenic animal model of amyotrophic lateral sclerosis. J Neurosci 22(16):6920–6928. 20026668.
Suzuki M, McHugh J, Tork C, Shelley B, Klein SM, Aebischer P, Svendsen CN (2007) GDNF secreting human neural progenitor cells protect dying motor neurons but not their projection to muscle in a rat model of familial ALS. PLoS One 2(8):e689. https://doi.org/10.1371/journal.pone.0000689
Suzuki M, McHugh J, Tork C, Shelley B, Hayes A, Bellantuono I, Aebischer P, Svendsen CN (2008) Direct muscle delivery of GDNF with human mesenchymal stem cells improves motor neuron survival and function in a rat model of familial ALS. Mol Ther 16(12):2002–2010. https://doi.org/10.1038/mt.2008.197
Baudet C, Pozas E, Adameyko I, Andersson E, Ericson J, Ernfors P (2008) Retrograde signaling onto ret during motor nerve terminal maturation. J Neurosci 28(4):963–975. https://doi.org/10.1523/JNEUROSCI.4489-07.2008
Airaksinen MS, Saarma M (2002) The GDNF family: signalling, biological functions and therapeutic value. Nat Rev Neurosci 3(5):383–394. https://doi.org/10.1038/nrn812
Soler RM, Dolcet X, Encinas M, Egea J, Bayascas JR, Comella JX (1999) Receptors of the glial cell line-derived neurotrophic factor family of neurotrophic factors signal cell survival through the phosphatidylinositol 3-kinase pathway in spinal cord motoneurons. J Neurosci 19(21):9160–9169. https://doi.org/10.1523/JNEUROSCI.19-21-09160.1999
Jiang Y, McLennan IS, Koishi K, Hendry IA (2000) Transforming growth factor-beta 2 is anterogradely and retrogradely transported in motoneurons and up-regulated after nerve injury. Neuroscience 97(4):735–742. https://doi.org/10.1016/s0306-4522(00)00084-1
Si Y, Kim S, Cui X, Zheng L, Oh SJ, Anderson T, AlSharabati M, Kazamel M et al (2015) Transforming growth factor beta (TGF-β) is a muscle biomarker of disease progression in ALS and correlates with Smad expression. PLoS One 10(9):e0138425. https://doi.org/10.1371/journal.pone.0138425
Duque T, Gromicho M, Pronto-Laborinho AC, de Carvalho M (2020) Transforming growth factor-β plasma levels and its role in amyotrophic lateral sclerosis. Med Hypotheses 139:109632. https://doi.org/10.1016/j.mehy.2020.109632
Peters S, Zitzelsperger E, Kuespert S, Iberl S, Heydn R, Johannesen S, Petri S, Aigner L et al (2017) The TGF-β system as a potential pathogenic player in disease modulation of amyotrophic lateral sclerosis. Front Neurol 8:669. https://doi.org/10.3389/fneur.2017.00669
Iłzecka J, Stelmasiak Z, Dobosz B (2002) Transforming growth factor-beta 1 (Tgf-Beta 1) in patients with amyotrophic lateral sclerosis. Cytokine 20(5):239–243. https://doi.org/10.1006/cyto.2002.2005
Houi K, Kobayashi T, Kato S, Mochio S, Inoue K (2002) Increased plasma TGF-Beta1 in patients with amyotrophic lateral sclerosis. Acta Neurol Scand 106(5):299–301. https://doi.org/10.1034/j.1600-0404.2002.01301.x
Meroni M, Crippa V, Cristofani R, Rusmini P, Cicardi ME, Messi E, Piccolella M, Tedesco B et al (2019) Transforming growth factor beta 1 signaling is altered in the spinal cord and muscle of amyotrophic lateral sclerosis mice and patients. Neurobiol Aging 82:48–59. https://doi.org/10.1016/j.neurobiolaging.2019.07.001
Gonzalez D, Contreras O, Rebolledo DL, Espinoza JP, van Zundert B, Brandan E (2017) ALS skeletal muscle shows enhanced TGF-β signaling fibrosis and induction of fibro/adipogenic progenitor markers. PLoS One 12(5):e0177649. https://doi.org/10.1371/journal.pone.0177649
Galbiati M, Crippa V, Rusmini P, Cristofani R, Messi E, Piccolella M, Tedesco B, Ferrari V et al (2020) Multiple roles of transforming growth factor beta in amyotrophic lateral sclerosis. Int J Mol Sci 21(12):E4291. https://doi.org/10.3390/ijms21124291
Taetzsch T, Tenga MJ, Valdez G (2017) Muscle fibers secrete FGFBP1 to slow degeneration of neuromuscular synapses during aging and progression of ALS. J Neurosci 37(1):70–82. https://doi.org/10.1523/JNEUROSCI.2992-16.2016
Day WA, Koishi K, Nukuda H, McLennan IS (2005) Transforming growth factor-beta 2 causes an acute improvement in the motor performance of transgenic ALS mice. Neurobiol Dis 19(1–2):323–330. https://doi.org/10.1016/j.nbd.2005.01.010
Fong SW, McLennan IS, McIntyre A, Reid J, Shennan KIJ, Bewick GS (2010) TGF-Beta2 alters the characteristics of the neuromuscular junction by regulating presynaptic quantal size. Proc Natl Acad Sci U S A 107(30):13515–13519. https://doi.org/10.1073/pnas.1001695107
Zheng C, Sköld MK, Li J, Nennesmo I, Fadeel B, Henter JI (2007) VEGF reduces astrogliosis and preserves neuromuscular junctions in ALS transgenic mice. Biochem Biophys Res Commun 363(4):989–993. https://doi.org/10.1016/j.bbrc.2007.09.088
Azzouz M, Ralph GS, Storkebaum E, Walmsley LE, Mitrophanous KA, Kingsman SM, Carmeliet P, Mazarakis ND (2004) VEGF delivery with retrogradely transported lentivector prolongs survival in a mouse ALS model. Nature 429(6990):413–417. https://doi.org/10.1038/nature02544
Krakora D, Mulcrone P, Meyer M, Lewis C, Bernau K, Gowing G, Zimprich C, Aebischer P et al (2013) Synergistic effects of GDNF and VEGF on lifespan and disease progression in a familial ALS rat model. Mol Ther 21(8):1602–1610. https://doi.org/10.1038/mt.2013.108
Torres-Aleman I, Barrios V, Berciano J (1998) The peripheral insulin-like growth factor system in amyotrophic lateral sclerosis and in multiple sclerosis. Neurology 50(3):772–776. https://doi.org/10.1212/wnl.50.3.772
Lunetta C, Serafini M, Prelle A, Magni P, Dozio E, Ruscica M, Sassone J, Colciago C et al (2012) Impaired expression of insulin-like growth factor-1 system in skeletal muscle of amyotrophic lateral sclerosis patients. Muscle Nerve 45(2):200–208. https://doi.org/10.1002/mus.22288
Nagel G, Peter RS, Rosenbohm A, Koenig W, Dupuis L, Rothenbacher D, Ludolph AC (2020) Association of insulin-like growth factor 1 concentrations with risk for and prognosis of amyotrophic lateral sclerosis - results from the ALS Registry Swabia. Sci Rep 10(1):736. https://doi.org/10.1038/s41598-020-57744-x
Kaspar BK, Lladó J, Sherkat N, Rothstein JD, Gage FH (2003) Retrograde Viral Delivery of IGF-1 Prolongs Survival in a Mouse ALS Model. Science 301(5634):839–842. https://doi.org/10.1126/science.1086137
Dobrowolny G, Giacinti C, Pelosi L, Nicoletti C, Winn N, Barberi L, Molinaro M, Rosenthal N et al (2005) Muscle expression of a local Igf-1 isoform protects motor neurons in an ALS mouse model. J Cell Biol 168(2):193–199. https://doi.org/10.1083/jcb.200407021
Wen D, Cui C, Duan W, Wang W, Wang Y, Liu Y, Li Z, Li C (2019) The role of insulin-like growth factor 1 in ALS cell and mouse models: a mitochondrial protector. Brain Res Bull 144:1–13. https://doi.org/10.1016/j.brainresbull.2018.09.015
Mantilla CB, Stowe JM, Sieck DC, Ermilov LG, Greising SM, Zhang C, Shokat KM (1985) Sieck GC (2014) TrkB kinase activity maintains synaptic function and structural integrity at adult neuromuscular junctions. J Appl Physiol 117(8):910–920. https://doi.org/10.1152/japplphysiol.01386.2013
Santafé MM, Garcia N, Tomàs M, Obis T, Lanuza MA, Besalduch N, Tomàs J (2014) The interaction between tropomyosin-related kinase B receptors and serine kinases modulates acetylcholine release in adult neuromuscular junctions. Neurosci Lett 561:171–175. https://doi.org/10.1016/j.neulet.2013.12.073
Nishio T, Sunohara N, Furukawa S (1998) Neutrophin switching in spinal motoneurons of amyotrophic lateral sclerosis. NeuroReport 9(7):1661–1665. https://doi.org/10.1097/00001756-199805110-00073
Küst BM, Copray JCVM, Brouwer N, Troost D, Boddeke HWGM (2002) Elevated levels of neurotrophins in human biceps brachii tissue of amyotrophic lateral sclerosis. Exp Neurol 177(2):419–427. https://doi.org/10.1006/exnr.2002.8011
Mutoh T, Sobue G, Hamano T, Kuriyama M, Hirayama M, Yamamoto M, Mitsuma T (2000) Decreased phosphorylation levels of TrkB neurotrophin receptor in the spinal cords from patients with amyotrophic lateral sclerosis. Neurochem Res 25(2):239–245. https://doi.org/10.1023/a:1007575504321
Xu L, Tian D, Li J, Chen L, Tang L, Fan D (2017) The analysis of two BDNF polymorphisms G196A/C270T in Chinese sporadic amyotrophic lateral sclerosis. Front Aging Neurosci 9:135. https://doi.org/10.3389/fnagi.2017.00135
Just-Borràs L, Hurtado E, Cilleros-Mañé V, Biondi O, Charbonnier F, Tomàs M, Garcia N, Lanuza MA et al (2019) Overview of impaired BDNF signaling their coupled downstream serine-threonine kinases and SNARE/SM complex in the neuromuscular junction of the amyotrophic lateral sclerosis model SOD1-G93A Mice. Mol Neurobiol 56(10):6856–6872. https://doi.org/10.1007/s12035-019-1550-1
Just-Borràs L, Hurtado E, Cilleros-Mañé V, Biondi O, Charbonnier F, Tomàs M, Garcia N, Tomàs J et al (2020) Running and swimming prevent the deregulation of the BDNF/TrkB neurotrophic signalling at the neuromuscular junction in mice with amyotrophic lateral sclerosis. Cell Mol Life Sci 77(15):3027–3040. https://doi.org/10.1007/s00018-019-03337-5
Vijayalakshmi K, Alladi PA, Sathyaprabha TN, Subramaniam JR, Nalini A, Raju TR (2009) Cerebrospinal fluid from sporadic amyotrophic lateral sclerosis patients induces degeneration of a cultured motor neuron cell line. Brain Res 1263:122–133. https://doi.org/10.1016/j.brainres.2009.01.041
Shruthi S, Sumitha R, Varghese AM, Ashok S, Chandrasekhar Sagar BK, Sathyaprabha TN, Nalini A, Kramer BW et al (2017) Brain-derived neurotrophic factor facilitates functional recovery from ALS-cerebral spinal fluid-induced neurodegenerative changes in the NSC-34 motor neuron cell line. Neurodegener Dis 17(1):44–58. https://doi.org/10.1159/000447559
Deepa P, Shahani N, Alladi PA, Vijayalakshmi K, Sathyaprabha TN, Nalini A, Ravi V, Raju TR (2011) Down regulation of trophic factors in neonatal rat spinal cord after administration of cerebrospinal fluid from sporadic amyotrophic lateral sclerosis patients. J Neural Transm (Vienna) 118(4):531–538. https://doi.org/10.1007/s00702-010-0520-6
Kovanda A, Leonardis L, Zidar J, Koritnik B, Dolenc-Groselj L, Ristic Kovacic S, Curk T, Rogelj B (2018) Differential expression of microRNAs and other small RNAs in muscle tissue of patients with ALS and healthy age-matched controls. Sci Rep 8(1):5609. https://doi.org/10.1038/s41598-018-23139-2
Dardiotis E, Aloizou AM, Siokas V, Patrinos GP, Deretzi G, Mitsias P, Aschner M, Tsatsakis A (2018) The role of MicroRNAs in patients with amyotrophic lateral sclerosis. J Mol Neurosci 66(4):617–628. https://doi.org/10.1007/s12031-018-1204-1
Russell AP, Wada S, Vergani L, Hock MB, Lamon S, Léger B, Ushida T, Cartoni R et al (2013) Disruption of skeletal muscle mitochondrial network genes and MiRNAs in amyotrophic lateral sclerosis. Neurobiol Dis 49:107–117. https://doi.org/10.1016/j.nbd.2012.08.015
Emde A, Eitan C, Liou L, Libby RT, Rivkin N, Magen I, Reichenstein I, Oppenheim H et al (2015) Dysregulated MiRNA biogenesis downstream of cellular stress and ALS-causing mutations: a new mechanism for ALS. EMBO J 34(21):2633–2651. https://doi.org/10.15252/embj.201490493
Chen JF, Mandel EM, Thomson JM, Wu Q, Callis TE, Hammond SM, Conlon FL, Wang DZ (2006) The role of MicroRNA-1 and MicroRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet 38(2):228–233. https://doi.org/10.1038/ng1725
Chen JF, Tao Y, Li J, Deng Z, Yan Z, Xiao X, Wang DZ (2010) MicroRNA-1 and MicroRNA-206 regulate skeletal muscle satellite cell proliferation and differentiation by repressing Pax7. J Cell Biol 190(5):867–879. https://doi.org/10.1083/jcb.200911036
Pegoraro V, Merico A, Angelini C (2017) Micro-RNAs in ALS muscle: differences in gender age at onset and disease duration. J Neurol Sci 380:58–63. https://doi.org/10.1016/j.jns.2017.07.008
Dobrowolny G, Bernardini C, Martini M, Baranzini M, Barba M, Musarò A (2015) Muscle Expression of SOD1(G93A) Modulates MicroRNA and MRNA transcription pattern associated with the myelination process in the spinal cord of transgenic mice. Front Cell Neurosci 9:463. https://doi.org/10.3389/fncel.2015.00463
Valdez G, Heyer MP, Feng G, Sanes JR (2014) The role of muscle MicroRNAs in repairing the neuromuscular junction. PLoS One 9(3):e93140. https://doi.org/10.1371/journal.pone.0093140
Maimon R, Ionescu A, Bonnie A, Sweetat S, Wald-Altman S, Inbar S, Gradus T, Trotti D et al (2018) MiR126-5p downregulation facilitates axon degeneration and NMJ disruption via a non-cell-autonomous mechanism in ALS. J Neurosci 38(24):5478–5494. https://doi.org/10.1523/JNEUROSCI.3037-17.2018
Williams AH, Valdez G, Moresi V, Qi X, McAnally J, Elliott JL, Bassel-Duby R, Sanes JR et al (2009) MicroRNA-206 delays ALS progression and promotes regeneration of neuromuscular synapses in mice. Science 326(5959):1549–1554. https://doi.org/10.1126/science.1181046
Toivonen JM, Manzano R, Oliván S, Zaragoza P, García-Redondo A, Osta R (2014) MicroRNA-206: A potential circulating biomarker candidate for amyotrophic lateral sclerosis. PLoS One 9(2):e89065. https://doi.org/10.1371/journal.pone.0089065
Patten SA, Aggad D, Martinez J, Tremblay E, Petrillo J, Armstrong GA, La Fontaine A, Maios C et al (2017) Neuroleptics as therapeutic compounds stabilizing neuromuscular transmission in amyotrophic lateral sclerosis. JCI Insight 2:(22). https://doi.org/10.1172/jci.insight.97152.
Pozzi S, Thammisetty SS, Julien JP (2018) Chronic administration of pimozide fails to attenuate motor and pathological deficits in two mouse models of amyotrophic lateral sclerosis. Neurotherapeutics 15(3):715–727. https://doi.org/10.1007/s13311-018-0634-3
Bose P, Tremblay E, Maois C, Narasimhan V, Armstrong GAB, Liao M, Parker JA, Robitaille R et al (2019) The novel small molecule TRVA242 stabilizes neuromuscular junction defects in multiple animal models of amyotrophic lateral sclerosis. Neurotherapeutics 16(4):1149–1166. https://doi.org/10.1007/s13311-019-00765-w
Boillée S, Vande Velde C, Cleveland DW (2006) ALS: A disease of motor neurons and their nonneuronal neighbors. Neuron 52(1):39–59. https://doi.org/10.1016/j.neuron.2006.09.018
Di Giorgio FP, Carrasco MA, Siao MC, Maniatis T, Eggan K (2007) Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS Model. Nat Neurosci 10(5):608–614. https://doi.org/10.1038/nn1885
Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, Przedborski S (2007) Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci 10(5):615–622. https://doi.org/10.1038/nn1876
Vats A, Gourie-Devi M, Ahuja K, Sharma A, Wajid S, Ganguly NK, Taneja V (2018) Expression analysis of protein homeostasis pathways in the peripheral blood mononuclear cells of sporadic amyotrophic lateral sclerosis patients. J Neurol Sci 387:85–91. https://doi.org/10.1016/j.jns.2018.01.035
Liu JX, Brännström T, Andersen PM, Pedrosa-Domellöf F (2011) Different impact of ALS on laminin isoforms in human extraocular muscles versus limb muscles. Invest Ophthalmol Vis Sci 52(7):4842–4852. https://doi.org/10.1167/iovs.10-7132
Charcot J, Joffroy A (1869) Deux cas d’atrophie musculaire progressive avec lesion de la substance grise et des faisceaux antero-lateraux de la moelle epiniere. Arch Physiol Neurol Pathol 2:744–754
Prout AJ, Eisen AA (1994) The cortical silent period and amyotrophic lateral sclerosis. Muscle Nerve 17(2):217–223. https://doi.org/10.1002/mus.880170213
Mills KR (1995) Motor neuron disease. studies of the corticospinal excitation of single motor neurons by magnetic brain stimulation. Brain 118(Pt 4):971–982. https://doi.org/10.1093/brain/118.4.971
Eisen A, Weber M (2001) The motor cortex and amyotrophic lateral sclerosis. Muscle Nerve 24(4):564–573. https://doi.org/10.1002/mus.1042
Vucic S, Cheah BC, Yiannikas C, Kiernan MC (2011) Cortical excitability distinguishes ALS from mimic disorders. Clin Neurophysiol 122(9):1860–1866. https://doi.org/10.1016/j.clinph.2010.12.062
Menon P, Kiernan MC, Vucic S (2014) Cortical dysfunction underlies the development of the split-hand in amyotrophic lateral sclerosis. PLoS One 9(1):e87124. https://doi.org/10.1371/journal.pone.0087124
Geevasinga N, Menon P, Özdinler PH, Kiernan MC, Vucic S (2016) Pathophysiological and diagnostic implications of cortical dysfunction in ALS. Nat Rev Neurol 12(11):651–661. https://doi.org/10.1038/nrneurol.2016.140
Menon P, Geevasinga N, van den Bos M, Yiannikas C, Kiernan MC, Vucic S (2017) Cortical hyperexcitability and disease spread in amyotrophic lateral sclerosis. Eur J Neurol 24(6):816–824. https://doi.org/10.1111/ene.13295
Acknowledgements
The authors thank Bikish Pegu for designing the illustrations and Ananya Chopra for critical reading of the manuscript.
Funding
SV is supported by the Council of Scientific and Industrial Research for SRF-NET scholarship (09/591(0150)/2018-EMR-I). AV, SK, and BS are supported by scholarship from the Indian Council of Medical Research (3/1/3/JRF-2012/HRD, BMS/Adhoc/BIOCHEM/2015–0430 and BIC/05 (09)/Indo-Russian/2016, respectively).
Author information
Authors and Affiliations
Contributions
SV, MG, and VT: conceptualization. SV and SK: carried out literature search and wrote the original draft and illustrations. AV and BS: reviewed and edited text and figures. NG, PC, MG, and VT: critical reviewing and editing of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Conflict of Interest
The authors have no conflicts of interest to declare that are relevant to the content of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Verma, S., Khurana, S., Vats, A. et al. Neuromuscular Junction Dysfunction in Amyotrophic Lateral Sclerosis. Mol Neurobiol 59, 1502–1527 (2022). https://doi.org/10.1007/s12035-021-02658-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-021-02658-6