
Abstract
Prenatal glucocorticoid (GC) overexposure impacts fetal hippocampal neural stem cells (NSCs) and increases the risk for relative cognitive and mood disorders in offspring. However, the precise underlying mechanisms remain elusive. Here, we treated mouse hippocampal NSCs with dexamethasone (DEX) in vitro and found that DEX inhibited cell proliferation and Sirt7 expression. In addition, prenatal mouse overexposure to DEX induced the suppression of Sirt7 in the hippocampus of offspring. Sirt7 knockdown significantly decreased the percentage of proliferating cells but did not further reduce the NSC proliferation rate in the presence of DEX, whereas Sirt7 overexpression rescued DEX-induced inhibition of hippocampal NSC proliferation. Moreover, DEX inhibited Sirt7 expression through the glucocorticoid receptor (GR), and p21 was found to mediate the functional effect of DEX-induced Sirt7 suppression. In conclusion, our data demonstrate for the first time the effect of DEX on the Sirt7-p21 pathway in hippocampal NSCs, identifying a new potential therapeutic target for prenatal GC overexposure–related neurodevelopmental disorders in offspring.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
During late gestation in most mammalian species, there is a physiological surge in maternal glucocorticoids (GCs) that is critical for normal fetal organ development (Cole 2006; Moisiadis and Matthews 2014). Usually, synthetic GCs are administered to pregnant women at risk of preterm birth to promote the maturation of fetal lungs (Neilson 2007). Maternal GC exposure of the fetus is tightly controlled by placental 11β-hydroxysteroid dehydrogenase (11β-HSD), which inactivates glucocorticoids and prevents prematuration of fetal organs. However, pathological conditions that impair placental functions or exogenous GC levels above the limit of placental 11β-HSD might increase fetal exposure to GCs (Holmes et al. 2006; Wyrwoll et al. 2011). Epidemiologic studies have demonstrated that prenatal GC overexposure induces harmful long-term effects on a child’s development, such as fetal growth restriction, mood changes, cognitive deficiencies, and induction of increased anxiety (Kino 2015; Lautarescu et al. 2020; Scheinost et al. 2017). DEX is a potent synthetic glucocorticoid that is widely used in the late gestation of animals to validate GC-mediated intrauterine programming (Kawata 1995; Seckl 2001). Evidence from animal studies, including those involving rodents and nonhuman primates, has also indicated that prenatal GC overexposure is associated with cognitive and mood disorders (Hauser et al. 2009; Hauser et al. 2008).
The hippocampus is the main brain region associated with cognitive and mood processes and responds to GC overexposure–induced changes (Eichenbaum and Cohen 2014; Lucassen et al. 2001; MacQueen and Frodl 2011). Hippocampal neurogenesis, in which neural stem cells (NSCs) in the dentate gyrus (DG) proliferate and differentiate into neurons and astrocytes, occurs during early development and persists throughout life (Anacker and Hen 2017). Glucocorticoids bind the GR and regulate essential physiological functions, including energy homeostasis, inflammatory processes, and the stress response (G and M 2020; Lu and Cidlowski 2006). Functional GRs have been shown to be expressed in hippocampal NSCs in vivo and in vitro. Importantly, glucocorticoids influence the activation of NSCs, decreasing proliferation, differentiation, and/or cell survival, which may underlie the negative effects of these hormones on the functions of the hippocampus, including cognition and mood (Choy et al. 2008; de los Angeles et al. 2016). Although excess glucocorticoids are harmful to hippocampal NSCs, the underlying molecular mechanism remains unclear.
Sirtuins are a family of NAD+-dependent protein lysine deacetylases. Seven homologs have been identified in mammals, namely SIRT1–7. In recent years, crosstalk with sirtuins and glucocorticoid signaling has emerged. Sirt1 physically interacts with GR and enhances its transcriptional activity, and glucocorticoids inhibit the expression of Sirt1 (Poulsen et al. 2014; Suzuki et al. 2018; Xu et al. 2018). Previous studies indicated that Sirt1 is associated with gene regulation and the response to DNA damage in NSCs (Ma et al. 2014; Wang et al. 2018). In addition, Sirt1 is involved in regulating memory and plasticity (Gao et al. 2010). Studies have shown that Sirt2 may have important effects on myelin genesis and the myelin-axon interaction (Li et al. 2007). Sirt2 plays roles in synaptic plasticity and cognitive function (Diaz-Perdigon et al. 2020; Wang et al. 2017). Studies have also shown that Sirt7 is associated with consolidating memory of fear in mice (Islam et al. 2018). However, it is unclear whether members of the Sirtuin family are involved in GC-mediated suppression of NSC proliferation. Here, we discovered that the Sirt7-p21 signaling pathway plays an important role in the effect of DEX on NSC proliferation, providing potential new targets for improving reduced adult cognition and mood caused by prenatal stress.
Materials and Methods
Experimental Animals
All mice were maintained and handled according to Tongji University institutional guidelines. The mice (C57BL/6J) were purchased from the SLAC Laboratory Animal Company (Shanghai, China). Pregnancy in the mice was confirmed by the presence of a vaginal plug the next morning after coitus, which was designated 0.5 days postcoitus (dpc). The pregnant mice in the DEX group were intraperitoneally injected with DEX (1 mg/kg/day, Selleck, S1322) at 14.5 dpc for five consecutive days; the mice in the NORMAL group received the same volume of normal saline on the same schedule. Considering the dose conversion rate (0.08) between mice and humans (Reagan-Shaw et al. 2008), treatment with DEX in pregnant mice at 1 mg/kg is comparable with that prescribed in human pregnancy for preterm labor at 0.1–0.5 mg/kg (Peaceman et al. 2006). Prenatal exposure to this DEX dose induces fetal growth retardation in offspring (Zhang et al. 2016). Mice were housed under pathogen-free conditions and maintained in a temperature- and humidity-controlled room with a 12-h light/12-h dark cycle.
Plasmids and Lentiviral Packaging
Specific short hairpin RNAs (shRNAs) (Sirt7/p21) and nontargeting control shRNA were cloned into the AgeI and EcoRI sites of the pLKO.1 RNAi plasmid (Addgene plasmid #10878) to generate gene knockdown plasmids. To generate gene overexpression plasmids, the cDNA sequences of HA-tagged human Sirt7 (NM_016538.3) and p21 (NM_007669.5) were isolated using KOD DNA polymerase (GenView) and subsequently cloned into the FUGW lentiviral vector (Addgene plasmid #14883). A luciferase plasmid encoding the HA tag was constructed as a control vector. For lentivirus packaging, pLKO.1 RNAi or FUGW plasmids were cotransfected with the packaging plasmids. Pax2 (0.9 μg) and Vsvg (0.6 μg) were transfected into 293FT cells (1-well of a 6-well plate) using FuGENE HD transfection reagent (Roche). After 72 h, lentivirus-containing medium was filtered to remove cellular debris and concentrated through virus precipitation solution (ExCell Bio, EMB810A-1). Harvested lentiviral particles used for cell infection were suspended in 100 μl of PBS.
Isolation and Treatment of Hippocampal NSCs
NSCs were isolated from the hippocampus of C57BL/6J mice on embryonic day 16.5 using a neural tissue dissociation kit (P, Miltenyi Biotec). NSCs were cultured in DMEM/F12 (Gibco) supplemented with 2% B27 supplement without vitamin A (Invitrogen), 1% GlutaMAX (Invitrogen), 1% NEAA (Invitrogen), 20 ng/ml fibroblast growth factor (bFGF, Sino Biological), and 20 ng/ml epidermal growth factor (EGF, Sino Biological). TrypLE™ (Invitrogen) was used to dissociate hippocampal NSCs. DEX (Selleck) and Sirt7 inhibitor ID: 97491 (Axon) were dissolved in DMSO (Sigma, St. Louis, MO), and the cells were treated with 50 μM DEX (Mutsaers and Tofighi 2012) or 5 μM ID: 97491 (Kim et al. 2019). For lentiviral infection, cells were passaged every 4 to 5 days, and suspensions were diluted to 2 × 105 cells per well in 6-well plates and incubated with 20 μl of lentivirus for 48 h. Then, the medium was replaced with fresh NSC medium, and the suspensions were diluted to 2 × 105 cells per well in 6-well plates. Additionally, after 48 h of infection, 0.5 μg/ml puromycin (Sigma) was added to generate knockdown cells.
Reverse Transcription and Quantitative PCR
Total RNA was extracted from cultured cell lines using RNAiso plus (TaKaRa) and reverse transcribed to produce complementary DNA (cDNA) using the PrimeScript™ RT reagent kit (TaKaRa). qPCR was performed using the TaKaRa Ex Taq PCR kit (TaKaRa) in the Stratagene Mx3000 qPCR system (Stratagene). The results were calculated using the 2−ΔΔCT method by normalizing against the GAPDH gene. The sequences of the primers used in this study are shown in Table S1.
Western Blotting
Cells were treated with RIPA buffer (Beyotime) containing PMSF and Halt™ Protease Inhibitor Cocktails (Thermo). Concentrations were measured using the Pierce BCA Protein Assay Kit (Thermo). Total protein samples from the cell lines were extracted and loaded onto 12.5% Tris-glycine gels, and the resolved proteins were transferred to polyvinylidene difluoride (PVDF) membranes (PerkinElmer Life Sciences). Blots were blocked by incubation with 3% BSA in TBST at RT for 1 h and probed with primary antibodies at 4 °C overnight in TBST, followed by incubation with the appropriate secondary antibodies and washing according to standard procedures. Primary antibodies against the following proteins were used: Sirt7 (ab78977, Abcam), Sirt6 (ab62739, Abcam), GR (ab3573, Abcam), and p21 (ab80633, Abcam). The protein bands were quantified and visualized using enhanced chemiluminescence (ECL Hakata, ImageQuant LAS 4000 Mini). The results were analyzed with Gel-Pro Analyzer 4.0 software (Media Cybernetics, USA) for quantification.
BrdU Incorporation Assay
A BrdU incorporation assay was performed to test the proliferation of NSCs. The cells were contained in a 24-well plate and exposed to the indicated treatment. The plates were fixed for 20 min at room temperature (RT) with 4% PFA and then washed three times with phosphate-buffered saline (PBS; pH 7.4). After DNA proteolysis (2 N HCl) for half an hour, the slides were permeabilized by incubation in 0.2% Triton X-100 for 8 min at RT and then washed three times with PBS. Nonspecific binding was blocked with 10% fetal bovine serum (FBS) for 1 h at room temperature, and the plates were then incubated with a primary antibody against BrdU (1:1000, MB6003, Bioworld) that was diluted with 10% FBS overnight at 4 °C. The plates were washed with PBS, and the secondary antibody was applied for 1 h at room temperature in the dark. BrdU-positive cells in each treatment group were counted, and the experiments were repeated three times. BrdU-positive cells were observed under a fluorescence microscope.
CFSE Staining
The CFSE (carboxyfluorescein diacetate succinimidyl ester) assay was used to assess cell proliferation by the Cell Tracer CFSE Kit (Invitrogen). NSCs were cultured in 6-well plates with media stained with CFSE (4 μM) for 48 h. Cells were washed with PBS and analyzed using flow cytometry, and the data were analyzed using FlowJo software. The results of the CFSE assay are shown in Supplementary Figure 1.
Cell Apoptosis Assay
Apoptosis analysis was conducted using an Annexin V/PI double-staining kit (KeyGen Biotech). NSCs after treatments were stained with Annexin V and PI in the dark for 15 min before analysis by flow cytometry. The results were analyzed using FlowJo software.
Statistical Analyses
Statistical results were obtained by using data analysis by GraphPad Prism 5.0 (GraphPad Software), and the data are presented as the means ± standard errors of the means (SEMs) for at least three independent experiments. Comparisons among the means were performed by one-way analysis of variance (ANOVA) followed by Bonferroni’s multiple comparison test for comparisons of multiple (> 2) groups or Student’s t test for comparisons of two means from independent samples. Differences were considered to be significant at P < 0.05, P < 0.01, and P < 0.001.
Results
DEX Inhibited NSC Proliferation and Suppressed Sirt7 Expression
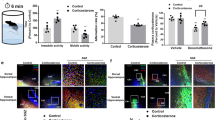
We treated hippocampal NSCs with DEX and observed that DEX significantly reduced NSC proliferation, as revealed by a significant decrease in the percentage of BrdU+ cells (Fig. 1a and b). CFSE analysis also indicated the inhibition of NSC proliferation by DEX treatment (Fig. S1a). Next, we studied whether DEX could induce cell death in NSCs using the Annexin V/PI double-staining experiment. Figure 2c demonstrates that exposure to 50 μM DEX does not induce cell death (Fig. 1c). In addition, when the Sirtuin family was examined upon exposure to DEX, determination of the mRNA and protein levels revealed that Sirt7 was significantly downregulated in the NSCs upon DEX exposure (Fig. 1d and e). Then, we examined changes in the level of Sirt7 in hippocampal DGs from NORMAL- and DEX-treated mice at postnatal week 3 (P3W). The results showed that prenatal DEX overexposure induced a decrease in the expression of Sirt7 in the hippocampus of the offspring (Fig. 1f). In the DEX-treated hippocampal NSCs, a decrease in NSC proliferation was accompanied by reductions in Sirt7 mRNA and protein levels. Taken together, these results indicate that the reduced NSC proliferation caused by DEX may be due to a decrease in Sirt7 expression.

DEX inhibits Sirt7 expression in murine NSCs. a Immunostaining for BrdU in DMSO- or DEX-treated neural stem cells (NSCs). b Quantification of the percentage of BrdU+ cells among DMSO- or DEX-treated NSCs. c Cell apoptosis assay evaluated by Annexin V/PI double-staining experiment in DMSO- or DEX-treated NSCs. d qPCR analysis of Sirt1, Sirt2, Sirt6, and Sirt7 expression levels in DMSO- or DEX-treated NSCs. e WB analysis of Sirt6 and Sirt7 expression levels in DMSO- or DEX-treated NSCs. f WB analysis of Sirt7 expression level in the hippocampal DGs at postnatal week 3 (P3W) (NORMAL, n = 5; FGR, n = 4). Scale bar, 100 μm. The data are shown as the mean ± SEM. Student’s t test. *P < 0.05; **P < 0.01; ***P < 0.001

Sirt7 knockdown decreases NSC proliferation. a qPCR analysis of Sirt7 expression level in NSCs following Sirt7 knockdown. b WB analysis of Sirt7 expression level in NSCs following Sirt7 knockdown. c Immunostaining for BrdU in Sirt7-knockdown and control NSCs treated with DMSO or DEX. d Quantification of the percentage of BrdU+ cells in Sirt7-knockdown and control NSCs treated with DMSO or DEX. e Immunostaining for BrdU in NSCs treated with DMSO, DEX, Sirt7 inhibitor 97491, and Sirt7 inhibitor 97491+DEX NSCs. f Quantification of the percentage of BrdU+ cells in NSCs treated with DMSO, DEX, Sirt7 inhibitor 97491, and Sirt7 inhibitor 97491+DEX NSCs. Scale bar, 100 μm. The data are shown as the mean ± SEM. Student’s t test. ns, not significant. *P < 0.05; **P < 0.01; ***P < 0.001
Knockdown of Sirt7 Decreased NSC Proliferation
We hypothesized that reduced Sirt7 expression contributed to the DEX-induced decrease in NSC proliferation. To test this hypothesis, we knocked down Sirt7 expression in the NSCs using the lentiviral-mediated short hairpin RNA (shRNA) approach. To exclude possible off-targeting effects, we designed a nontargeting shRNA (shCtrl) and two specific shRNAs targeting the mouse Sirt7 gene (shSirt7-1, shSirt7-2) and validated their efficiency in downregulating endogenous Sirt7 mRNA and protein levels in the NSCs (Fig. 2a and b). Using BrdU incorporation and CFSE assays, we found that Sirt7 knockdown significantly decreased the percentage of proliferating cells but did not further reduce the NSC proliferation rate in the presence of DEX, suggesting that Sirt7 serves as an important downstream target in hippocampal NSCs that decreases proliferation in response to DEX (Fig. 2c and d; Supplementary Fig. 1b). Moreover, we conducted an experiment with a specific Sirt7 inhibitor (ID: 97491) and found that the effect of the Sirt7 inhibitor on NSC proliferation was in line with the shRNA approach (Fig. 2e and f).
Overexpression of Sirt7 Reversed the DEX-Induced Inhibition of NSC Proliferation
To further examine whether Sirt7 was responsible for the DEX-induced inhibition of NSC proliferation, we overexpressed HA-tagged human SIRT7 (HA-SIRT7) or luciferase (HA-Luc) in NSCs via a lentiviral approach. The overexpression of Sirt7 was validated at the mRNA and protein levels (Fig. 3a and b). We found that Sirt7 overexpression rescued the DEX-induced inhibition of hippocampal NSC proliferation, but this was not observed in the control groups (Fig. 3c and d). We also found that the overexpression of Sirt7 reversed the decreased Sirt7 expression in the NSCs induced by DEX (Fig. 3e). Thus, our results indicated the critical role of Sirt7 in the DEX-induced decrease in the proliferation of hippocampal NSCs.

Sirt7 overexpression prevents the inhibition of NSC proliferation by DEX. a qPCR analysis of Sirt7 expression levels in NSCs overexpressing HA-Luc and HA-SIRT7. b WB analysis of Sirt7 expression levels in NSCs overexpressing HA-Luc and HA-SIRT7. c Immunostaining for BrdU in NSCs of the DMSO+HA-Luc, DEX+HA-Luc, DMSO+HA-SIRT7, and DEX+HA-SIRT7 groups. d Quantification of the percentage of BrdU+ cells among NSCs of the DMSO+HA-Luc, DEX+HA-Luc, DMSO+HA-SIRT7, and DEX+HA-SIRT7 groups. e WB analysis of Sirt7 expression levels in NSCs of the DMSO+HA-Luc, DEX+HA-Luc, DMSO+HA-SIRT7, and DEX+HA-SIRT7 groups. Scale bar, 100 μm. The data are shown as the mean ± SEM. Student’s t test. *P < 0.05; **P < 0.01
DEX Suppressed the Expression of Sirt7 Via GR
To determine the mechanism of DEX-induced suppression of Sirt7 expression, we constructed two lentiviral vectors expressing shRNAs that specifically target GR (shGR-1, shGR-2) and validated their efficiency in downregulating endogenous GR mRNA and protein levels in NSCs (Fig. 4a and b). Then, we studied cell proliferation using a BrdU incorporation assay, and the results showed that knockdown of GR abrogated the DEX-induced decrease in NSC proliferation (Fig. 4c and d). The qPCR and WB results showed that knockdown of GR abolished the decrease in Sirt7 expression that was induced by DEX treatment in NSCs (Fig. 4e and f). Taken together, these results demonstrate that DEX decreases NSC proliferation and suppresses the expression of Sirt7 through GR.

DEX alters the expression of Sirt7 via interacting with GR. a qPCR analysis of GR expression levels in NSCs following GR knockdown. b WB analysis of GR expression levels in NSCs following GR knockdown. c Immunostaining for BrdU in GR-knockdown and control NSCs treated with DMSO or DEX. d Quantification of the percentage of BrdU+ cells among GR-knockdown and control NSCs treated with DMSO or DEX. e qPCR analysis of Sirt7 expression levels in GR-knockdown and control NSCs treated with DMSO or DEX. f WB analysis of Sirt7 expression levels in GR-knockdown and control NSCs treated with DMSO or DEX. Scale bar, 100 μm. The data are shown as the mean ± SEM. Student’s t test. **P < 0.01; ***P < 0.001
DEX Exposure and Reduced Sirt7 Affected Cell Cycle–Related Genes
To determine the mechanism by which a reduction in Sirt7 inhibits NSC proliferation, we tested the effect of Sirt7 knockdown on cell cycle–related gene expression in DMSO- and DEX-treated NSCs. The qPCR and WB results showed that DEX treatment increased the expression of p21 in the NSCs (Fig. 5a–c). The expression of p21 was also significantly increased in the Sirt7-knockdown NSCs and Sirt7 inhibitor–treated NSCs (Fig. 5d–g). In contrast, overexpression of Sirt7 decreased the expression of p21 in NSCs (Fig. 5h and i). These results indicate that DEX treatment and the expression level of Sirt7 affect p21 expression. Furthermore, we hypothesized that the Sirt7/p21 axis regulates NSC proliferation under DEX treatment.

DEX and a reduction in Sirt7 inhibit cell cycle–related genes. a qPCR analysis of negative regulators of cell cycle expression levels in DMSO- or DEX-treated NSCs. b qPCR analysis of positive regulators of cell cycle expression levels in DMSO- or DEX-treated NSCs. c WB analysis of p21 expression levels in DMSO- or DEX-treated NSCs. d qPCR analysis of p21 expression levels in NSCs following Sirt7 knockdown. e WB analysis of p21 expression levels in NSCs following Sirt7 knockdown. f qPCR analysis of p21 expression levels in NSCs following Sirt7 inhibitor 97491 treatment. g WB analysis of p21 expression levels in NSCs following Sirt7 inhibitor 97491 treatment. h qPCR analysis of p21 expression levels in NSCs overexpressing HA-Luc and HA-SIRT7. i WB analysis of p21 expression levels in NSCs overexpressing HA-Luc and HA-SIRT7. The data are shown as the mean ± SEM. Student’s t test. *P < 0.05; **P < 0.01
Sirt7 Mediates the Effect of DEX in NSCs by Targeting p21
To further examine the role of Sirt7 in regulating the expression of p21 under DEX intervention, we developed two lentiviral vectors that express shRNAs that specifically target p21 (shp21-1, shp21-2). The knockdown efficiency of the individual sequences was confirmed by the downregulated endogenous p21 mRNA and protein levels in NSCs (Fig. 6a and b). The BrdU incorporation and CFSE assay showed that knockdown of p21 rescued the DEX-induced inhibition of NSC proliferation, but this was not observed in the control groups (Fig. 6c and d; Supplementary Fig. 1c). In addition, the BrdU incorporation and CFSE assay showed that knockdown of p21 reversed the decreased NSC proliferation caused by a reduction in Sirt7 in the NSCs (Fig. 6e and f; Supplementary Fig. 1d). Next, we overexpressed HA-tagged p21 (HA-p21) or luciferase (HA-Luc) in NSCs via a lentiviral approach. Western blot analysis showed that the expression of p21 increased in the HA-p21 group compared with the control group (Fig. 6g). The BrdU incorporation and CFSE assay showed that overexpression of p21 abrogated the Sirt7 overexpression–induced rescue of DEX-treated NSC proliferation (Fig. 6h and i; Supplementary Fig. 1e). Taken together, our results indicate that Sirt7 mediates the DEX-induced decrease in NSC proliferation by upregulating p21 expression.

Sirt7 mediates the effect of DEX on the proliferation of NSCs through targeting p21. a qPCR analysis of p21 expression levels in NSCs following p21 knockdown. b WB analysis of p21 expression levels in NSCs following p21 knockdown. c Immunostaining for BrdU in p21-knockdown and control NSCs treated with DMSO or DEX. d Quantification of the percentage of BrdU+ cells among p21-knockdown and control NSCs treated with DMSO or DEX. e Immunostaining for BrdU in shCtrl- or shSirt7 (shSirt7-1, shSirt7-2)-transfected NSCs following p21 knockdown. f Quantification of the percentage of BrdU+ cells among shCtrl- or shSirt7 (shSirt7-1, shSirt7-2)-transfected NSCs following p21 knockdown. g WB analysis of p21 expression levels in NSCs overexpressing HA-Luc and HA-p21. h Immunostaining for BrdU in the DMSO+HA-Luc, DEX+HA-Luc, DEX+HA-SIRT7, DEX+HA-p21, and DEX+HA-SIRT7+HA-p21 groups. i Quantification of the percentage of BrdU+ cells among the DMSO+HA-Luc, DEX+HA-Luc, DEX+HA-SIRT7, DEX+HA-p21, and DEX+HA-SIRT7+HA-p21 groups. Scale bar, 100 μm. The data are shown as the mean ± SEM. Student’s t test. *P < 0.05; **P < 0.01; ***P < 0.001
Discussion
Prenatal excess GC hormones are well-known inhibitors of NSC proliferation, increasing the risk of cognition and mood deficits in offspring. However, the mechanism remains unclear. Here, we identified a new Sirt7-p21 pathway that mediates the inhibition of NSC proliferation by glucocorticoids.
The effect of glucocorticoids on target tissues is mediated by their interaction with the high-affinity mineralocorticoid receptor (MR) or the low-affinity GR. The GR is not activated under baseline conditions but is responsible for binding glucocorticoids during neurogenesis (Mirescu et al. 2004). GR activation by high levels of glucocorticoids consistently decreases NSC proliferation in vitro and in vivo (Bose et al. 2010; Kim et al. 2004). In agreement with this finding, we observed by BrdU labeling that DEX, a GR agonist, decreased the NSC proliferation rate. Emerging evidence indicates that epigenetic changes play an important role in the proliferation and fate decisions of NSCs (Mohamed Ariff et al. 2012). Among the Sirtuin family members, Sirt1 modulates the proliferation and self-renewal of NSCs in response to glucose (Fusco et al. 2016), and Sirt2 significantly reduces adult hippocampal NSC proliferation (Yoo et al. 2015). Although these studies have revealed that sirtuins regulate the proliferation of NSCs, evidence of their regulation of the effect of glucocorticoid hormones on NSC proliferation is limited. From this study, our results showed that DEX inhibits Sirt7 gene expression in vitro and in vivo with no effects on the expression of Sirt1, Sirt2, or Sirt6. With a gain or loss of Sirt7 function generated by the lentiviral approach, our results revealed that Sirt7 mediates the effects of DEX on NSC proliferation. Furthermore, by blocking GR through knockdown experiments, we could prevent the inhibition of Sirt7 by DEX, indicating new crosstalk between the glucocorticoid pathway and sirtuins. GR-mediated gene downregulation occurs in three different ways: direct GR binding of the negative GC-responsive elements (nGREs) of target genes, tethering of GR to AP-1/NF-kB sites, and GR-mediated recruitment of corepressors, such as GRIP1 (Uhlenhaut et al. 2013). We were not able to identify the GC-responsive regions or AP-1/NF-kB DNA-binding sites in the promoter sequence of Sirt7 by bioinformatic prediction. Thus, DEX-mediated inhibition of Sirt7 expression might occur through protein interactions between GR and corepressors.
P21 is an important member of the cyclin-dependent kinase (CDK) family that inhibits cell cycle progression (Bertoli et al. 2013). Previous studies reported the upregulation of p21 by DEX in NSCs, osteoblastic cells, and cancer cells (Bose et al. 2010; Cha et al. 1998; Cram et al. 1998; Han et al. 2018). However, the molecular mechanisms by which DEX upregulates the expression of p21 have not been well characterized. In rat hepatoma cells, GCs stimulated p21 gene expression via protein interactions between GR and C/EBPα through the direct binding of GC-responsive elements (GREs) and a C/EBP DNA-binding site in the p21 promoter sequence via a tethering mechanism (Cha et al. 1998; Cram et al. 1998). Our data showed that both DEX and Sirt7 knockdown induced the upregulation of p21, while Sirt7 overexpression inhibited p21 gene expression. Importantly, p21 shRNA attenuated Sirt7 shRNA–induced inhibition of NSC proliferation, while p21 overexpression abrogated the Sirt7 overexpression–induced rescue of the altered proliferation of DEX-treated NSCs. Thus, our findings identified a new mechanism for the upregulation of p21 by glucocorticoids in which DEX upregulates the expression of p21 in mouse NSCs in vitro by downregulating Sirt7, and this downregulation is mediated by GR. Similar to this indirect mechanism of regulation, DEX upregulates the expression of p27, which is another CDK inhibitor, by downregulating the phosphorylation of 4E-BP1 in human breast cancer cells (Eto 2010). Our new findings enrich the present understanding of the mechanisms by which glucocorticoids regulate cell cycle–related genes.
Taken together, our results revealed that Sirt7 mediates the inhibitory effect of DEX on NSC proliferation by targeting p21, which may provide potential new targets for improving the declines in adult cognition and mood caused by prenatal overexposure to GC hormones.
Change history
20 April 2021
A Correction to this paper has been published: https://doi.org/10.1007/s12640-021-00360-y
Abbreviations
- NSCs:
-
Neural stem cells
- DEX:
-
Dexamethasone
- DG:
-
Dentate gyrus
- GR:
-
Glucocorticoid receptor
- GC:
-
Glucocorticoid
- P3W:
-
Postnatal 3 week
- GREs:
-
GC-responsive elements
- CDK:
-
Cyclin-dependent kinase
- MR:
-
Mineralocorticoid receptor
References
Anacker C, Hen R (2017) Adult hippocampal neurogenesis and cognitive flexibility—linking memory and mood. Nat Rev Neurosci 18:335–346
Bertoli C, Skotheim JM, De Bruin RA (2013) Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol 14:518–528
Bose R, Moors M, Tofighi R, Cascante A, Hermanson O, Ceccatelli S (2010) Glucocorticoids induce long-lasting effects in neural stem cells resulting in senescence-related alterations. Cell Death Dis 1:e92–e92
Cha HH, Cram EJ, Wang EC, Huang AJ, Kasler HG, Firestone GL (1998) Glucocorticoids stimulate p21 gene expression by targeting multiple transcriptional elements within a steroid responsive region of the p21 waf1/cip1 promoter in rat hepatoma cells. J Biol Chem 273:1998–2007
Choy KHC, de Visser Y, Nichols NR, van den Buuse M (2008) Combined neonatal stress and young-adult glucocorticoid stimulation in rats reduce BDNF expression in hippocampus: effects on learning and memory. Hippocampus 18:655–667
Cole TJ (2006) Glucocorticoid action and the development of selective glucocorticoid receptor ligands. Biotechnol Annu Rev 12:269–300
Cram EJ, Ramos RA, Wang EC, Cha HH, Nishio Y, Firestone GL (1998) Role of the CCAAT/enhancer binding protein-α transcription factor in the glucocorticoid stimulation of p21 waf1/cip1 gene promoter activity in growth-arrested rat hepatoma cells. J Biol Chem 273:2008–2014
de los Angeles GAM, del Carmen ROM, Wendy PM, Socorro R-M (2016) Tactile stimulation effects on hippocampal neurogenesis and spatial learning and memory in prenatally stressed rats. Brain Res Bull 124:1–11
Diaz-Perdigon T, Belloch FB, Ricobaraza A, Elboray EE, Suzuki T, Tordera RM, Puerta E (2020) Early sirtuin 2 inhibition prevents age-related cognitive decline in a senescence-accelerated mouse model. Neuropsychopharmacology 45:347–357
Eichenbaum H, Cohen NJ (2014) Can we reconcile the declarative memory and spatial navigation views on hippocampal function? Neuron 83:764–770
Eto I (2010) Upstream molecular signaling pathways of p27 (Kip1) expression: effects of 4-hydroxytamoxifen, dexamethasone, and retinoic acids. Cancer Cell Int 10:3
Fusco S, Leone L, Barbati SA, Samengo D, Piacentini R, Maulucci G, Toietta G, Spinelli M, McBurney M, Pani G, Grassi C (2016) A CREB-Sirt1-Hes1 circuitry mediates neural stem cell response to glucose availability. Cell Rep 14:1195–1205
G V, M L (2020) Genetics in endocrinology: glucocorticoid resistance syndrome. Eur J Endocrinol 182:R15–R27
Gao J, Wang W-Y, Mao Y-W, Gräff J, Guan J-S, Pan L, Mak G, Kim D, Su SC, Tsai LH (2010) A novel pathway regulates memory and plasticity via SIRT1 and miR-134. Nature 466:1105–1109
Han D, Gao J, Gu X, Hengstler JG, Zhang L, Shahid M, Ali T, Han B (2018) P21 Waf1/Cip1 depletion promotes dexamethasone-induced apoptosis in osteoblastic MC3T3-E1 cells by inhibiting the Nrf2/HO-1 pathway. Arch Toxicol 92:679–692
Hauser J, Feldon J, Pryce CR (2009) Direct and dam-mediated effects of prenatal dexamethasone on emotionality, cognition and HPA axis in adult Wistar rats. Horm Behav 56:364–375
Hauser J, Knapman A, Zürcher NR, Pilloud S, Maier C et al (2008) Effects of prenatal dexamethasone treatment on physical growth, pituitary-adrenal hormones, and performance of motor, motivational, and cognitive tasks in juvenile and adolescent common marmoset monkeys. Endocrinology 149:6343–6355
Holmes MC, Abrahamsen CT, French KL, Paterson JM, Mullins JJ, Seckl JR (2006) The mother or the fetus? 11β-Hydroxysteroid dehydrogenase type 2 null mice provide evidence for direct fetal programming of behavior by endogenous glucocorticoids. J Neurosci 26:3840–3844
Islam MS, Wei F-Y, Ohta K, Shigematsu N, Fukuda T, Tomizawa K, Yoshizawa T, Yamagata K (2018) Sirtuin 7 is involved in the consolidation of fear memory in mice. Biochem Biophys Res Commun 495:261–266
Kawata M (1995) Roles of steroid hormones and their receptors in structural organization in the nervous system. Neurosci Res 24:1–46
Kim J-H, Kim D, Cho SJ, Jung K-Y, Kim J-H, Lee JM, Jung HJ, Kim KR (2019) Identification of a novel SIRT7 inhibitor as anticancer drug candidate. Biochem Biophys Res Commun 508:451–457
Kim JB, Ju JY, Kim JH, Kim T-Y, Yang B-H, Lee Y-S, Son H (2004) Dexamethasone inhibits proliferation of adult hippocampal neurogenesis in vivo and in vitro. Brain Res 1027:1–10
Kino T (2015) Stress, glucocorticoid hormones, and hippocampal neural progenitor cells: implications to mood disorders. Front Physiol 6:230
Lautarescu A, Pecheva D, Nosarti C, Nihouarn J, Zhang H, Victor S, Craig M, Edwards AD, Counsell SJ (2020) Maternal prenatal stress is associated with altered uncinate fasciculus microstructure in premature neonates. Biol Psychiatry 87:559–569
Li W, Zhang B, Tang J, Cao Q, Wu Y, Wu C, Guo J, Ling EA, Liang F (2007) Sirtuin 2, a mammalian homolog of yeast silent information regulator-2 longevity regulator, is an oligodendroglial protein that decelerates cell differentiation through deacetylating α-tubulin. J Neurosci 27:2606–2616
Lu NZ, Cidlowski JA (2006) Glucocorticoid receptor isoforms generate transcription specificity. Trends Cell Biol 16:301–307
Lucassen PJ, Müller MB, Holsboer F, Bauer J, Holtrop A, Wouda J, Hoogendijk WJG, de Kloet ER, Swaab DF (2001) Hippocampal apoptosis in major depression is a minor event and absent from subareas at risk for glucocorticoid overexposure. Am J Pathol 158:453–468
Ma C-y, Yao M-j, Zhai Q-w, J-w J, Yuan X-b, Poo M-m (2014) SIRT1 suppresses self-renewal of adult hippocampal neural stem cells. Development 141:4697–4709
MacQueen G, Frodl T (2011) The hippocampus in major depression: evidence for the convergence of the bench and bedside in psychiatric research? Mol Psychiatry 16:252–264
Mirescu C, Peters JD, Gould E (2004) Early life experience alters response of adult neurogenesis to stress. Nat Neurosci 7:841–846
Mohamed Ariff I, Mitra A, Basu A (2012) Epigenetic regulation of self-renewal and fate determination in neural stem cells. J Neurosci Res 90:529–539
Moisiadis VG, Matthews SG (2014) Glucocorticoids and fetal programming part 1: outcomes. Nat Rev Endocrinol 10:391–402
Mutsaers HA, Tofighi R (2012) Dexamethasone enhances oxidative stress-induced cell death in murine neural stem cells. Neurotox Res 22:127–137
Neilson J (2007) Antenatal corticosteroids for accelerating fetal lung maturation for women at risk of preterm birth. Obstet Gynecol 109:189–190
Peaceman A, Bajaj K, Kumar P, Grobman W (2006) The interval between a single course of antenatal steroids and delivery and its association with neonatal outcomes. Midirs Midwifery Digest 16:112–113
Poulsen RC, Watts AC, Murphy RJ, Snelling SJ, Carr AJ, Hulley PA (2014) Glucocorticoids induce senescence in primary human tenocytes by inhibition of sirtuin 1 and activation of the p53/p21 pathway: in vivo and in vitro evidence. Ann Rheum Dis 73:1405–1413
Reagan-Shaw S, Nihal M, Ahmad N (2008) Dose translation from animal to human studies revisited. FASEB J 22:659–661
Scheinost D, Sinha R, Cross SN, Kwon SH, Sze G, Constable RT, Ment LR (2017) Does prenatal stress alter the developing connectome? Pediatr Res 81:214–226
Seckl JR (2001) Glucocorticoid programming of the fetus; adult phenotypes and molecular mechanisms. Mol Cell Endocrinol 185:61–71
Suzuki S, Iben JR, Coon SL, Kino T (2018) SIRT1 is a transcriptional enhancer of the glucocorticoid receptor acting independently to its deacetylase activity. Mol Cell Endocrinol 461:178–187
Uhlenhaut NH, Barish GD, Ruth TY, Downes M, Karunasiri M et al (2013) Insights into negative regulation by the glucocorticoid receptor from genome-wide profiling of inflammatory cistromes. Mol Cell 49:158–171
Wang G, Li S, Gilbert J, Gritton HJ, Wang Z, Li Z, Han X, Selkoe DJ, Man HY (2017) Crucial roles for SIRT2 and AMPA receptor acetylation in synaptic plasticity and memory. Cell Rep 20:1335–1347
Wang G, Wang F, Ren J, Qiu Y, Zhang W, Gao S, Yang D, Wang Z, Liang A, Gao Z, Xu J (2018) SIRT1 involved in the regulation of alternative splicing affects the DNA damage response in neural stem cells. Cell Physiol Biochem 48:657–669
Wyrwoll CS, Holmes MC, Seckl JR (2011) 11β-Hydroxysteroid dehydrogenases and the brain: from zero to hero, a decade of progress. Front Neuroendocrinol 32:265–286
Xu D, Luo HW, Hu W, Hu SW, Yuan C, Wang GH, Zhang L, Yu H, Magdalou J, Chen LB, Wang H (2018) Intrauterine programming mechanism for hypercholesterolemia in prenatal caffeine-exposed female adult rat offspring. FASEB J 32:5563–5576
Yoo DY, Kim DW, Kim MJ, Choi JH, Jung HY, Nam SM, Kim JW, Yoon YS, Choi SY, Hwang IK (2015) Sodium butyrate, a histone deacetylase inhibitor, ameliorates SIRT2-induced memory impairment, reduction of cell proliferation, and neuroblast differentiation in the dentate gyrus. Neurol Res 37:69–76
Zhang X, Shang-Guan Y, Ma J, Hu H, Wang L, Magdalou J, Chen L, Wang H (2016) Mitogen-inducible gene-6 partly mediates the inhibitory effects of prenatal dexamethasone exposure on endochondral ossification in long bones of fetal rats. Br J Pharmacol 173:2250–2262
Funding
This work was supported by the National Natural Science Foundation of China (grants 81530042, 31830059, 31721003, 31871495, 31701110) and Fundamental Research Funds for the Central Universities (22120190149).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All mice were maintained and handled according to Tongji University institutional guidelines.
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
ESM 1
(DOC 121 kb)
Supplementary Figure 1.
Test of NSC proliferation by CFSE-based assay. (a) CFSE-based assay among DMSO- or DEX-treated NSCs. (b) CFSE-based assay in Sirt7-knockdown and control NSCs treated with DMSO or DEX. (c) CFSE-based assay in p21-knockdown and control NSCs treated with DMSO or DEX. (d) CFSE based-assay in shCtrl- or shSirt7 (shSirt7-1, shSirt7-2)-transfected NSCs following p21 knockdown. (e) CFSE-based assay in the DMSO+HA-Luc, DEX+HA-Luc, DEX+HA-SIRT7, DEX+HA-p21, and DEX+HA-SIRT7+HA-p21 groups. The data are shown as the mean ± SEM. Student’s t test. ***P<0.001. (JPG 706 kb)
Rights and permissions
About this article

{kind=link}
Cite this article
Alnoud, M.A.H., Chen, W., Liu, N. et al. Sirt7-p21 Signaling Pathway Mediates Glucocorticoid-Induced Inhibition of Mouse Neural Stem Cell Proliferation. Neurotox Res 39, 444–455 (2021). https://doi.org/10.1007/s12640-020-00294-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-020-00294-x