
Abstract
Glucocorticoids (GCs) are essential for normal brain development; however, there is consistent evidence that prenatal exposure of the fetal brain to excess GCs permanently modifies the phenotype of neuronal cells. In this paper, the murine-derived multipotent stem cell line C17.2 was used, as an in vitro model, to investigate the impact of GCs on neural stem cell survival. Our results indicate that dexamethasone (Dex) increases the sensitivity of murine neural stem cells (NSCs) to 2,3-methoxy-1,4-naphthoquinone-induced apoptosis, and this effect could be blocked by the glucocorticoid-receptor (GR) antagonist mifepristone, strongly suggesting the involvement of the GR. Furthermore, our results show that Dex decreases cell number and induces a G1-arrest. We hypothesized that the mitochondria are the main target of Dex. Interestingly, after treatment with Dex, 72% of the investigated genes involved in the mitochondrial respiratory chain are down-regulated, as well as 29% of the genes encoding for antioxidant enzymes. In conclusion, using the C17.2 cell line as a model to study developmental neurotoxicity in vitro, we have shown that GCs can increase cellular sensitivity to oxidative stress and alter the phenotype of NCSs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
There is increasing evidence that prenatal events can predispose to a wide variety of diseases during adult life. Glucocorticoids (GCs) exert their biochemical function mainly by binding to the glucocorticoid-receptor (GR), which is expressed to varying degrees in all cell types (Cole 2006). During fetal development, GCs play a key role in the maturation of terminal respiratory alveoli. Furthermore, GCs stimulate the production of surfactant in the fetal lung and activate gut enzyme systems, and they are essential for many aspects of normal brain development (Cole 2006; Seckl and Holmes 2007). Postnatal, GCs play a central role in the adaptation to acute physical stressors. In response to stress, the secretion of GCs is elevated resulting in increased blood glucose levels, enhanced cardiovascular tone and an improved cognition. Furthermore, stress-induced GCs cause a suppression of digestion, growth, reproduction and immune responses (Cole 2006; Sapolsky 1999). Because of their systemic effects, GCs are widely used as therapeutics, for example to treat inflammatory and autoimmune conditions including rheumatoid arthritis (Barnes 1998; Cole 2006; Czock et al. 2005). Moreover, antenatal GCs are administered to women at risk of preterm delivery in order to promote maturation of the fetal lung (Cole 2006; Roberts and Dalziel 2006). However, there is increasing evidence that prenatal events, including exposure to excess GCs, may predispose to diseases in later life, such as hypertension and type 2 diabetes (Barker 1995; Moritz et al. 2005; Seckl 2008; Seckl and Holmes 2007).
Fetal exposure to elevated GC levels can occur when maternal glucocorticoids are elevated in response to stress or when exogenous GCs, such as dexamethasone (Dex), are administered. Normally, the fetus is protected from maternal GCs by placental 11 beta-hydroxysteroid dehydrogenase (11 β-HSD). However, high levels of maternal GCs, exceeding the limit of placental 11 β-HSD or pathological conditions impairing placental functions, might lead to fetal exposure to excess GCs (Fowden and Forhead 2004; Moritz et al. 2005; Seckl and Holmes 2007).
Dex is a synthetic glucocorticoid, which is 25–30 times more potent than the natural steroid cortisol (Kawata 1995). Therefore, in many studies, Dex has been used as a model compound to investigate GC-mediated intrauterine programming. Seckl (2001) described that fetal exposure to high levels of GCs, via maternal administration of Dex, resulted in a reduced fetal growth in rats, rabbits, sheep, monkeys and humans. Also, it is well documented that exposure to high levels of GCs adversely affects normal brain and kidney morphology (Sapolsky 1985, 1996, 1999; Singh et al. 2007; Uno et al. 1994). Moreover, it has been demonstrated that in utero exposure to GCs leads to hypertension during adult life (MohanKumar et al. 2007; Seckl and Holmes 2007). In a variety of animal models, prenatal exposure to GCs resulted in an increased hypothalamic–pituitary–adrenal (HPA) system activity in the offspring (Buhl et al. 2007; de Vries et al. 2007), which is associated with neuropsychiatric disorders including anxiety-like behavior (Holmes et al. 2006; Seckl and Holmes 2007; Welberg et al. 2000), depressive behavior (Kapoor et al. 2006; Wilcoxon and Redei 2007) and altered cognitive behavior (Hauser et al. 2008; Sandi 2004; Wilcoxon and Redei 2007).
It was previously demonstrated, using an in vitro/ex vivo model, that prenatal and indirect exposure to high levels of GCs permanently modified the phenotype of rat neuronal cells and induced an increased susceptibility to oxidative stress (Ahlbom et al. 2000; Bose et al. 2010; Canlon et al. 2003). Furthermore, it has been proposed that Dex can regulate apoptotic cell death (Noguchi et al. 2008). The aim of the present study was to investigate the impact of Dex on the phenotype of murine-derived neural stem cells (NSCs) and the molecular mechanisms underlying Dex-mediated alterations.
Materials and Methods
Cell Culture and Experimental Treatment
The murine-derived multipotent neural stem cell line, C17.2, was cultured in Dulbecco’s modified eagle medium (Life Technologies, Gibco BRL, Grand Island, NY, USA) containing 10% (v/v) fetal bovine serum and 5% (v/v) horse serum (both Life Technologies) at 37°C in a 5% (v/v) CO2 atmosphere. Cells were routinely seeded at a density of 4,000 cells/cm2.
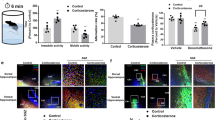
To study the lasting effects of Dex, cells were seeded in cell culture dishes and allowed to adhere and grow for 48 h. Subsequently, the cells were exposed to the solvent dimethyl sulfoxide (final concentration 0.1% v/v; control cells) or to 50 μM Dex (Sigma-Aldrich, St. Louis, MO, USA) for 24 h. This concentration of Dex did not affect cell viability in the model. When used, the well-known GR antagonist mifepristone (50 μM; Sigma) was added 30 min prior to Dex exposure (Cole 2006; Heikinheimo et al. 1987). After 24 h exposure to Dex, the cell medium was changed, and new medium was added containing no Dex or DMSO. After additional 48 h, cells were harvested for experiments using trypsin–EDTA (Life Technologies). Oxidative stress was induced using the redox-cycling chemical 2,3-methoxy-1,4-naphthoquinone (DMNQ; Sigma), which exerts its toxic effect via continuous generation of superoxide and the subsequent dismutation product H2O2. Moreover, DMNQ does not react with free thiol groups and is nonalkylating and non-adduct forming (Gant et al. 1988; Henry and Wallace 1995; Tchivilev et al. 2008). A schematic overview of the experimental exposure is presented in Fig. 1.

Experimental exposure. *When used, mifepristone was added 30 min prior to dexamethasone exposure
Detection of Cell Death Using the Trypan Blue Exclusion Assay
Control and Dex-treated cells were cultured and allowed to adhere before being exposed to 10 μM DMNQ during 24 h. Afterward, supernatant was collected, and cells were harvested using trypsin–EDTA followed by centrifugation at 1,000×g for 5 min. Subsequently, cells were resuspended in 250 μL PBS, and a small aliquot of the cell suspension was mixed with 0.4% (v/v) trypan blue solution (1:1; Sigma). Both blue stained (dead) and unstained cells (healthy and early apoptotic) were counted under a phase-contrast microscope using a Neubauer improved counting chamber.
Assessment of Apoptotic Nuclear Morphology
To evaluate nuclear morphology, control and Dex-treated cells were cultured on poly-l-lysine coated coverslips. After exposure to 10 μM DMNQ for 16 h, cells were washed with PBS and stained with Hoechst 33342 (1 μg/mL) for 5 min at room temperature (RT). Afterward, coverslips were immediately examined using an Olympus BX60 fluorescence microscope (Olympus, Tokyo, Japan). The percentage of apoptotic nuclei (nuclei with smaller size and high intensity of chromatin) was determined by scoring at least 100 cells, which were randomly selected on each coverslip.
To assess nuclear morphology using confocal microscopy, control and Dex-treated cells were cultured on coverslips and exposed to DMNQ as described above. Following treatment, cells were washed with PBS and fixed with ice-cold 80% (v/v) methanol for 30 min at −20°C. Subsequently, cells were stained with propidium iodide (PI; 2.5 μg/mL) for 5 min at RT. Afterward, cells were examined using a Zeiss LSM 510 Meta confocal microscope (Zeiss, Jena, Germany).
Odyssey Western Protocol
The effect of DMNQ treatment on the cleavage of caspase 3 precursor protein and poly (ADP-ribose) polymerase (PARP) was studied using the Odyssey Western blotting technique. Control and Dex-exposed cells were cultured and allowed to adhere before being exposed to 10 μM DMNQ for 24 h. After treatment, cells were harvested using trypsin–EDTA and resuspended in 150-μL protease inhibitor mix (10 mM EDTA, 10 mM DTT and 0.24 μg/mL Petablock in PBS), followed by sonication for 10 s. The protein content of samples was determined using the BCA protein assay (Pierce, Rockford, IL, USA). Total protein (50 μg) was separated via SDS/PAGE using 12% (w/v) gels and blotted onto nitrocellulose membranes using a BioRad wet blotting system (1 h/150 V). Afterward, the membrane was blocked using Odyssey Blocking Buffer, (1:1 diluted with PBS; LI-COR Biosciences, Lincoln, NE, USA) during 1 h at RT. The membrane was then incubated overnight at 4°C with rabbit polyclonal caspase 3 antibody (1:500) or rabbit polyclonal PARP antibody (1:1,000; both Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). Mouse monoclonal β-actin antibody (1:10,000; Sigma) was simultaneously incubated to serve as a protein loading control. Antibodies were diluted in Odyssey Blocking Buffer containing 0.1% (v/v) Tween-20. Afterward, the membrane was thoroughly washed three times during 10 min with PBS containing 0.1% (v/v) Tween-20. The secondary antibodies, goat anti mouse Alexa Fluor 680 (1:20,000; Invitrogen) and goat anti rabbit IRDye 800 (1:20,000; Rockland, Gilbertsville, PA, USA), were incubated for 1 h at RT in Odyssey blocking buffer containing 0.1% (v/v) Tween-20 and 0.01% (v/v) SDS. The membrane was thoroughly washed, as described above, and then scanned using the Odyssey infrared imaging system (LI-COR Biotechnology).
Caspase Activity
To evaluate caspases 3 activity, the cleavage of DEVD-MCA (Peptide Institute, Osaka, Japan) was assessed as previously described (Dare et al. 2002). In short, control and Dex-treated cells were cultured and allowed to adhere before being exposed to 10 μM DMNQ during 24 h. Afterward, supernatant was collected, and cells were harvested using a cell scraper. Next, the caspase substrate DEVD-MCA was added, and the formation of 4-methyl-coumaryl-7-amide (excitation 355 nm, emission 460 nm) was measured at 37°C using a Fluoroscan II (Labsystem AB, Stockholm, Sweden). Fluorescence units were converted to picomoles 4-methyl-coumaryl-7-amide and subsequently related to protein content of the samples.
Determination of Cell Cycle Arrest Using Flow Cytometry
Flow cytometry was used to analyze the effect of Dex on the cell cycle using PI staining. Control and Dex-exposed cells were cultured and allowed to grow for 48 h. Subsequently, supernatant was collected, and cells were harvested using trypsin–EDTA followed by centrifugation at 1,000×g for 5 min. Cells were then washed once with PBS, and subsequently, 250 μL ice-cold 99.5% (v/v) ethanol was slowly added to the samples, while vortexing. The samples were then stored at −20°C for at least 24 h. Afterward, samples were centrifuged (1,000×g for 5 min), and the cells were resuspended in 300 μL PBS containing PI (5 μg/mL) and RNAse A (0.5 μg/mL), followed by 1-h incubation at RT. Quantification of PI binding was done with FACScan (Becton–Dickinson) using channel FL-3. Cellquest Pro was used to perform histogram analysis.
Gene Expression Using Quantitative PCR
The effects of treatment with Dex on gene expression were studied using quantitative PCR. Control and Dex-exposed cells were seeded and allowed to grow for 48 h. Afterward, cells were harvested, and total RNA was isolated using a RNeasy Mini kit in combination with the RNase-free DNase set (both Qiagen AB, Solna, Sweden) according to the manufacturers recommendations. Subsequently, cDNA was generated using Superscrip II Reverse Transcriptase (Invitrogen) according to the manufacturers recommendations. Following cDNA-synthesis, a PCR mix was prepared containing the SYBR green PCR Master Mix (Applied biosystems, Stockholm, Sweden), 1.5 μL 10 μM forward primer, 1.5 μL 10 μM reverse primer, 1 μL 10–50 ng cDNA and RNAse-free Mili-Q to make a total volume of 25 μL. See Online Resource 1 for the primer sequences. The PCR was performed using an ABI Prism 7000 (Applied Biosystems) as follows: Taq activation at 95°C for 10 min; 40 cycles of denaturing at 95°C for 15 s and annealing and extension at 60°C for 1 min (or 56°C, see Online Resource 1 for annealing and extension temperature per primer pair); followed by a dissociation curve to analyze the PCR product. Quantification of gene expression was performed using the ABI Prism 7000 SDS software. Hypoxanthine phosphoribosyltransferase (HPRT) was used as housekeeping gene, and relative expression levels were calculated using the 2−ΔΔCT method, as described previously (Bose et al. 2010). Final PCR-product size was examined on a 1.5% (w/v) agarose gel.
Statistics
Statistics were performed using GraphPad Prism 5 via a one-way analysis of variance (ANOVA) followed by the Bonferroni’s multiple comparison test or a two-way ANOVA followed by Bonferroni post-tests. Differences between groups were stated to be statistically significant when P < 0.05.
Results
Dexamethasone Increases the Susceptibility to DMNQ-Induced Cell Death via GR-Activation
It was previously demonstrated that cerebellar granule cells prepared from rats exposed to Dex in utero were more prone to oxidative stress-induced cell death (Ahlbom et al. 2000). Here, we investigated whether the same effect could be observed in our in vitro model. Phase-contrast microscopy revealed that treatment with 10 μM DMNQ for 24 h induced abnormal morphology and caused the cells to detach from the culture dish (Fig. 2b, d). Moreover, the total number of cells is decreased in the Dex-treated group (Fig. 2c), compared to the control cells (Fig. 2a). Next, we studied whether DMNQ could induce cell death in NSCs using the trypan blue exclusion assay. Figure 2e demonstrates that in the control group, exposure to 10 μM DMNQ during 24 h significantly increased the number of trypan blue positive cells from 4.8 to 36.1% (P < 0.001). Subsequently, we studied whether Dex-treated cells were more susceptible to DMNQ-induced cell death. As shown in Fig. 2e, the percentage of dead cells after DMNQ exposure is 1.3 times higher in the Dex-treated group (46.3%), compared to the control cells (36.1%). These results are in agreement with the data shown in Fig. 2d, where there is an obvious increase in the number of rounded and detached cells in the Dex group treated with DMNQ. Moreover, treatment with Dex and mifepristone significantly decreased the percentage of trypan blue positive cells to 26% after DMNQ treatment, compared to the cells that were solely exposed to Dex (P < 0.001). Thus, our results showed that Dex increased the susceptibility to DMNQ-induced cell death in NSCs in vitro, via the activation of GR.

Effect of dexamethasone and mifepristone on DMNQ-induced cell death. Control, Dex-treated and DexMife-treated cells were cultured at a density of 11.5 × 104 cells per 60-mm cell culture dish and allowed to adhere before being exposed to 10 μM DMNQ for 24 h. Phase contrast images of control and Dex-treated cells unexposed (a, c) or exposed to DMNQ (b, d). e Following treatment, supernatant was collected, and cells were harvested using trypsin–EDTA. Next, cell suspension was mixed with 0.4% (v/v) Trypan blue solution (1:1), and cells were counted under a phase-contrast microscope using a Neubauer improved counting chamber. Statistical analysis was performed via a one-way ANOVA followed by a Bonferroni’s multiple comparison Test. Bars represent mean + SEM of one representative experiment performed in triplicate. *P < 0.001
Apoptotic Nuclear Morphology Following DMNQ Exposure
Several studies have described that DMNQ induces apoptotic cell death in NSCs (Ceccatelli et al. 2004, 2007; Tamm et al. 2004, 2008). Therefore, we investigated whether the observed cell death after DMNQ-exposure, as described above, is apoptosis by studying nuclear morphology at an earlier time point than 24 h. First, a time-curve was established using fluorescence microscopy by nuclear staining with the DNA-selective dye Hoechst. As shown in Fig. 3a, treatment with DMNQ for 16 h resulted in an increased percentage of condensed nuclei in both the control cells (13.7%) and the Dex-treated cells (31.8%), compared to the 0 h time-point. Moreover, there is a significant difference in the percentage of condensed nuclei between the control cells and the Dex-treated cells after 16 h of DMNQ-exposure (P < 0.001). Results in Figs. 2 and 3 pointed to an increased toxicity of DMNQ in the Dex-treated group. Confocal microscopy was used to study apoptotic nuclear morphology via staining with the DNA-selective dye PI. Figure 3c, e shows that treatment with 10 μM DMNQ during 16 h resulted in condensed and fragmented nuclei, further indicating that DMNQ induced apoptosis in NSCs.

Time-dependent effect of DMNQ on nuclear morphology. Control and Dex-treated cells were cultured at a density of 7.2 × 104 cells per poly-l-lysine-coated glass coverslip and allowed to adhere before being exposed to 10 μM DMNQ during 0–16 h. a Afterward, cells were stained with Hoechst and immediately viewed using an Olympus BX60 fluorescence microscope. Cells were counted in 4 microscopic fields, randomly selected on each coverslip. Statistical analysis was performed via a two-way ANOVA. Bars represent mean + SEM of one representative experiment performed in triplicate. * P < 0.001. Confocal images of control and Dex-treated cells unexposed (b, d) or exposed to DMNQ for 16 h (c, e). Following treatment, cells were fixed with 80% (v/v) methanol and stained with PI. Apoptotic nuclei were detected as condensed (arrows) and fragmented nuclei (arrowhead). Cells were viewed using a Zeiss LSM 510 Meta confocal microscope equipped with the 60× objective
DMNQ-Induced Cell Death is Associated with Caspase Activation, Which is Altered by Dexamethasone
Caspase 3 is a key executioner protein that can cause cleavage of DNA and DNA repair proteins, including PARP, during apoptosis (Ashe and Berry 2003; Jin and El Deiry 2005; Lakhani et al. 2006; Scharstuhl et al. 2009). Therefore, we investigated whether this executioner protein was involved in DMNQ-induced cell death by studying protein expression. Following induction of apoptosis, precursor caspase 3 (32 kD) is cleaved into a 17-kD (p17) and a 11-kD (p11) subunit, which are needed to form the activated caspase 3 protein (a heterodimer composed of two p17 and two p11 subunits). After treatment with 10 μM DMNQ for 24 h, precursor caspase 3 (32 kD) expression was absent in both control cells and Dex-treated cells (Fig. 4a), suggesting caspase 3 activation. In addition, exposure to DMNQ induced cleavage of full-length PARP (116 kD) into a specific fragment (89 kD) in both the control and Dex-treated group (Fig. 4b). These results imply that DMNQ-induced apoptosis is caspase-mediated.

DMNQ induces cell death via an apoptotic mechanism. Control and Dex-exposed cells were cultured and treated with 10 μM DMNQ for 24 h. Proteins, obtained through lysis, were separated via SDS/PAGE using a 12% (w/v) gel and were blotted onto nitrocellulose membranes. Rabbit-α-caspase 3 or rabbit-α-PARP and mouse-α-β-actin were used as the primary antibodies. Goat-α-mouse Alexa Fluor 680 and goat-α-rabbit IRDye 800 were used as the secondary antibodies. The membrane was scanned using the Odyssey infrared imaging system (LI-COR Biotechnology) using channels 700 and 800. a Membrane showing precursor caspase 3 (32 kD) and β-actin (42 kD). b Membrane showing PARP (116 kD), cleavage fragment (89 kD) and β-actin (42 kD). c Following treatment for 24 h, supernatant was collected, and cells were harvested using a cell scraper. Next, DEVD-MCA was added, and cleavage of the fluorogenic substrate was measured using a fluorometric assay. Statistical analysis was performed via a one-way ANOVA followed by a Bonferroni’s multiple comparison test. Bars represent mean + SEM of one representative experiment performed in triplicate. * P < 0.0001
As previously mentioned, it has been suggested that Dex can modulate apoptotic cell death. As shown in Fig. 4c, Dex significantly increased DMNQ-induced caspase activity at 24 h with 180%, compared to control cells (P < 0.0001). Furthermore, there is no difference in caspase activity between control cells and Dex-treated cells in the absence of DMNQ-exposure. These results suggest that Dex can modify the apoptotic machinery in NSCs.
Dexamethasone Induces a G1-Arrest in NSCs
It is well established that Dex can induce cell cycle alterations (Sundberg et al. 2006, Moors et al. 2011). We investigated whether the same effect could be observed in our model. Microscopy revealed that the number of cells is lower after Dex-exposure, compared to the control (Figs. 2c, 3d). These results suggest that Dex decreased cell proliferation; therefore, the cell cycle was studied in more detail using flow cytometry. As shown in Fig. 5, treatment with Dex significantly increased the percentage of cells in the G1-phase from 63 to 84%, compared to control cells (P < 0.001). In addition, the percentage of cells in the G2/M-phase significantly decreased from 20 to 2% following Dex treatment compared to the control cells (P < 0.001). Thus, also in murine-derived NSCs, Dex is able to induce changes in the cell cycle process.

Impact of dexamethasone on the cell cycle. Control and Dex-exposed cells were cultured and allowed to grow for 48 h. Afterward, cells were collected, fixed overnight with ice cold 99.5% (v/v) ethanol and stained with PI. Quantification of PI binding was done with a flow cytometer using channel FL-3. Representative histograms of control (a) or Dex-treated (b) cells. Cellquest Pro was used to perform histogram analysis (c). Statistical analysis was performed via a two-way ANOVA followed by Bonferroni’s post-tests. Bars represent mean + SEM of minimal four experiments. * P < 0.001
Dexamethasone Alters Gene Expression
We have shown that Dex increased the susceptibility of NSCs to oxidative stress. However, the intracellular mechanism remains unclear. Therefore, we studied the effect of Dex on genes that are known to be targets of GCs and are involved in the mitochondrial respiratory chain, protection against oxidative stress and reactive oxygen species (ROS) metabolism (Bose et al. 2010; Datson et al. 2008). Table 1 shows that 86% of the selected genes involved in the mitochondrial respiratory chain are modulated after treatment with Dex. Moreover, 72% of these genes are down-regulated, viz.: NADH dehydrogenase (ND)1, ND4, cytochrome b (Cyt B), cyclooxygenase (Cox)-2 and Cox-3, whereas Cox-1 gene expression is up-regulated, compared to the control cells. Furthermore, 43% of the genes encoding for antioxidant enzymes show an altered expression. Catalase and superoxide dismutase (SOD)1 are markedly down-regulated, and uncoupling protein 3 is up-regulated, compared to the control cells. Additionally, Table 1 shows that thioredoxin-interacting protein, one of the genes involved in ROS metabolism, is down-regulated. These results suggest that Dex modulates the expression of mitochondrial encoded genes, possibly resulting in functional changes of the mitochondrial respiratory chain.
Discussion
GCs are essential for normal brain development; however, there is consistent evidence that exposure of the fetal brain to excess GCs can have life-long effects on the nervous system. Our group previously demonstrated, using an in vitro/ex vivo model, that prenatal exposure to high levels of GCs permanently modified the phenotype of neuronal cells and increased the susceptibility to oxidative stress; however, the mechanism remains unclear (Bose et al. 2010). Therefore, the aim of the present study was to investigate the impact of Dex on murine NSCs and to elucidate the intracellular events leading to neural damage in more detail.
To validate our in vitro model, we tried to reproduce effects that have been described in the literature after prenatal exposure to Dex and after direct Dex-exposure. Ahlbom et al. injected pregnant rats with 0.1 mg/kg Dex daily, from day 14 post conception. Subsequently, primary cultures of cerebellar granule cells (CGCs) were prepared from pups exposed to Dex. Confocal microscopy revealed that exposure of Dex-treated CGCs to the ROS-inducers, hydrogen peroxide and methyl mercury, resulted in a significantly higher number of cells with apoptotic morphology, compared to control cells (Ahlbom et al. 2000). Furthermore, it has been described by Canlon et al. (2003) that prenatal exposure to Dex decreased the potential of the cochlea to recover from oxidative stress induced by acoustic trauma. In this study, we have shown, using several techniques, that a single exposure to 50 μM Dex increased the sensitivity of NSCs to oxidative stress induced by DMNQ.
Our results show that exposure to Dex resulted in a decreased cell number and induced a G1-arrest. Previously, Sundberg et al. reported that treatment of embryonic rat NSCs with 1 μM Dex resulted in a significant decrease in cell proliferation, accompanied by a decrease in cyclin D1 (Sundberg et al. 2006). In addition, Bose et al. reported that exposure of primary rat embryonic NSCs to Dex caused a significant up-regulation of the cell cycle regulating genes p16 and p21, which are associated with cellular senescence (Bose et al. 2010). Moreover, this effect could be abolished via pre-treatment with mifepristone, indicating a GR-mediated mechanism.
The concentration of Dex used in this study is higher compared to previous studies in which glucocorticoid concentrations ranging from 0.1 to 10 μM are demonstrated to enhance oxidative stress-induced neural cell death (Behl et al. 1997). Yet, Budni et al. (2011) previously reported that it was necessary to expose the human neuroblastoma SH-SY5Y cell line to 1 mM of Dex to induce a 40% reduction in cell viability. The same group also demonstrated that 50 μM of Dex did not induce cell death, which is in agreement with our findings. Furthermore, Tamm et al. (2006) previously described that C17.2 cells were less sensitive to methylmercury-induced neurotoxicity compared to primary rat embryonic cortical NSCs. Taken together, sensitivity to neurotoxicants varies between different neural cell models, and more research is needed to fully characterize the action of glucocorticoids in C17.2 cells.
Cell death can occur via an intracellular program (i.e. apoptosis) or as an acute response to stress (i.e. necrosis) (Jin and El Deiry 2005). Here, we report that, in NSCs, treatment with the ROS-inducer DMNQ resulted in nuclear condensation, a hallmark of apoptosis. Furthermore, our results indicated that exposure to DMNQ leads to activation of caspase 3 and is accompanied by cleavage of the DNA repair protein PARP. These results strongly suggest that DNMQ induces apoptosis in NSCs. Our observations are in line with the study of Tamm et al. (2004) showing that treatment of NSCs with DMNQ results in cytochrome c release, activation of caspase-3 and -9 and chromatin condensation.
Our findings revealed that Dex by itself does not induce cell death in NSCs, which is in line with previous studies (Bose et al. 2010). In contrast, it is widely known that Dex can induce cell death in cancer cells, and GCs are generally used in the treatment of malignant tumors (Budni et al. 2011; Laane et al. 2009). In the study by Laane et al.(2009), Dex-induced cell death was studied in acute lymphoblastic leukemia cells, and they demonstrated that Dex initiated autophagy prior to the onset of apoptosis. Following 24 h exposure to 100 nM Dex, autophagosomes became apparent, while the nuclues and cell morphology remained unaffected. After 36 h, Dex treatment resulted in the exposure of phosphatidylserine on the plasma membrane (Annexin V-positivity) and caspase 3 activity (Laane et al. 2009). Our results demonstrated that Dex enhanced DMNQ-induced caspase 3 activity in NSCs, indicating an altered apoptotic machinery. Thus, it is apparent that Dex can be involved in either the initiation or modulation of cell death processes depending on cell type.
Using our in vitro model, we investigated the intracellular mechanism of Dex-mediated increase in susceptibility to ROS in more detail, focusing on genes related to mitochondrial functions and antioxidant defense. In this study, we demonstrated that Dex mainly decreased the gene expression of mitochondrial-encoded respiratory chain enzymes. These results are in agreement with the study by Bose and coworkers, demonstrating that exposure of primary rat embryonic NSCs to Dex resulted in a decreased expression of ND3 and Cyt B (Bose et al. 2010). Ahlbom et al. (2000) described that, in CGCs prenatally exposed to Dex, the rate of mitochondrial Ca2+ uptake was markedly decreased. Furthermore, they showed a decrease in mitochondrial oxygen consumption in Dex-exposed CGCs, which was accompanied by a decrease in ATP levels. Moreover, Desquiret et al. described that, in HepG2 cells, already after 10 min of Dex treatment, the activity of complex (Com) I and II of the mitochondrial respiratory chain was decreased. Furthermore, they showed that after treatment with Dex for 8 h, Com I and II activity remained lowered. On the other hand, complex IV activity increased (Desquiret et al. 2008). In addition, Desquiret et al. (2008) showed that treatment with Dex for 8 h decreased mitochondrial oxygen consumption, suggesting a decrease in ATP-production. Thus, Dex modifies mitochondrial functioning, which might be the pathophysiological pathway underlying the observed increase in susceptibility to DMNQ following Dex exposure.
As described above, the mitochondria seem to be the main target of Dex. However, it is unclear whether Dex acts on the mitochondria via a genomic (i.e. receptor-mediated) or non-genomic (i.e. direct) pathway. We have shown that treatment with the GR-antagonist mifepristone completely attenuates Dex-induced increase in susceptibility to DMNQ, strongly suggesting that Dex affects the mitochondria via a genomic mechanism. Additionally, it has been described that the GR is present in mitochondria in several cell types, further supporting our hypothesis (Desquiret et al. 2008; Psarra et al. 2006; Scheller et al. 2000).
Antioxidant enzymes are essential for the protection of cells to ROS (Mates et al. 1999). Therefore, we investigated whether the increased susceptibility to DMNQ of Dex-treated cells was due to decreased antioxidant defenses. Our results show that treatment with Dex resulted in a decreased gene expression of the antioxidant enzymes catalase and SOD1, possibly suggesting a decreased activity of these enzymes. These results are in agreement with the study of Ahlbom et al. (2000), which demonstrated that prenatal exposure of CGCs to Dex resulted in a decrease of catalase activity. Thus, next to mitochondrial dysfunctioning, a decreased antioxidant defense also renders GC-exposed cells more prone to oxidative stress.
We observed that a single treatment with Dex has lasting effect on NSCs, suggesting that Dex alters the methylation status of target genes resulting in epigenetic changes. This hypothesis is supported by previous work from our laboratory demonstrating that, in primary cultures of rat embryonic NSCs, treatment with Dex resulted in a decreased global DNA methylation concurrent with a decreased expression of DNA methyltransferases (Bose et al. 2010). Further research is needed to fully elucidate which genes are targeted by Dex-mediated epigenetic reprogramming.
The present study is based on the premise that glucocorticoid activity is similar in an in vitro model compared to the in vivo situation. In a recent meta-analysis, studying the relationship between GCs and oxidative stress, Costantini et al. (2011) reported that there is a positive correlation between the duration of glucocorticoid treatment and the induction of oxidative stress. Furthermore, it was demonstrated that the brain is the main target of GCs. These findings corroborate our results demonstrating that a short exposure of murine NSCs to Dex for 24 h did not affect cell viability, but highly influenced the expression of a myriad of genes involved in redox physiology. Costantini et al. (2011) also noted that Dex may not be a perfect substitute to mimic high GCs levels, due to different affinities or kinetics with the mineralcorticoid and glucocorticoid receptor than natural GCs. Thus, although similar effects of GCs are obtained in the C17.2 model compared to in vivo models, caution is always needed when extrapolating results between species.
Taken together, we report that a single exposure to Dex lastingly increased the susceptibility of murine NSCs to DMNQ-induced apoptosis via activation of the GR and decreased cell proliferation, probably via the induction of a G1-arrest. Furthermore, we demonstrated that Dex alters the gene expression of multiple mitochondrial encoded respiratory chain enzymes and several antioxidant enzymes. Thus, we have established a novel and valid in vitro model that can be used to study the effects of prenatal exposure to Dex on NSCs. Moreover, this model can be used as an alternative for the in vitro/ex vivo model that is normally used to study the effects of intrauterine exposure to chemicals, and thereby decreasing the number of laboratory animals used in developmental toxicity studies.
References
Ahlbom E, Gogvadze V, Chen M, Celsi G, Ceccatelli S (2000) Prenatal exposure to high levels of glucocorticoids increases the susceptibility of cerebellar granule cells to oxidative stress-induced cell death. Proc Natl Acad Sci USA 97:14726–14730
Ashe PC, Berry MD (2003) Apoptotic signaling cascades. Prog Neuropsychopharmacol Biol Psychiatry 27:199–214
Barker DJ (1995) Intrauterine programming of adult disease. Mol Med Today 1:418–423
Barnes PJ (1998) Anti-inflammatory actions of glucocorticoids: molecular mechanisms. Clin Sci (Lond) 94:557–572
Behl C, Lezoualc’h F, Trapp T, Widmann M, Skutella T, Holsboer F (1997) Glucocorticoids enhance oxidative stress-induced cell death in hippocampal neurons in vitro. Endocrinology 138:101–106
Bose R, Moors M, Tofighi R, Cascante A, Hermanson O, Ceccatelli S (2010) Glucocorticoids induce long-lasting effects in neural stem cells resulting in senescence-related alterations. Cell Death Dis 1:e92
Budni J, Romero A, Molz S, Martin-de-Saavedra MD, Egea J, Del BL, Tasca CI, Rodrigues AL, Lopez MG (2011) Neurotoxicity induced by dexamethasone in the human neuroblastoma SH-SY5Y cell line can be prevented by folic acid. Neuroscience 190:346–353
Buhl ES, Neschen S, Yonemitsu S, Rossbacher J, Zhang D, Morino K, Flyvbjerg A, Perret P, Samuel V, Kim J, Cline GW, Petersen KF (2007) Increased hypothalamic–pituitary–adrenal axis activity and hepatic insulin resistance in low-birth-weight rats. Am J Physiol Endocrinol Metab 293:E1451–E1458
Canlon B, Erichsen S, Nemlander E, Chen M, Hossain A, Celsi G, Ceccatelli S (2003) Alterations in the intrauterine environment by glucocorticoids modifies the developmental programme of the auditory system. Eur J Neurosci 17:2035–2041
Ceccatelli S, Tamm C, Sleeper E, Orrenius S (2004) Neural stem cells and cell death. Toxicol Lett 149:59–66
Ceccatelli S, Tamm C, Zhang Q, Chen M (2007) Mechanisms and modulation of neural cell damage induced by oxidative stress. Physiol Behav 92:87–92
Cole TJ (2006) Glucocorticoid action and the development of selective glucocorticoid receptor ligands. Biotechnol Annu Rev 12:269–300
Costantini D, Marasco V, Moller AP (2011) A meta-analysis of glucocorticoids as modulators of oxidative stress in vertebrates. J Comp Physiol B 181:447–456
Czock D, Keller F, Rasche FM, Haussler U (2005) Pharmacokinetics and pharmacodynamics of systemically administered glucocorticoids. Clin Pharmacokinet 44:61–98
Dare E, Tofighi R, Vettori MV, Momoi T, Poli D, Saido TC, Mutti A, Ceccatelli S (2002) Styrene 7,8-oxide induces caspase activation and regular DNA fragmentation in neuronal cells. Brain Res 933:12–22
Datson NA, Morsink MC, Meijer OC, de Kloet ER (2008) Central corticosteroid actions: search for gene targets. Eur J Pharmacol 583:272–289
de Vries A, Holmes MC, Heijnis A, Seier JV, Heerden J, Louw J, Wolfe-Coote S, Meaney MJ, Levitt NS, Seckl JR (2007) Prenatal dexamethasone exposure induces changes in nonhuman primate offspring cardiometabolic and hypothalamic–pituitary–adrenal axis function. J Clin Invest 117:1058–1067
Desquiret V, Gueguen N, Malthiery Y, Ritz P, Simard G (2008) Mitochondrial effects of dexamethasone imply both membrane and cytosolic-initiated pathways in HepG2 cells. Int J Biochem Cell Biol 40:1629–1641
Fowden AL, Forhead AJ (2004) Endocrine mechanisms of intrauterine programming. Reproduction 127:515–526
Gant TW, Rao DN, Mason RP, Cohen GM (1988) Redox cycling and sulphydryl arylation; their relative importance in the mechanism of quinone cytotoxicity to isolated hepatocytes. Chem Biol Interact 65:157–173
Hauser J, Knapman A, Zurcher NR, Pilloud S, Maier C, Diaz-Heijtz R, Forssberg H, Dettling A, Feldon J, Pryce CR (2008) Effects of prenatal dexamethasone treatment on physical growth, pituitary–adrenal hormones, and performance of motor, motivational and cognitive tasks in juvenile and adolescent common marmoset monkeys. Endocrinology 149:6343–6355
Heikinheimo O, Kontula K, Croxatto H, Spitz I, Luukkainen T, Lahteenmaki P (1987) Plasma concentrations and receptor binding of RU 486 and its metabolites in humans. J Steroid Biochem 26:279–284
Henry TR, Wallace KB (1995) The role of redox cycling versus arylation in quinone-induced mitochondrial dysfunction: a mechanistic approach in classifying reactive toxicants. SAR QSAR Environ Res 4:97–108
Holmes MC, Abrahamsen CT, French KL, Paterson JM, Mullins JJ, Seckl JR (2006) The mother or the fetus? 11beta-hydroxysteroid dehydrogenase type 2 null mice provide evidence for direct fetal programming of behavior by endogenous glucocorticoids. J Neurosci 26:3840–3844
Jin Z, El Deiry WS (2005) Overview of cell death signaling pathways. Cancer Biol Ther 4:139–163
Kapoor A, Dunn E, Kostaki A, Andrews MH, Matthews SG (2006) Fetal programming of hypothalamo–pituitary–adrenal function: prenatal stress and glucocorticoids. J Physiol 572:31–44
Kawata M (1995) Roles of steroid hormones and their receptors in structural organization in the nervous system. Neurosci Res 24:1–46
Laane E, Tamm KP, Buentke E, Ito K, Kharaziha P, Oscarsson J, Corcoran M, Bjorklund AC, Hultenby K, Lundin J, Heyman M, Soderhall S, Mazur J, Porwit A, Pandolfi PP, Zhivotovsky B, Panaretakis T, Grander D (2009) Cell death induced by dexamethasone in lymphoid leukemia is mediated through initiation of autophagy. Cell Death Differ 16:1018–1029
Lakhani SA, Masud A, Kuida K, Porter GA Jr, Booth CJ, Mehal WZ, Inayat I, Flavell RA (2006) Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science 311:847–851
Mates JM, Perez-Gomez C, Nunez dC I (1999) Antioxidant enzymes and human diseases. Clin Biochem 32:595–603
MohanKumar SM, King A, Shin AC, Sirivelu MP, MohanKumar PS, Fink GD (2007) Developmental programming of cardiovascular disorders: focus on hypertension. Rev Endocr Metab Disord 8:115–125
Moors M, Bose R, Johansson-Haque K, Edoff K, Okret S, Ceccatelli S (2011) Dickkopf 1 mediates glucocorticoid-induced changes in human neural progenitor cell proliferation and differentiation. Toxicol Sci. doi: 10.1093/toxsci/kfr304
Moritz KM, Boon WM, Wintour EM (2005) Glucocorticoid programming of adult disease. Cell Tissue Res 322:81–88
Noguchi KK, Walls KC, Wozniak DF, Olney JW, Roth KA, Farber NB (2008) Acute neonatal glucocorticoid exposure produces selective and rapid cerebellar neural progenitor cell apoptotic death. Cell Death Differ 15:1582–1592
Psarra AM, Solakidi S, Sekeris CE (2006) The mitochondrion as a primary site of action of steroid and thyroid hormones: presence and action of steroid and thyroid hormone receptors in mitochondria of animal cells. Mol Cell Endocrinol 246:21–33
Roberts D, Dalziel S (2006) Antenatal corticosteroids for accelerating fetal lung maturation for women at risk of preterm birth. Cochrane Database Syst Rev 3:CD004454
Sandi C (2004) Stress, cognitive impairment and cell adhesion molecules. Nat Rev Neurosci 5:917–930
Sapolsky RM (1985) Glucocorticoid toxicity in the hippocampus: temporal aspects of neuronal vulnerability. Brain Res 359:300–305
Sapolsky RM (1996) Why stress is bad for your brain. Science 273:749–750
Sapolsky RM (1999) Glucocorticoids, stress, and their adverse neurological effects: relevance to aging. Exp Gerontol 34:721–732
Scharstuhl A, Mutsaers HA, Pennings SW, Szarek WA, Russel FG, Wagener FA (2009) Curcumin-induced fibroblast apoptosis and in vitro wound contraction are regulated by antioxidants and heme oxygenase: implications for scar formation. J Cell Mol Med 13:712–725
Scheller K, Sekeris CE, Krohne G, Hock R, Hansen IA, Scheer U (2000) Localization of glucocorticoid hormone receptors in mitochondria of human cells. Eur J Cell Biol 79:299–307
Seckl JR (2001) Glucocorticoid programming of the fetus; adult phenotypes and molecular mechanisms. Mol Cell Endocrinol 185:61–71
Seckl JR (2008) Glucocorticoids, developmental ‘programming’ and the risk of affective dysfunction. Prog Brain Res 167:17–34
Seckl JR, Holmes MC (2007) Mechanisms of disease: glucocorticoids, their placental metabolism and fetal ‘programming’ of adult pathophysiology. Nat Clin Pract Endocrinol Metab 3:479–488
Singh RR, Moritz KM, Bertram JF, Cullen-McEwen LA (2007) Effects of dexamethasone exposure on rat metanephric development: in vitro and in vivo studies. Am J Physiol Renal Physiol 293:F548–F554
Sundberg M, Savola S, Hienola A, Korhonen L, Lindholm D (2006) Glucocorticoid hormones decrease proliferation of embryonic neural stem cells through ubiquitin-mediated degradation of cyclin D1. J Neurosci 26:5402–5410
Tamm C, Robertson JD, Sleeper E, Enoksson M, Emgard M, Orrenius S, Ceccatelli S (2004) Differential regulation of the mitochondrial and death receptor pathways in neural stem cells. Eur J Neurosci 19:2613–2621
Tamm C, Duckworth J, Hermanson O, Ceccatelli S (2006) High susceptibility of neural stem cells to methylmercury toxicity: effects on cell survival and neuronal differentiation. J Neurochem 97:69–78
Tamm C, Zhivotovsky B, Ceccatelli S (2008) Caspase-2 activation in neural stem cells undergoing oxidative stress-induced apoptosis. Apoptosis 13:354–363
Tchivilev I, Madamanchi NR, Vendrov AE, Niu XL, Runge MS (2008) Identification of a protective role for protein phosphatase 1cgamma1 against oxidative stress-induced vascular smooth muscle cell apoptosis. J Biol Chem 283:22193–22205
Uno H, Eisele S, Sakai A, Shelton S, Baker E, DeJesus O, Holden J (1994) Neurotoxicity of glucocorticoids in the primate brain. Horm Behav 28:336–348
Welberg LA, Seckl JR, Holmes MC (2000) Inhibition of 11beta-hydroxysteroid dehydrogenase, the foeto-placental barrier to maternal glucocorticoids, permanently programs amygdala GR mRNA expression and anxiety-like behaviour in the offspring. Eur J Neurosci 12:1047–1054
Wilcoxon JS, Redei EE (2007) Maternal glucocorticoid deficit affects hypothalamic–pituitary–adrenal function and behavior of rat offspring. Horm Behav 51:321–327
Acknowledgments
This work was supported by the Netherlands Brain Foundation, the Swedish Research Council (10815) and Karolinska Institutet. The authors would like to thank Dr.Sandra Ceccatelli for fruitful discussions and careful reading of the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Mutsaers, H.A.M., Tofighi, R. Dexamethasone Enhances Oxidative Stress-Induced Cell Death in Murine Neural Stem Cells. Neurotox Res 22, 127–137 (2012). https://doi.org/10.1007/s12640-012-9308-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-012-9308-9