
Abstract
Different animal and human studies from last two decades in the case of Parkinson’s disease (PD) have concentrated on oxidative stress due to increased inflammation and cytokine-dependent neurotoxicity leading to induction of dopaminergic (DA) degeneration pathway in the nigrostriatal region. Chronic inflammation, the principle hallmark of PD, forms the basis of neurodegeneration. Aging in association with activation of glia due to neuronal injury, perhaps because of immune alterations and genetic predispositions, leads to deregulation of inflammatory pathways premising the onset of PD. A family of inducible transcription factors, nuclear factor-κB (NF-κB), is found to show expression in various cells and tissues, such as microglia, neurons, and astrocytes which play an important role in activation and regulation of inflammatory intermediates during inflammation. Both canonical and non-canonical NF-κB pathways are involved in the regulation of the stimulated cells. During the prodromal/asymptomatic stage of age-associated neurodegenerative diseases (i.e., PD and AD), chronic neuroinflammation may act silently as the driver of neuronal dysfunction. Though research has provided an insight over age-related neurodegeneration in PD, elaborative role of NF-κB in neuroinflammation is yet to be completely understood and thus requires more investigation. Polyphenols, a group of naturally occurring compound in medicinal plants, have gained attention because of their anti-oxidative and anti-neuroinflammatory properties in neurodegenerative diseases. In this aspect, this review highlights the role of NF-κB and the possible therapeutic roles of polyphenols in NF-κB-mediated neuroinflammation in PD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
About 2% of the human population over 65 years of age are affected by Parkinson’s disease (PD), which is the second most common neurodegenerative disorder (Nuytemans et al. 2010). PD is characterized by the selective loss of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNpc) and reduction in the DA levels within the corpus striatum of the brain. Due to the loss of DA regulation in the basal ganglia, circuitries get disturbed which results into motor symptoms such as postural (Aggarwal and Shishodia 2004) instability, bradykinesia, rigidity, and resting tremor along with some non-motor symptoms such as cognitive deficits, depression, and sleep disturbance (Rodriguez-Oroz et al. 2009). During the pathogenesis of the disease, loss of about 50–70% of the neurons takes place which further worsens the disease condition. Exact etiology of PD is still indefinable and the mechanism behind its pathogenesis is needed to be identified (Adler et al. 2007; Poewe 2008; Rodriguez et al. 2010). Several experimental evidences have convinced the researchers about the crucial role of oxidative stress, mitochondrial dysfunction, and inflammation in the initiation and progression of PD (Luo et al. 2015; Tansey and Goldberg 2010). Currently available treatments are only helpful in managing fewer symptoms and show only sub-optimal efficacy related to the duration of the disease. Thus, an efficacious neuroprotective or disease-modifying treatment is still needed in order to deal with the worsening symptoms of PD (Goetz and Pal 2014; Pilleri and Antonini 2015). As chronic neuroinflammation has been found to play a critical role in the PD pathogenesis; thus, the neuroinflammatory signaling pathways in the central nervous system (CNS) are of interest as potential pharmacotherapy targets (More et al. 2013; Russo et al. 2014; Zhang et al. 2012a). Activated glial cells, mainly microglia and astrocytes are the key player in the progression of neuroinflammation. They play important role in initiating the immune response against damage and produce inflammatory mediators (Block et al. 2007). In different PD models such as 6-hydroxydopamine (OHDA), 1-methyl-4-phenylpyridinium (MPTP), and rotenone-induced PD animal models, increased activation of microglia has been found in the substantia nigra (SN) of the brain (Kitamura et al. 1994; Kurkowska-Jastrzębska et al. 1999). Numerous pro-inflammatory cytokines are also produced by abruptly activated astrocytes and microglia, contributing in making the pathobiology of PD more complex (Cunningham and Su 2002; Marchetti et al. 2013; Waak et al. 2009). Nuclear factor-κB (NF-κB) plays vital role in making milieu of neuroinflammation by regulating the expression of genes that encode inducible nitric oxide synthase, chemokines (IL-8, macrophage inflammatory protein [MIP]-1α, monocyte chemo-attractant protein [MCP]-1) (Lan et al. 2011; Qian et al. 2015), pro-inflammatory cytokines (IL-1β, TNF-α, IL-12/23), subunits p47 and p67 of NADPH oxidase and cell adhesion molecules, intercellular adhesion molecule [ICAM]-1, vascular cell adhesion molecule [VCAM], and E-selectin (Chen and Manning 1995; Gauss et al. 2007; Tak and Firestein 2001). It is evident through various studies that numerous polyphenolic flavonoids and non-flavonoids found in nature possess anti-inflammatory properties thus illustrate beneficial effects in preventing neurodegeneration mediated by inflammatory damage (Ebrahimi and Schluesener 2012). Besides staging anti-oxidant property, various polyphenolic compounds are observed to alter diverse signaling pathways by targeting specific molecules that yield multiple cellular effects. Anti-inflammatory effects through inhibition of NF-κB pathway by these polyphenols or their derivative compounds is one such example. Hence, it might be suggested that NF-κB activity can be targeted to have a control on the chronic inflammation and its inhibition in glial cells may offer an effective treatment of PD.
Oxidative Stress Mediated Neuroinflammation in PD
Inflammation normally helps the individual to show natural defense mechanism against invading pathogens linked with different pathogenic diseases (viral and microbial), allergens exposure, obesity, tobacco consumption, alcohol abuse, chronic autoimmune diseases, and exposure to hazardous chemicals or radiations. Studies have also shown that oxidative stress is closely associated to chronic inflammatory diseases. Higher production of reactive oxygen species (ROS) leads to oxidation of various biomolecules causing the accumulation of oxidized proteins and lipid peroxides, resulting in the degeneration of neurons (Berlett and Stadtman 1997).
DA can also act as a source of oxidative stress during degeneration of the DA neurons in SNpc (Segura-Aguilar et al. 2014). Synthesis of DA takes place from tyrosine by the enzymes namely tyrosine hydroxylase (TH) and aromatic amino acid decarboxylase. Synthesized DA is then taken up by the vesicular monoamine transporter-2 (VMAT2) and stored within the synaptic vesicles. However, after the L-DOPA treatment, excess DA found in the cytosol of damaged neurons is either metabolized by monoamine oxidase (MAO) or is auto-oxidized by cytosolic ROS (Zucca et al. 2014). The excess amount of production of DA in normal condition as well as after the L-DOPA treatment is required to maintain the synthesis and accumulation of neuromelanin in the autolysosomes which further averts the formation of neurotoxic DA quinones and protects the neurons of SN from iron-induced oxidative stress (Monzani et al. 2019; Sulzer et al. 2018). DA-mediated toxicity was found to be associated with lowered VMAT2 expression, resulting in loss of DA neurons (Fig. 1) (Caudle et al. 2007).

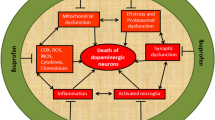
Oxidative stress and neuroinflammation in PD. Generation of oxidative stress and neuroinflammation mediated neurodegeneration of dopaminergic (DA) neurons. Degenerating neurons along with auto-oxidation of dopamine generated dopamine metabolites like DOPAC, HVA, and dopamine quinines create reactive oxygen species (ROS). Mutation in PD associated genes like α-synuclein, parkin, PINK1, and DJ-1 inhibits the complex I and complex III of mitochondrial electron transport chain and triggers the formation of ROS which further creates the oxidative stress inside the cell; neuroinflammation is the downstream event of oxidative stress. This oxidative stress causes the overactivation of microglia and astrocytes; these cells are the mediator of neuroinflammation and ultimately cause the degeneration of the DA neurons
Increased ROS production is seen to be closely related to mitochondrial dysfunction in PD (Schapira 2008). Major unfavorable neuronal apoptosis is due to complex I deficiencies of the electron transport system and acts as the primary source of ROS generation in PD (Fig. 1). Complex I activity has been seen to be reduced in sporadic PD cases (Hattingen et al. 2009; Hattori et al. 1991; Schapira et al. 1990). Moreover, in fibroblast (Mytilineou et al. 1994), skeletal muscle (Blin et al. 1994), lymphocytes (Haas et al. 1995; Yoshino et al. 1992), blood platelets (Taylor et al. 1994), and different brain regions (Blandini and Greenamyre 1998; Mizuno et al. 1989; Parker et al. 2008), mitochondrial complex I deficiency was found in different PD patients. Also, complex I inhibitors like 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) or rotenone are found to be toxic to the DA neurons (Blesa and Przedborski 2014). Mechanism of MPTP oxidation to1-methyl-4-phenylpyridinium (MPP+) and its transfer across the blood–brain barrier has been well studied (Blesa and Przedborski 2014). Mitochondrial dysfunction and oxidative damage has been found to be linked with familial forms of PD as seen through the mutations in genes encoding DJ-1, PINK, parkin, or α-synuclein. The relationship of all these proteins with mitochondrial dynamics reveal their roles in mediating the mitochondrial dysfunction and resulting oxidative stress response which might help us in understanding the pathophysiology of PD (Fig. 1) (Norris et al. 2015; van der Merwe et al. 2015; Zuo and Motherwell 2013).
Signaling processes might also get affected due to the production of increased reactive radical species and as a result cell-damaging pathways get stimulated such as increased expression of pro-inflammatory genes, senescence, and apoptosis. Researchers have shown the active involvement of this inflammatory cascade during aging and the pathology is typical of age-linked neurodegenerative disorders, particularly PD (Vitale et al. 2013). Thus, both oxidative stress and inflammation are simultaneously responsible for maintaining the pathogenesis of PD (Harman 1992). ROS has been seen to activate the transcription factors NF-κB and AP-1 responsible for mediating inflammation directly or indirectly (Harman 2006). Notably, transcription factors such as NF-κB are equally responsible for mediating the pro-survival and anti-oxidant cellular response. However, the increase and decrease in level of anti-oxidant response during aging are still under study but the level of ROS is particularly found to be increased during aging, thus disturbing the redox balance (Vitale et al. 2013). Inflammatory pathways gets activated because of oxidative stress due to reduced level of anti-oxidants and primarily NF-κB-mediated pathway was seen to be activated in animal model of PD (Lee et al. 2010b). Hence, elevated levels of IL-1β, IL-6, and TNF-α were found due to resulting activation of microglia and astrocytes (Lee et al. 2010b).
Glial Cells and Neuroinflammation in PD
Chronic neuroinflammation, which is primarily controlled by microglia, by the resident immune cells of the brain, and to a lesser extent by astrocytes and oligodendrocytes, is responsible for the loss of neurons in the case of PD (Blesa et al. 2015). Both familial and sporadic patients were having abruptly increased microglial activation in SNpc and olfactory bulb (Doorn et al. 2014a, 2014b; Lawson et al. 1990). Also, microglial activation has been seen to be linked with genes or proteins associated with PD pathogenesis such as α-synuclein or LRRK2 in SNpc and striatum of PD animal models (Daher et al. 2014; Pisanu et al. 2014; Sacino et al. 2014; Stott and Barker 2014). Microglia-derived pro-inflammatory cytokines, TNF-α, IL-6, IL-1β, and pro-oxidant NOS expressions were seen to be elevated in putamen, cerebrospinal fluid (CSF), SN, and serum of PD patients, thus indicating the role of microglia in the induction of pro-inflammatory and pro-oxidant effects (Brown and Neher 2010; Knott et al. 2000). Consequently, ROS including superoxide radicals and hydrogen peroxides are released, known as respiratory burst. The resultant inflammatory mediators produced due to microglial activation such as interleukin-1β (IL-1β, cathepsin-B) (Arai et al. 2004; Mogi et al. 1994) and glutamate (Caudle and Zhang 2009; Choi 1988). ROS, nitric oxide (NO), tumor necrosis factor-α (TNF-α), and IL-8 (Thirumangalakudi et al. 2007) have actively triggered apoptosis in neuronal cell cultures. Moreover, many of these inflammatory mediators (NO, IL-6, TNF-α, PGE2, IL-1β) were found to be in highly elevated level in SNpc tissue of post mortem PD patients as well as in CSF of PD patients (Imamura et al. 2003). Interestingly, activation of NF-κB is necessary for microglial cells to produce these mediators. Microglial cells get activated on DA neuronal loss resulting from immune damage or by the effect of DA neurotoxins such as MPTP or 6-OHDA. Even after 1 year of MPTP administration, intranigral and/or plasma TNF-α levels were found to be elevated in MPTP-treated rodents or nonhuman primates (Barcia et al. 2005). One of the several interconnected pathways leading to PD pathogenesis involves the activation of microglia as a result of release of neuromelanin from the degenerating neurons which further leads to the death of neurons. This simultaneously stimulates the production of neuromelanin causing the self-activating cascade of neurodegeneration to begin (Zecca et al. 2008; Zhang et al. 2011). It is reported that an acute MPTP subjection can lead to continued neurodegenerative effect for a prolonged duration resulting from MPTP-induced parkinsonism even without any further exposure to the toxin (Langston et al. 1999). Progressive neurodegeneration in PD is induced by chronic neuroinflammation which sustains in the brain due to activation of microglia which release neurotoxic inflammatory mediators causing loss of DA neurons further resulting in reactive microgliosis (Venkateshappa et al. 2012). In this way, chronic nature of the disease is maintained with neuronal damage over a longer period of time. Hence, NF-κB inhibitors can be used to hinder the microglial activation at initial stages and thus to stop the reactive microgliosis, i.e., the major hallmark of chronic neuroinflammation in PD.
Another type of glial cells, i.e., astrocytes, also show important role in regulating PD’s neuroinflammation (Ben Haim et al. 2015). These cells show prominent function in both protecting and helping in survival of DA neurons, both by removing toxic metabolites from extracellular space and by releasing the anti-oxidants (Mena and García de Yébenes 2008). But, a study (Zhang and Barres 2010) has shown its role in the amplification of inflammatory responses produced by microglia (Chinta et al. 2013; Lee et al. 2010a). Also, under pathological conditions, these astrocytes have been seen to release pro-inflammatory cytokines. Recently, a study has shown the removal of extracellular α-synuclein released from neurons by astrocyte. The uptake and degradation of α-synuclein aggregates by the astrocytes conferring the protection of DA neurons, but as the capacity of astrocytes to degrade the aggregated α-synuclein gets exceeded, they get accumulated within astrocytes too. It was first reported in a study where a lysosomal inhibitor, bafilomycin A1, was shown to increase the formation of detergent-insoluble α-synuclein in astrocytes (Lee et al. 2010a). Also, due to this pathological condition, transcripts of inflammatory cytokines such as IL-1β, IL-1α and IL-6 were upregulated and release of TNF-α and IL-6 was seen (Doo et al. 2010). Thus, nigral death of DA neurons was found to be related with α-synuclein toxicity and presence of its aggregates within the astrocytes (Wakabayashi et al. 2000). Moreover, astrocytes in addition to the release of pro-inflammatory cytokines also get activated by cytokines such as TNF-α and IL-1β from microglia resulting in the production of reactive oxygen and nitrogen species. A recent study has suggested that the enhancement of microglial inflammatory responses by astrocytes is mediated by NF-κB-dependent pathway, resulting in more DA toxicity (Saijo et al. 2009).
NF-κB: Location in the Brain and Its Biology
The inflammatory mediators that majorly contribute in causing chronic inflammation and DA neuronal loss in PD show common feature of being regulated by NF-κB. NF-κB was first of all identified and described as a transcription factor by David Baltimore’s group in 1986, playing an essential role in the expression of mouse kappa light chain genes (Sen and Baltimore 1986b). Evidences have now suggested that NF-κB acts as a “master switch” for inflammatory gene expression (Tsoulfas and Geller 2001). The transcription factor NF-κB found in all nucleated cell types appears in multiple forms and consists of five proteins, p105/p50 (NF-κB1), p100/52 (NF-κB2), p65 (RelA), RelB, and c-Rel. These form distinct combinations of transcriptionally active homo- and heterodimeric complexes in mammals. Although, all these complexes have a common Rel homology domain (RHD) which is a conserved 300 amino acid long amino-terminal (Baldwin 1996). The RHD consists of sequences essential for interaction with IκB, nuclear translocation, and dimerization, where carboxy-terminal part of the RHD plays an important role, whereas the amino-terminal part of the RHD mediates specific DNA binding to the IκB consensus sequence present in regulatory elements of NF-κB target genes (5′ GGGPuNNPyPyCC-3′). The role of RHD in DNA binding is studied by the crystal structures of p50 homo- and p50/p65 heterodimers bound to DNA (Chen et al. 1998; Kaltschmidt and Kaltschmidt 2009; Müller et al. 1995). Glutamatergic neurons have been seen to show constitutive NF-κB expression. Glial cells have basal NF-κB activity but are highly inducible, playing decisive role in causing chronic inflammation in the brain (Kaltschmidt and Kaltschmidt 2009). NF-κB transcription factors express abundantly in the brain and their basal level is found to be higher in the peripheral tissues (Kaltschmidt et al. 1993, 1994, 1995). Complexes of c-Rel/p65, p50/p65 heterodimer, and p50 homodimers are among the members of NF-κB, seen in the developing rat brain (Bakalkin et al. 1993). Among all the complexes, inactive dimer combinations of p50 and p65 subunits are the most prevalent and are present in the cell nucleus and hence can be studied to see the distribution of NF-κB in neurons. Moreover, p50/p65 heterodimers have been found to show constitutive expression in the adult brain (Kaltschmidt et al. 2005; Meffert and Baltimore 2005), whereas the studies conducted on developed rodent brain reveal the transformation of p50/p65 heterodimers into the major κB-binding complex (Meffert and Baltimore 2005; Schmidt-Ullrich et al. 1996). NF-κB can be activated also by neuromelanin in microglia, via phosphorylation and degradation of the inhibitor protein κB inducing upregulation of tumor necrosis factor-α, interleukin-6, and nitric oxide (Wilms et al. 2003). Studies also suggest that the constitutive expression of NF-κB is controlled by physiological basal synaptic transmission. Although, the inducible NF-κB is found to be present in synapses, retrograde transport of p65 proteins activated by glutamatergic stimulation from synapses to the cell nucleus (Kaltschmidt et al. 1993; Meberg et al. 1996; Meffert et al. 2003). Thus, changes in the gene expression can persist by the translation of short-lasting synaptic signals by NF-κB (Meffert et al. 2003). Effect of NF-κB is seen on almost all cell types present in body and it plays significant role in immune responses, cell cycle, inflammation, and cell survival pathways (Kaltschmidt et al. 2005; Ledoux and Perkins 2014; Li and Verma 2002; Mattson 2005; Sen and Baltimore 1986a). p50 and p65 subunits are the heterodimer of NF-κB, known to be the potent activator of gene transcription (Schmitz and Baeuerle 1991). The transcription factor NF-κB has been studied to be activated in response to various external agents including lipopolysaccharide (LPS), inflammatory stimuli, UV light, free radicals, carcinogens, tumor promoters, cigarette smoke, cytokines, and various mitogens (Baeuerle and Henkel 1994; Baldwin 1996). On activation, NF-κB plays an important role in regulating numerous different genes, such as cell cycle regulatory molecules, enzymes like 5-lipoxygenase (LOX), inducible NO synthase (iNOS), and cyclooxygenase (COX)-2, adhesion molecules, angiogenic factors, cytokines including chemokines, interleukin (IL)-1, IL-6, IL-8, and tumor necrosis factor (TNF) (Duh et al. 1989; Gupta et al. 2010a; Gupta et al. 2010b; Kaltschmidt et al. 1993; Wang et al. 2005). Various human diseases like AIDS, atherosclerosis, Alzheimer’s disease (AD), rheumatoid arthritis, PD, cancer, osteoporosis, and diabetes are linked with constitutive expression of NF-κB (Table 1) (Gupta et al. 2010b; Vallabhapurapu and Karin 2009).
Signaling Pathways of NF-κB
The classical or canonical pathway and the alternate or non-canonical are the two major pathways involved in the activation of NF-κB. Dimer of Rel proteins p50 and p65 form found complexed with inhibitory complex IκBα in the cytosol and get activated and regulate the production of pro-inflammatory cytokines, known as the classical pathway (Fig. 2) (Lawrence 2009). The canonical pathway is the most extensively studied pathway of NF-κB activation (Noort et al. 2015), triggered in response to pro-inflammatory molecules such as TNF, LPS, IL-1, and T cell receptor or B cell receptor and other cell surface receptors like Toll-like receptors (TLRs), TNF receptor, and IL-1 receptor. A natural biological inhibitor IκB is found to be associated with NF-κB, keeping it in inactive state inside the cytoplasm (Baeuerle and Baltimore 1996; Verma et al. 1995). IκB family, consisting of IκBα, IκBβ, p105/IκBγ (precursor of p50), p100 (precursor of p52), and IκBε are proteins that contain an ankyrin repeated sequence which help in keeping the NF-κB in an inactive state (Fig. 2) (Li and Nabel 1997; Whiteside et al. 1997) while keeping the nuclear localization signal (NLS) as well as the DNA binding domain masked. Upon activation of IκB kinase complex through a diverse range of stimuli, it causes the phosphorylation of two serine residues, serine32 and 36 of IκBα or serine19 and 23 of IκBβ (DiDonato et al. 1997; Karin et al. 1997; Mercurio et al. 1997; Régnier et al. 1997), thereby targeting IκB for ubiquitin-mediated proteasomal degradation through the 26S proteasome complex mediating the nuclear translocation of NF-κB (Finco and Baldwin 1995; Thanos and Maniatis 1995). Hence, the expression of NF-κB occurs ubiquitously in almost all cell types and on activation, it gets translocated from the cytosol to the nucleus where it binds with κB site of promoter. Inhibitor of κB kinase (IKK) β is necessary for NF-κB activation, while inhibitor of κB kinase (IKK) α is redundant in the classical pathway (Tak et al. 2001; Vallabhapurapu and Karin 2009). Both acute as well as chronic inflammations require the activation of NF-κB through canonical pathway which is essential in cell survival and proliferation as is evident by active NF-κB signaling in different tissues (Ben-Neriah and Karin 2011).

The NF-κB-mediated neuroinflammatory cascade. Pro-inflammatory factors like TNF-α, IL-1β, IL-6, and CD-40L activate the cell surface receptor. The cell surface receptor consists of two pathways, first is canonical pathway in which inhibitor of κB kinase (IKK) β is necessary for NF-κB activation that phosphorylates IκB, while NF-κB essential modulator (NEMO) is regulatory subunit of IKK complex. IκB undergoes proteasomal degradation in the cytosol and then phosphorylated heterodimer of NF-κB (p50-p65) goes to the nucleus via nuclear membrane and binds to the NF-κB response element activating the associated pro-inflammatory mediators like TNF-α, IL-1β, IL-6, iNOS, and ICAM and ultimately causes NF-κB-mediated neuroinflammation that leads to the progressive degeneration of the DA neurons in PD. In alternate or non-canonical pathway, NEMO along with NF-κB inducing kinase (NIK) phosphorylates IKK-α and causes the proteasomal degradation along with the proteasomal processing of p100 which is a subunit of NF-κB heterodimer and forms the p52-RELB active heterodimer. The p52-RELB active heterodimer goes into the nucleus through nuclear membrane and similar to the canonical pathway binds to the NF-κB response element regulating the expression of pro-inflammatory factors like TNF-α, IL-1β, IL-6, iNOS, and ICAM, ultimately causing the NF-κB-mediated neuroinflammation via progressive neurodegeneration in PD
The other pathway of NF-κB activation is non-canonical pathway that involves the heterodimers of Rel proteins p100/RelB that too show transcriptional activity but is majorly involved in regulation of cellular activation and differentiation rather than in inflammation. The non-canonical NF-κB pathway is activated in response to various stimuli including members of TNF receptor superfamily such as B cell activating factor (BAF), receptor activator of NF-κB (RANK), lymphotoxin β (LTβ) receptor, and CD40. These receptors simultaneously activate the canonical pathway too. Only, IKKα homodimers are responsible for the non-canonical activation of NF-κB pathway, unlike to that of IKKβ or IKKγ involved in canonical pathway for IκB phosphorylation.
Non-canonical activation of NF-κB needs the synthesis and accumulation of NF-κB inducing kinase (NIK) which is the central signaling component of the non-canonical pathway, whereas canonical NF-κB activation is quick and does not require the protein synthesis, thereby slowing the kinetics of the non-canonical pathway (Sun 2011; Vallabhapurapu and Karin 2009). The canonical NF-κB-dependent gene expression requires enzymatic action with the IKKα for its regulation by controlling the promoter-associated histone phosphorylation exposed to cytokines (Anest et al. 2003; Yamamoto et al. 2003). Immune cells were seen to have suppressed basal non-canonical signaling by activation of canonical pathway and initiation of NF-κB-mediated signal transduction (Gray et al. 2014). Remarkably, non-canonical pathway also gets activated in several specific cell types under certain circumstances and other stimuli including TNF (Zhang et al. 2014) and IKKα play an important role in the production of interferon-α induced by TLR 7 and 9 (Hoshino et al. 2006). Moreover, distinct genes are regulated in response to various stimuli by both the classical and alternative pathways (Pomerantz and Baltimore 2002). Post mortem PD brains and SN of animals undergoing DA neurodegeneration have shown the highly activated canonical pathways (Hunot et al. 1997; Mogi et al. 2007). However, regenerating DA neurons from rats treated with glial-derived neurotrophic factor (GDNF) have shown the active involvement of non-canonical NF-κB pathway, whereas the canonical p65/p50 pathway was concurrently reduced, indicating the role of non-canonical NF-κB pathway in regeneration of DA neurons within the SN (Cao et al. 2008).
At the molecular level, inflammation is regulated by alterations in redox balance which in turn activates various molecules and factors, including adhesion molecules like, intercellular adhesion molecule (ICAM-1), endothelial-leukocyte adhesion molecule (ELAM)-1, and vascular cell adhesion molecule (VCAM)-1; chemokines such as monocyte chemo-attractant protein 1, IL-8; cytokines including IL-1, IL-2, IL-6, IL-12, TNF-α, and TNF-β; signal transducer and activator of transcription (STAT)-3; pro-inflammatory enzymes, namely, COX-2, 5-LOX, 12-LOX, and matrix metalloproteinases (MMPs); prostate-specific antigen (PSA); C-reactive protein, vascular endothelial growth factor (VEGF); and pro-inflammatory transcription factors NF-κB (Aggarwal and Shishodia 2004). Thus, the fundamental regulator of inflammation is NF-κB (Fig. 2) (Aggarwal and Shishodia 2004; Ahn and Aggarwal 2005; Lukiw and Bazan 1998).
Recent researches conducted in the area of NF-κB-mediated neurodegeneration along with the significance of each article are mentioned in Table 1.
Regulation of NF-κB Activity
Studies have suggested dysregulation of NF-κB in neurodegenerative mechanisms occurred during trauma or ischemia (Schneider et al. 1999) and also in the brain of patients suffering from PD (Ghosh et al. 2007; Hunot et al. 1997). Neuroinflammatory molecules play important role in these CNS diseases. Differential activation of NF-κB dimers confers the response of neurons against external stimuli. Contradictory effects on the survival of neurons are produced by expression of Rel A or c-Rel (Pizzi et al. 2002; Pizzi et al. 2005; Sarnico et al. 2009b). Neurodegenerative processes induced by ischemic insults (Inta et al. 2006; Sarnico et al. 2009a, 2009b), Aβ toxicity (Inta et al. 2006; Lanzillotta et al. 2010; Pizzi et al. 2009), or glutamate (Pizzi et al. 2002) are initiated by the RelA subunit (a member of NF-κB), composing the activated p50/RelA dimer, and its posttranscriptional modifications.
Neuronal cell death is seen to be due to RelA while cell death is limited by overexpression of c-Rel factor. Oxygen–glucose deprivation (OGD) reduces the level of c-Rel factor in neurons, whereas neuronal loss in cortical neurons exposed to OGD is prevented by overexpression of c-Rel, as a result of this protective effect the transcription of Bcl-xL gene increases (Sarnico et al. 2009a). By inducing transcription of manganese superoxide dismutase (MnSOD), overexpression of c-Rel promotes anti-apoptotic effects in cultured neurons (Bernard et al. 2001; Chen et al. 2000; Pizzi et al. 2005). An age-related behavioral Parkinsonism in mice with degeneration of DA neurons and generation of PD-like neuropathology is induced by the deficiency of c-Rel (Baiguera et al. 2012). Evidences have also proven that the systemic and brain aging processes in mice occur due to activation of NF-κB (Adler et al. 2007; Zhang et al. 2013a). Thus, data from different studies have suggested that the composition of NF-κB complex is responsible for the diverse functions of NF-κB transcription factor (Lanzillotta et al. 2015). Taken together, all these studies suggest the potential of NF-κB to serve as an excellent therapeutic target in preventing DA neurodegeneration, and also research is required to govern the finest approach and agent appropriate for the treatment of PD.
Polyphenols and Its Bioavailability in the Brain
Polyphenols are a large group of naturally occurring plant chemicals produced as secondary metabolites, extensively found in variety of natural products including fruits and vegetables. All polyphenols are basically characterized by the presence of phenolic hydroxyl groups in their structure. On the basis of the number of phenol rings present and their properties, these compounds are broadly classified into two groups: flavonoids and non-flavonoids. Flavonoids are extensively distributed phytochemicals and are a type of phenylpropanoids consisting of 15 carbons arranged as a three carbon bridge (C6–C3–C6) connected to two aromatic rings. Flavanols and flavanones have similar structures based on 2,3-dihydro-2-phenylchromen-4-one skeleton and thus are characterized on the basis of the site of hydroxylation. Flavanols are differentiated due to the hydroxylation on carbon-3 of the phenyl ring. These act as the intermediary and diverge into various classes of flavonoids. Different classes of flavonoids are formed by the diversification of the pathway from these central intermediates as side branches. Non-flavonoids are further divided into two groups: the phenolic acids which include the hydroxycinnamic acids (HCAs; C3–C6 skeleton) and hydroxybenzoic acids (HBAs; C1–C3 skeleton), and the stilbenes (C6–C2–C6 skeleton) (Renaud and Martinoli 2019; Vauzour 2012).
Various in vitro studies have revealed that the lipophilic nature of a polyphenol determines its permeability through the blood–brain barrier (BBB) making the less polar polyphenols (i.e., O-methylated derivatives) more permeable in comparison to the highly polar metabolites (i.e., sulfated and glucuronidated derivatives) (Youdim et al. 2003). Similar studies have also been performed in vivo models validating that irrespective of their mode of administration, the polyphenols can translocate through the BBB. A study on bioavailability of polyphenols in the brain reported the presence of epigallocatechin gallate, epicatechin, and anthocyanins in the brain after the oral administration, whereas naringenin was detected following the intravenous administration (El Mohsen et al. 2006; El Mohsen et al. 2002; Peng et al. 1998; Suganuma et al. 1998). Although the data suggests low availability of polyphenols in the brain and plasma, i.e., below 1 nmol/g tissue by Schaffer and Halliwell with no specific region to be targeted for accumulation (Schaffer and Halliwell 2012; Williams et al. 2008; Zecca et al. 1982), in fact studies performed on rat and pig brains (Kalt et al. 2008; Milbury and Kalt 2010) show the presence of polyphenols in various regions of the brain. In an experiment by Janle et al. with 14C-labeled grape polyphenols, it was indicated that the 14C was uniformly distributed among all the regions of the brain (Janle et al. 2010). Similarly, multiple in vivo studies have validated the bioavailability of curcumin (oral administration) and have shown it to penetrate through the BBB suggesting its therapeutic importance. A study by Suresh and Srinivasan in rat model exhibited that following the curcumin administration (500 mg/kg body weight) in conjugation with piperine (20 mg/kg body weight), a concentration of 1.84 ± 0.33 mg/whole tissue was measured at 24 h interval in the brain which was later found to have reached to 5.87 ± 0.38 mg/whole tissue at 48 h. In a study by Wang and colleagues, the intraperitoneal administration (30 mg/kg body weight) of curcumin in gerbils was detected to be 0.15 ng/mg protein in the brain at 1 h (Suresh and Srinivasan 2010; Wang et al. 2005), whereas Begum et al. reported different routes of administration of curcumin and tetrahydrocurcumin (TC) including oral, intraperitoneal, and intramuscular and quantified their significant level in the brain (Begum et al. 2008). Thus, these results prove the permeability of polyphenols through the BBB and their uniform distribution among various regions of the brain.
Anti-neuroinflammatory Role of Polyphenols in PD
Dietary polyphenols (Table 2) are widely studied for their anti-inflammatory and anti-oxidant properties and have been documented to contain anti-cancerous properties and help in the prevention of cardiovascular disease and neurodegenerative diseases (Liu 2004; Patil et al. 2014; Tsao 2010). Polyphenols have been shown to modulate various cellular signaling pathways including alterations at two specific sites in the pathway leading from receptors to NF-κB. They suppress the degradation of IκB by restraining phosphorylation or ubiquitination which arrests the transcription of pro-inflammatory cytokines by inhibiting the NF-κB translocation in to the nucleus (Ruiz and Haller 2006; Wang et al. 2017). Furthermore, polyphenols also perform anti-inflammatory action by retarding the interaction of NF-κB subunits with target DNA (Ruiz and Haller 2006). Both modes of action offer indirect protection by preventing various NF-κB regulated proinflammatory proteins (cytokines, chemokines) and enzymes (iNOS, COX-2) to express.
Dietary polyphenols responsible in regulating various pro-inflammatory gene expression are a group of secondary metabolites including curcumin (Jobin et al. 1999; Zhou et al. 2015), resveratrol (Kundu et al. 2006), Chlorogenic acid (Hwang et al. 2015; Singh et al. 2018). and baicalein (Lee et al. 2014).
Chlorogenic Acid
Chlorogenic acid (CGA) is a phenolic compound and the second major component after caffeine found in green coffee beans. It is a thermally unstable ester which is readily broken down to form caffeic acid and quinic acid (Feng et al. 2005). CGA demonstrates multiple biological properties including anti-oxidant (Feng et al. 2005), anti-inflammatory effect (Hwang et al. 2015; Hwang et al. 2014), and neuroprotective (Kwon et al. 2010) properties which have gained its huge popularity in the scientific world. Various studies working on the mechanism of action of CGA have confirmed that it minimizes the overactivation of glial cells and downregulation of expression of pro-inflammatory factors such as TNF-α, IL-1β, and iNOS, thus protecting the degeneration of DA neurons (Shen et al. 2012); (Singh et al. 2018). A study conducted in MPTP-intoxicated mice confirmed that CGA downregulated NF-κB-mediated neuroinflammatory pathway in PD, ultimately protecting the loss of the DA neurons in SN (Singh et al. 2018) (Table 2).
Curcumin
Curcumin (diferuloylmethane) is a polyphenolic compound found in the plant Curcuma longa which is a widely used spice in the preparation of food and medicines in Southeast Asia, China, and India (Aggarwal et al. 2007) is proving to be effective in neuroprotection as it illustrates anti-oxidant, anti-inflammatory, and anti-cancer activities and is able to pass through the blood–brain barrier (Lee et al. 2013). In recent studies conducted on in vitro and in vivo models of PD, curcumin has been observed to have neuroprotective effect against neurotoxicity induced by LPS and α-synuclein aggregation preventing dopamine loss and altering the oxidative stress, as well as mitochondrial dysfunction (Rajeswari and Sabesan 2008; Wang and Xu 2009). Curcumin as a result of its anti-inflammatory activity has been found to neutralize microglial activation, enzymes like cyclooxygenase-2 (COX-2), lipoxygenase, inducible nitric oxide synthase (iNOS), and pro-inflammatory cytokines (Guo et al. 2012; Tripanichkul and Jaroensuppaperch 2013; Yu et al. 2010). In addition to the above activity, curcumin has been suggested to restrain the nuclear translocation and NF-κB activity along with abbreviation in the levels of TNF-α and IL-1β (Guo et al. 2012). Another study suggested that curcumin alleviated NF-κB in the microglial cytoplasm as a result of the stimulation with LTA in BV-2 microglial cells which caused inhibition of phosphorylation and degradation of IκBα, as well as nuclear translocation of p65 (Yu et al. 2018). The study showed that curcumin inhibits NF-κB and p38 MAPK activation confirming its anti-inflammatory activity in LTA-stimulated microglial cells (Table 2).
Resveratrol
Resveratrol is a plant compound present in the covering of grapes, blueberries, raspberries, mulberries, peanuts, and red wine. It is a naturally occurring polyphenol that wields neuroprotective activity. It is soluble in water and is capable of crossing the blood–brain barrier (Fu et al. 2015). Studies have reported that resveratrol is capable of scavenging free radicals, repressing glial activation, and minimizing the production of pro-inflammatory factors and is also observed to enhance the release of anti-inflammatory IL-10 in MPTP-based PD mouse models (Cianciulli et al. 2015; Lofrumento et al. 2014; Renaud et al. 2014; Song et al. 2014; Yang et al. 2016) (Table 2). Various studies in the past have demonstrated inhibition of signaling cascades, NF-κB translocation, and ROS production as result of the mechanism of action of resveratrol (Capiralla et al. 2012; Ren et al. 2013; Zhang et al. 2013b). Apart from this, resveratrol is reported to promote microglial polarization to the M2 phenotype by increasing PGC-1α expression and acting against inflammatory damage (Yang et al. 2017). It is also noticed that along with the advancement of microglial polarization through the activation of the STAT6 and STAT3 pathways to the M2 phenotype, PGC-1α also inhibited NF-κB activity ultimately reducing the activation LPS-induced M1. These studies conclude that resveratrol, by targeting NF-κB pathway, can be used as potential anti-inflammatory drug in the treatment of PD (Yang et al. 2017).
Baicalein
Baicalein is a type of flavonoid, a component of traditional Chinese herbal remedy known as Scutellaria baicalensis Georgi (Huangqin). It has been reported that baicalein shows anti-inflammatory and anti-oxidative activities that facilitate neuroprotective effects in PD model (Mu et al. 2009; Wang et al. 2013; Zhang et al. 2012b) (Table 2). Previous studies in vitro have revealed that α-synuclein aggregation cannot only be inhibited using baicalein but also the already formed wild-type α-synuclein oligomers can be disaggregated (Hong et al. 2008). For several decades, research has been determined to evaluate the neuroprotective activity of baicalein in various chemically induced mice models and the results revealed that the anti-oxidative property of baicalein, along with increasing the number of tyrosine hydroxylase (TH) neurons, also attenuated muscle tremors in 6-hydroxydopamine (OHDA)-lesioned rats (Cheng et al. 2008; Mu et al. 2011). Furthermore, baicalein inhibited mitochondrial oxidation along with up regulating DJ-1 protein expression in 6-OHDA-induced SH-SY5Y cells resulting in the inhibition of mitochondrial dysfunction (Wang et al. 2013). Additionally, recent studies conducted on the MPTP-induced PD model have published the role of baicalein in the reduction of astroglial activation by inhibiting the activation of NF-κB, extracellular-signal regulated kinases (ERK)1/2, and Jun-amino-terminal kinase (JNK), while showing neuroprotective activity in 6-OHDA-induced mice model via activation of the Kelch-like ECH-associated protein 1 (Keap1)/NF-E2-related factor 2 (NRF-2)/heme oxygenase-1 (HO-1) and phosphoinositide 3-kinase (PI3K)/AKT signaling pathways (Lee et al. 2014; Zhang et al. 2012b).
Conclusion
NF-κB-mediated neuroinflammation plays a major role in the PD pathogenesis, thus is being extensively studied to target the pathways involving its activation in neurons. Hence, inhibiting the overactivation of NF-κB may serve as a potential therapeutic mechanism in the prevention of PD progression. Previous studies on animal and cell models have indicated the beneficial effects of dietary supplements comprising of polyphenolic compounds and have suggested their use in the treatment and prevention of inflammatory mediated neurodegeneration of DA neurons. More comprehensive analysis about the mechanism of action of these polyphenolic compounds is required for the designing of novel treatment having advanced therapeutic properties in vivo for PD. Nevertheless, the requirement for the conduction of clinical trial to establish the effectiveness of the phenolic compounds as a treatment for PD by preventing NF-κB pathway is still required.
Abbreviations
- NF-κB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- PD:
-
Parkinson’s disease
- DA:
-
Dopaminergic
- TNF-α:
-
Tumor necrosis factor
- MPTPq:
-
1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- CNS:
-
Central nervous system
- SNpc:
-
Substantia nigra pars compacta
- CGA:
-
Chlorogenic acid
- ROS:
-
Reactive oxygen species
- TH:
-
Tyrosine hydroxylase
References
Adler AS, Sinha S, Kawahara TL, Zhang JY, Segal E et al (2007) Motif module map reveals enforcement of aging by continual NF-κB activity. Genes & Development 21:000–000
Aggarwal BB, Shishodia S (2004) Suppression of the nuclear factor-κB activation pathway by spice-derived phytochemicals: reasoning for seasoning. Annals of the New York Academy of Sciences 1030:434–441
Aggarwal BB, Surh YJ, Shishodia S (Eds.) (2007) The molecular targets and therapeutic uses of curcumin in health and disease (Vol. 595). Springer Science & Business Media
Ahn KS, Aggarwal BB (2005) Transcription factor NF-κB: a sensor for smoke and stress signals. Annals of the New York Academy of Sciences 1056:218–233
Anest V, Hanson JL, Cogswell PC, Steinbrecher KA, Strahl BD et al (2003) A nucleosomal function for IκB kinase-α in NF-κB-dependent gene expression. Nature 423:659
Arai H, Furuya T, Yasuda T, Miura M, Mizuno Y et al (2004) Neurotoxic effects of lipopolysaccharide on nigral dopaminergic neurons are mediated by microglial activation, interleukin-1β, and expression of caspase-11 in mice. Journal of Biological Chemistry 279:51647–51653
Baeuerle PA, Baltimore D (1996) NF-κB: ten years after. Cell 87:13–20
Baeuerle PA, Henkel T (1994) Function and activation of NF-kappaB in the immune system. Annual Review of Immunology 12:141–179
Bahar E, Kim J-Y, Yoon H (2017) Quercetin attenuates manganese-induced neuroinflammation by alleviating oxidative stress through regulation of apoptosis, iNOS/NF-κB and HO-1/Nrf2 pathways. International Journal of Molecular Sciences 18:1989
Baiguera C, Alghisi M, Pinna A, Bellucci A, De Luca MA et al (2012) Late-onset Parkinsonism in NFκB/c-Rel-deficient mice. Brain 135:2750–2765
Bakalkin GY, Yakovleva T, Terenius L (1993) NF-κB-like factors in the murine brain. Developmentally-regulated and tissue-specific expression. Molecular Brain Research 20:137–146
Baldwin AS Jr (1996) The NF-κB and IκB proteins: new discoveries and insights. Annu Rev Immunol 14:649–681
Barcia C, de Pablos V, Bautista-Hernández V, Sánchez-Bahillo Á, Bernal I et al (2005) Increased plasma levels of TNF-α but not of IL1-β in MPTP-treated monkeys one year after the MPTP administration. Parkinsonism & Related Disorders 11:435–439
Begum AN, Jones MR, Lim GP, Morihara T, Kim P et al (2008) Curcumin structure-function, bioavailability, and efficacy in models of neuroinflammation and Alzheimer’s disease. J Pharmacol Exp Ther 326:196–208
Ben Haim L, Carrillo-de Sauvage M-A, Ceyzériat K, Escartin C (2015) Elusive roles for reactive astrocytes in neurodegenerative diseases. Frontiers in Cellular Neuroscience 9:278
Ben-Neriah Y, Karin M (2011) Inflammation meets cancer, with NF-κB as the matchmaker. Nat Immunol 12:715
Berlett BS, Stadtman ER (1997) Protein oxidation in aging, disease, and oxidative stress. J Biol Chem 272:20313–20316
Bernard D, Quatannens B, Begue A, Vandenbunder B, Abbadie C (2001) Antiproliferative and antiapoptotic effects of cRel may occur within the same cells via the up-regulation of manganese superoxide dismutase. Cancer Research 61:2656–2664
Birla H, Rai SN, Singh SS, Zahra W, Rawat A et al (2019) Tinospora cordifolia suppresses neuroinflammation in parkinsonian mouse model. Neuromolecular Medicine 21:42–53
Blandini F, Greenamyre J (1998) Prospects of glutamate antagonists in the therapy of Parkinson’s disease. Fundamental & Clinical Pharmacology 12:4–12
Blesa J, Przedborski S (2014) Parkinson’s disease: animal models and dopaminergic cell vulnerability. Frontiers in Neuroanatomy 8:155
Blesa J, Trigo-Damas I, Quiroga-Varela A, Jackson-Lewis VR (2015) Oxidative stress and Parkinson’s disease. Frontiers in Neuroanatomy 9:91
Blin O, Desnuelle C, Rascol O, Borg M, Saint Paul HP et al (1994) Mitochondrial respiratory failure in skeletal muscle from patients with Parkinson’s disease and multiple system atrophy. Journal of the Neurological Sciences 125:95–101
Block ML, Zecca L, Hong J-S (2007) Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nature Reviews Neuroscience 8:57
Brown GC, Neher JJ (2010) Inflammatory neurodegeneration and mechanisms of microglial killing of neurons. Molecular Neurobiology 41:242–247
Cao JP, Wang HJ, Yu JK, Liu HM, Gao DS (2008) The involvement of NF-κB p65/p52 in the effects of GDNF on DA neurons in early PD rats. Brain Res Bull 76:505–511
Capiralla H, Vingtdeux V, Zhao H, Sankowski R, Al-Abed Y et al (2012) Resveratrol mitigates lipopolysaccharide-and Aβ-mediated microglial inflammation by inhibiting the TLR4/NF-κB/STAT signaling cascade. Journal of Neurochemistry 120:461–472
Caudle WM, Zhang J (2009) Glutamate, excitotoxicity, and programmed cell death in Parkinson disease. Experimental Neurology 220:–230
Caudle WM, Richardson JR, Wang MZ, Taylor TN, Guillot TS et al (2007) Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. Journal of Neuroscience 27:8138–8148
Chen C, Manning A (1995) Transcriptional regulation of endothelial cell adhesion molecules: a dominant role for NF-kappa B. Agents and Actions Supplements 47:135–141
Chen FE, Huang D-B, Chen Y-Q, Ghosh G (1998) Crystal structure of p50/p65 heterodimer of transcription factor NF-κB bound to DNA. Nature 391:–410
Chen C, Edelstein LC, Gélinas C (2000) The Rel/NF-κB family directly activates expression of the apoptosis inhibitor Bcl-xL. Molecular and Cellular Biology 20:2687–2695
Chen G, Liu J, Jiang L, Ran X, He D et al (2018) Peiminine protects dopaminergic neurons from inflammation-induced cell death by inhibiting the ERK1/2 and NF-κB signalling pathways. International Journal of Molecular Sciences 19:821
Chen Y, Wu T, Li H, Li X, Li Q et al (2019) Dl-3-n-butylphthalide exerts dopaminergic neuroprotection through inhibition of neuroinflammation. Frontiers in Aging Neuroscience 11
Cheng C, Zhu X (2019) Cordycepin mitigates MPTP-induced Parkinson’s disease through inhibiting TLR/NF-κB signaling pathway. Life Sciences 223:120–127
Cheng Y, He G, Mu X, Zhang T, Li X, Hu J, Xu B, Du G (2008) Neuroprotective effect of baicalein against MPTP neurotoxicity: behavioral, biochemical and immunohistochemical profile. Neurosci Lett 441:16–20
Chinta SJ, Lieu CA, DeMaria M, Laberge RM, Campisi J et al (2013) Environmental stress, ageing and glial cell senescence: a novel mechanistic link to Parkinson’s disease? J Intern Med 273:429–436
Choi DW (1988) Glutamate neurotoxicity and diseases of the nervous system. Neuron 1:623–634
Choi JH, Jang M, Cho I-H (2018) Neuroprotective effects of a traditional multi-herbal medicine Kyung-Ok-Ko in an animal model of Parkinson’s disease: inhibition of MAPKs and NF-κB pathways and activation of Keap1-Nrf2 pathway. Frontiers in Pharmacology 9:1444
Cianciulli A, Dragone T, Calvello R, Porro C, Trotta T et al (2015) IL-10 plays a pivotal role in anti-inflammatory effects of resveratrol in activated microglia cells. International Immunopharmacology 24:369–376
Cunningham LA, Su C (2002) Astrocyte delivery of glial cell line-derived neurotrophic factor in a mouse model of Parkinson’s disease. Experimental Neurology 174:230–242
Daher JP, Volpicelli-Daley LA, Blackburn JP, Moehle MS, West AB (2014) Abrogation of α-synuclein-mediated dopaminergic neurodegeneration in LRRK2-deficient rats. Proceedings of the National Academy of Sciences 111:9289–9294
DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M (1997) A cytokine-responsive IκB kinase that activates the transcription factor NF-κB. Nature 388:548
Doo A-R, Kim S-N, Park J-Y, Cho KH, Hong J et al (2010) Neuroprotective effects of an herbal medicine, Yi-Gan San on MPP+/MPTP-induced cytotoxicity in vitro and in vivo. Journal of Ethnopharmacology 131:433–442
Doorn KJ, Goudriaan A, Blits-Huizinga C, Bol JG, Rozemuller AJ et al (2014a) Increased amoeboid microglial density in the olfactory bulb of Parkinson’s and Alzheimer’s patients. Brain Pathology 24:152–165
Doorn KJ, Moors T, Drukarch B, van de Berg WD, Lucassen PJ et al (2014b) Microglial phenotypes and toll-like receptor 2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson’s disease patients. Acta Neuropathologica Communications 2:90
Dou F, Chu X, Zhang B, Liang L, Lu G et al (2018) EriB targeted inhibition of microglia activity attenuates MPP+ induced DA neuron injury through the NF-κB signaling pathway. Molecular Brain 11:75
Duh EJ, Maury WJ, Folks TM, Fauci AS, Rabson AB (1989) Tumor necrosis factor alpha activates human immunodeficiency virus type 1 through induction of nuclear factor binding to the NF-kappa B sites in the long terminal repeat. Proceedings of the National Academy of Sciences 86:5974–5978
Ebrahimi A, Schluesener H (2012) Natural polyphenols against neurodegenerative disorders: potentials and pitfalls. Ageing Research Reviews 11:329–345
El Mohsen MMA, Kuhnle G, Rechner AR, Schroeter H, Rose S et al (2002) Uptake and metabolism of epicatechin and its access to the brain after oral ingestion. Free Radical Biology and Medicine 33:1693–1702
El Mohsen MA, Marks J, Kuhnle G, Moore K, Debnam E et al (2006) Absorption, tissue distribution and excretion of pelargonidin and its metabolites following oral administration to rats. Br J Nutr 95:51–58
Feng R, Lu Y, Bowman LL, Qian Y, Castranova V, Ding M (2005) Inhibition of activator protein-1, NF-κB, and MAPKs and induction of phase 2 detoxifying enzyme activity by chlorogenic acid. J Biol Chem 280:27888–27895
Finco TS, Baldwin AS (1995) Mechanistic aspects of NF-κB regulation: the emerging role of phosphorylation and proteolysis. Immunity 3:263–272
Fu W, Zhuang W, Zhou S, Wang X (2015) Plant-derived neuroprotective agents in Parkinson’s disease. American Journal of Translational Research 7:1189
Gauss KA, Nelson-Overton LK, Siemsen DW, Gao Y, DeLeo FR et al (2007) Role of NF-κB in transcriptional regulation of the phagocyte NADPH oxidase by tumor necrosis factor-α. Journal of Leukocyte Biology 82:729–741
Ghosh A, Roy A, Liu X, Kordower JH, Mufson EJ et al (2007) Selective inhibition of NF-κB activation prevents dopaminergic neuronal loss in a mouse model of Parkinson’s disease. Proceedings of the National Academy of Sciences 104:18754–18759
Goetz CG, Pal G (2014) Initial management of Parkinson’s disease. BMJ 349:g6258
Gray CM, Remouchamps C, McCorkell KA, Solt LA, Dejardin E et al (2014) Noncanonical NF-κB signaling is limited by classical NF-κB activity. Sci Signal 7:ra13–ra13
Guo Y, Yang B, Shi L, Gu J, Chen H (2012) Anti-inflammation mechanism of curcumin in mice with lipopolysaccharide-induced Parkinson’s disease. Journal of Medical Postgraduates 25:582–587
Gupta SC, Kim JH, Prasad S, Aggarwal BB (2010a) Regulation of survival, proliferation, invasion, angiogenesis, and metastasis of tumor cells through modulation of inflammatory pathways by nutraceuticals. Cancer Metastasis Rev 29:405–434
Gupta SC, Sundaram C, Reuter S, Aggarwal BB (2010b) Inhibiting NF-κB activation by small molecules as a therapeutic strategy, Biochimica et Biophysica Acta (BBA)-Gene Regulatory Mechanisms. 1799:775–787
Haas RH, Nasirian F, Nakano K, Ward D, Pay M et al (1995) Low platelet mitochondrial complex I and complex II/III activity in early untreated Parkinson’s disease. Annals of Neurology 37:714–722
Harman D (1992) Free radical theory of aging. Mutation Research/DNAging 275:257–266
Harman D (2006) Free radical theory of aging: an update. Ann N Y Acad Sci 1067:10–21
Hattingen E, Magerkurth J, Pilatus U, Mozer A, Seifried C et al (2009) Phosphorus and proton magnetic resonance spectroscopy demonstrates mitochondrial dysfunction in early and advanced Parkinson’s disease. Brain 132:3285–3297
Hattori N, Tanaka M, Ozawa T, Mizuno Y (1991) Immunohistochemical studies on complexes I, II, III, and IV of mitochondria in Parkinson’s disease. Annals of Neurology 30:563–571
Hong D-P, Fink AL, Uversky VN (2008) Structural characteristics of α-synuclein oligomers stabilized by the flavonoid baicalein. Journal of Molecular Biology 383:214–223
Hoshino K, Sugiyama T, Matsumoto M, Tanaka T, Saito M et al (2006) IκB kinase-α is critical for interferon-α production induced by Toll-like receptors 7 and 9. Nature 440:949
Hunot S, Brugg B, Ricard D, Michel PP, Muriel M-P et al (1997) Nuclear translocation of NF-κB is increased in dopaminergic neurons of patients with Parkinson disease. Proceedings of the National Academy of Sciences 94:7531–7536
Hwang SJ, Kim Y-W, Park Y, Lee H-J, Kim K-W (2014) Anti-inflammatory effects of chlorogenic acid in lipopolysaccharide-stimulated RAW 264.7 cells. Inflammation Research 63:81–90
Hwang SJ, Jun SH, Park Y, Cha S-H, Yoon M et al (2015) Green synthesis of gold nanoparticles using chlorogenic acid and their enhanced performance for inflammation nanomedicine: nanotechnology. Biology and Medicine 11:1677–1688
Imamura K, Hishikawa N, Sawada M, Nagatsu T, Yoshida M et al (2003) Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathologica 106:518–526
Inta I, Paxian S, Maegele I, Zhang W, Pizzi M et al (2006) Bim and Noxa are candidates to mediate the deleterious effect of the NF-κB subunit RelA in cerebral ischemia. Journal of Neuroscience 26:12896–12903
Janle EM, Lila MA, Grannan M, Wood L, Higgins A et al (2010) Pharmacokinetics and tissue distribution of 14C-labeled grape polyphenols in the periphery and the central nervous system following oral administration. Journal of Medicinal Food 13:926–933
Jobin C, Bradham CA, Russo MP, Juma B, Narula AS et al (1999) Curcumin blocks cytokine-mediated NF-κB activation and proinflammatory gene expression by inhibiting inhibitory factor I-κB kinase activity. The Journal of Immunology 163:3474–3483
Kalt W, Blumberg JB, McDonald JE, Vinqvist-Tymchuk MR, Fillmore SA et al (2008) Identification of anthocyanins in the liver, eye, and brain of blueberry-fed pigs. Journal of Agricultural and Food Chemistry 56:705–712
Kaltschmidt B, Kaltschmidt C (2009) NF-κB in the nervous system. Cold Spring Harbor Perspectives in Biology 1:a001271
Kaltschmidt C, Kaltschmidt B, Baeuerle PA (1993) Brain synapses contain inducible forms of the transcription factor NF-κB. Mechanisms of Development 43:135–147
Kaltschmidt C, Kaltschmidt B, Neumann H, Wekerle H, Baeuerle PA (1994) Constitutive NF-kappa B activity in neurons. Molecular and Cellular Biology 14:3981–3992
Kaltschmidt C, Kaltschmidt B, Baeuerle PA (1995) Stimulation of ionotropic glutamate receptors activates transcription factor NF-kappa B in primary neurons. Proceedings of the National Academy of Sciences 92:9618–9622
Kaltschmidt B, Widera D, Kaltschmidt C (2005) Signaling via NF-κB in the nervous system. Biochimica et Biophysica Acta (BBA)-Molecular. Cell Research 1745:287–299
Karin M, Liu Z-G, Zandi E (1997) AP-1 function and regulation. Curr Opin Cell Biol 9:240–246
Kim N, Do J, Bae J-S, Jin HK, Kim J-H et al (2018) Piperlongumine inhibits neuroinflammation via regulating NF-κB signaling pathways in lipopolysaccharide-stimulated BV2 microglia cells. Journal of Pharmacological Sciences 137:195–201
Kitamura Y, Itano Y, Kubo T, Nomura Y (1994) Suppressive effect of FK-506, a novel immunosuppressant, against MPTP-induced dopamine depletion in the striatum of young C57BL/6 mice. Journal of Neuroimmunology 50:221–224
Knott C, Stern G, Wilkin G (2000) Inflammatory regulators in Parkinson’s disease: iNOS, lipocortin-1, and cyclooxygenases-1 and-2. Molecular and Cellular Neuroscience 16:724–739
Kundu JK, Shin YK, Kim SH, Surh Y-J (2006) Resveratrol inhibits phorbol ester-induced expression of COX-2 and activation of NF-κB in mouse skin by blocking IκB kinase activity. Carcinogenesis 27:1465–1474
Kurkowska-Jastrzębska I, Wrońska A, Kohutnicka M, Członkowski A, Członkowska A (1999) The inflammatory reaction following 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine intoxication in mouse. Exp Neurol 156:50–61
Kwon S-H, Lee H-K, Kim J-A, Hong S-I, Kim H-C et al (2010) Neuroprotective effects of chlorogenic acid on scopolamine-induced amnesia via anti-acetylcholinesterase and anti-oxidative activities in mice. European Journal of Pharmacology 649:210–217
Lan X, Liu R, Sun L, Zhang T, Du G (2011) Methyl salicylate 2-O-β-D-lactoside, a novel salicylic acid analogue, acts as an anti-inflammatory agent on microglia and astrocytes. J Neuroinflammation 8:98
Langston J, Forno L, Tetrud J, Reeves A, Kaplan J et al (1999) Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine exposure. Annals of Neurology 46:598–605
Lanzillotta A, Sarnico I, Ingrassia R, Boroni F, Branca C et al (2010) The acetylation of RelA in Lys310 dictates the NF-κB-dependent response in post-ischemic injury. Cell Death & Disease 1:e96
Lanzillotta A, Porrini V, Bellucci A, Benarese M, Branca C et al (2015) NF-κB in innate neuroprotection and age-related neurodegenerative diseases. Frontiers in Neurology 6:98
Lawrence T (2009) The nuclear factor NF-κB pathway in inflammation. Cold Spring Harbor Perspectives in Biology 1:a001651
Lawson L, Perry V, Dri P, Gordon S (1990) Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 39:151–170
Ledoux AC, Perkins ND (2014) NF-κB and the cell cycle. 76–81
Lee J-Y, Nagano Y, Taylor JP, Lim KL, Yao T-P (2010a) Disease-causing mutations in parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. J Cell Biol 189:671–679
Lee M, Cho T, Jantaratnotai N, Wang YT, McGeer E et al (2010b) Depletion of GSH in glial cells induces neurotoxicity: relevance to aging and degenerative neurological diseases. The FASEB Journal 24:2533–2545
Lee W-H, Loo C-Y, Bebawy M, Luk F, Mason RS, Rohanizadeh R (2013) Curcumin and its derivatives: their application in neuropharmacology and neuroscience in the 21st century. Curr Neuropharmacol 11:338–378
Lee E, Park HR, Ji ST, Lee Y, Lee J (2014) Baicalein attenuates astroglial activation in the 1-methyl-4-phenyl-1, 2, 3, 4-tetrahydropyridine-induced Parkinson's disease model by downregulating the activations of nuclear factor-κB, ERK, and JNK. Journal of Neuroscience Research 92:130–139
Li Z, Nabel GJ (1997) A new member of the I kappaB protein family, I kappaB epsilon, inhibits RelA (p65)-mediated NF-kappaB transcription. Molecular and Cellular Biology 17:6184–6190
Li Q, Verma IM (2002) NF-κB regulation in the immune system. Nature Reviews Immunology 2:725
Lim H-S, Kim YJ, Kim B-Y, Park G, Jeong S-J (2018) The anti-neuroinflammatory activity of tectorigenin pretreatment via downregulated NF-κB and ERK/JNK pathways in BV-2 microglial and microglia inactivation in mice with lipopolysaccharide. Frontiers in Pharmacology 9:462
Liu RH (2004) Potential synergy of phytochemicals in cancer prevention: mechanism of action. The Journal of Nutrition 134:3479S–3485S
Lofrumento DD, Nicolardi G, Cianciulli A, Nuccio FD, Pesa VL et al (2014) Neuroprotective effects of resveratrol in an MPTP mouse model of Parkinson’s-like disease: possible role of SOCS-1 in reducing pro-inflammatory responses. Innate Immunity 20:249–260
Lukiw WJ, Bazan NG (1998) Strong nuclear factor-κB-DNA binding parallels cyclooxygenase-2 gene transcription in aging and in sporadic Alzheimer’s disease superior temporal lobe neocortex. J Neurosci Res 53:583–592
Luo Y, Hoffer A, Hoffer B, Qi X (2015) Mitochondria: a therapeutic target for Parkinson’s disease? Int J Mol Sci 16:20704–20730
Lv R, Du L, Liu X, Zhou F, Zhang Z et al (2019) Rosmarinic acid attenuates inflammatory responses through inhibiting HMGB1/TLR4/NF-κB signaling pathway in a mouse model of Parkinson’s disease. Life Sciences 223:158–165
Marchetti B, L’episcopo F, Morale MC, Tirolo C, Testa N et al (2013) Uncovering novel actors in astrocyte–neuron crosstalk in P arkinson’s disease: the W nt/β-catenin signaling cascade as the common final pathway for neuroprotection and self-repair. European Journal of Neuroscience 37:1550–1563
Mattson MP (2005) NF-κB in the survival and plasticity of neurons. Neurochem Res 30:883–893
Meberg PJ, Kinney WR, Valcourt EG, Routtenberg A (1996) Gene expression of the transcription factor NF-κ B in hippocampus: regulation by synaptic activity. Molecular Brain Research 38:179–190
Meffert MK, Baltimore D (2005) Physiological functions for brain NF-κB. Trends in Neurosciences 28:37–43
Meffert MK, Chang JM, Wiltgen BJ, Fanselow MS, Baltimore D (2003) NF-κB functions in synaptic signaling and behavior. Nature Neuroscience 6:1072
Mena MA, García de Yébenes J (2008) Glial cells as players in parkinsonism: the “good,” the “bad,” and the “mysterious” glia. The Neuroscientist 14:544–560
Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL et al (1997) IKK-1 and IKK-2: cytokine-activated IκB kinases essential for NF-κB activation. Science 278:860–866
Milbury PE, Kalt W (2010) Xenobiotic metabolism and berry flavonoid transport across the blood–brain barrier. J Agric Food Chem 58:3950–3956
Mizuno Y, Ohta S, Tanaka M, Takamiya S, Suzuki K et al (1989) Deficiencies in complex I subunits of the respiratory chain in Parkinson’s disease. Biochemical and Biophysical Research Communications 163:1450–1455
Mogi M, Harada M, Riederer P, Narabayashi H, Fujita K et al (1994) Tumor necrosis factor-α (TNF-α) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neuroscience Letters 165:208–210
Mogi M, Kondo T, Mizuno Y, Nagatsu T (2007) p53 protein, interferon-γ, and NF-κB levels are elevated in the parkinsonian brain. Neuroscience Letters 414:94–97
Monzani E, Nicolis S, Dell'Acqua S, Capucciati A, Bacchella C, Zucca FA, Mosharov EV, Sulzer D, Zecca L, Casella L (2019) Dopamine, oxidative stress and protein–quinone modifications in parkinson's and other neurodegenerative diseases. Angewandte Chemie International Edition 58:6512–6527
More SV, Kumar H, Kim IS, Song S-Y, Choi D-K (2013) Cellular and molecular mediators of neuroinflammation in the pathogenesis of Parkinson’s disease. Mediators of Inflammation 2013:952375
Mu X, He G, Cheng Y, Li X, Xu B et al (2009) Baicalein exerts neuroprotective effects in 6-hydroxydopamine-induced experimental parkinsonism in vivo and in vitro. Pharmacology Biochemistry and Behavior 92:642–648
Mu X, He G-R, Yuan X, Li X-X, Du G-H (2011) Baicalein protects the brain against neuron impairments induced by MPTP in C57BL/6 mice. Pharmacology Biochemistry and Behavior 98:286–291
Müller CW, Rey FA, Sodeoka M, Verdine GL, Harrison SC (1995) Structure of the NF-κB p50 homodimer bound to DNA. Nature 373:311
Mytilineou C, Werner P, Molinari S, Di Rocco A, Cohen G et al (1994) Impaired oxidative decarboxylation of pyruvate in fibroblasts from patients with Parkinson’s disease. Journal of Neural Transmission-Parkinson’s Disease and Dementia Section 8:223–228
Noort AR, Tak PP, Tas SW (2015) Non-canonical NF-κB signaling in rheumatoid arthritis: Dr Jekyll and Mr Hyde? Arthritis Research & Therapy 17:15
Norris KL, Hao R, Chen L-F, Lai C-H, Kapur M et al (2015) Convergence of parkin, PINK1 and α-synuclein on stress-induced mitochondrial morphological remodelling. Journal of Biological Chemistry M114:634063
Nuytemans K, Theuns J, Cruts M, Van Broeckhoven C (2010) Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: a mutation update. Human Mutation 31:763–780
Parker WD Jr, Parks JK, Swerdlow RH (2008) Complex I deficiency in Parkinson’s disease frontal cortex. Brain Research 1189:215–218
Patil SP, Jain PD, Sancheti JS, Ghumatkar PJ, Tambe R et al (2014) Neuroprotective and neurotrophic effects of apigenin and luteolin in MPTP induced parkinsonism in mice. Neuropharmacology 86:192–202
Peng H, Cheng F, Huang Y, Chen C, Tsai T (1998) Determination of naringenin and its glucuronide conjugate in rat plasma and brain tissue by high-performance liquid chromatography. Journal of Chromatography B: Biomedical Sciences and Applications 714:369–374
Pilleri M, Antonini A (2015) Therapeutic strategies to prevent and manage dyskinesias in Parkinson’s disease. Expert Opinion on Drug Safety 14:281–294
Pisanu A, Lecca D, Mulas G, Wardas J, Simbula G et al (2014) Dynamic changes in pro-and anti-inflammatory cytokines in microglia after PPAR-γ agonist neuroprotective treatment in the MPTPp mouse model of progressive Parkinson’s disease. Neurobiology of Disease 71:280–291
Pizzi M, Boroni F, Bianchetti A, Moraitis C, Sarnico I et al (2002) Expression of functional NR1/NR2B-type NMDA receptors in neuronally differentiated SK-N-SH human cell line. European Journal of Neuroscience 16:2342–2350
Pizzi M, Sarnico I, Boroni F, Benetti A, Benarese M et al. (2005) Inhibition of IκBα phosphorylation prevents glutamate-induced NF-κB activation and neuronal cell death. In: Re-engineering of the damaged brain and spinal cord. Springer, pp 59–63
Pizzi M, Sarnico I, Lanzillotta A, Battistin L, Spano P (2009) Post-ischemic brain damage: NF-κB dimer heterogeneity as a molecular determinant of neuron vulnerability. The FEBS Journal 276:27–35
Poewe W (2008) Non-motor symptoms in Parkinson’s disease. Eur J Neurol 15:14–20
Pomerantz JL, Baltimore D (2002) Two pathways to NF-κB. Molecular Cell 10:693–695
Qian Y, Cao L, Guan T, Chen L, Xin H, Li Y, Zheng R, Yu D (2015) Protection by genistein on cortical neurons against oxidative stress injury via inhibition of NF-kappaB, and ERK signaling pathway. JNK Pharmaceutical Biology 53:1124–1132
Rabie MA, El Fattah MAA, Nassar NN, El-Abhar HS, Abdallah DM (2018) Angiotensin 1-7 ameliorates 6-hydroxydopamine lesions in hemiparkinsonian rats through activation of MAS receptor/PI3K/Akt/BDNF pathway and inhibition of angiotensin II type-1 receptor/NF-κB axis. Biochem Pharmacol 151:126–134
Rai SN, Dilnashin H, Birla H, Singh SS, Zahra W, Rathore AS, Singh BK, Singh SP (2019a) The role of PI3K/Akt and ERK in neurodegenerative disorders. Neurotox Res 35:775–795
Rai SN, Zahra W, Singh SS, Birla H, Keswani C et al. (2019b) Anti-inflammatory activity of ursolic acid in MPTP-induced parkinsonian mouse model. Neurotox Res 1–11
Rajeswari A, Sabesan M (2008) Inhibition of monoamine oxidase-B by the polyphenolic compound, curcumin and its metabolite tetrahydrocurcumin, in a model of Parkinson’s disease induced by MPTP neurodegeneration in mice. Inflammopharmacology 16:96–99
Régnier CH, Song HY, Gao X, Goeddel DV, Cao Z et al (1997) Identification and characterization of an IκB kinase. Cell 90:373–383
Ren Z, Wang L, Cui J, Huoc Z, Xue J et al (2013) Resveratrol inhibits NF-κB signaling through suppression of p65 and IB kinase activities. Die Pharmazie-An International Journal of Pharmaceutical Sciences 68:689–694
Renaud J, Martinoli M-G (2019) Considerations for the use of polyphenols as therapies in neurodegenerative diseases. International Journal of Molecular Sciences 20:1883
Renaud J, Bournival J, Zottig X, Martinoli M-G (2014) Resveratrol protects DAergic PC12 cells from high glucose-induced oxidative stress and apoptosis: effect on p53 and GRP75 localization. Neurotoxicity Research 25:110–123
Rodriguez M, Hirsch E, Farrer M, Schapira A, Halliday G (2010) Missing pieces in the Parkinson’s disease puzzle. Nat Med 16:653661
Rodriguez-Oroz MC, Jahanshahi M, Krack P, Litvan I, Macias R et al (2009) Initial clinical manifestations of Parkinson’s disease: features and pathophysiological mechanisms. The Lancet Neurology 8:1128–1139
Ruiz PA, Haller D (2006) Functional diversity of flavonoids in the inhibition of the proinflammatory NF-κB, IRF, and Akt signaling pathways in murine intestinal epithelial cells. The Journal of Nutrition 136:664–671
Russo I, Bubacco L, Greggio E (2014) LRRK2 and neuroinflammation: partners in crime in Parkinson’s disease? J Neuroinflammation 11:52
Sacino AN, Brooks M, Thomas MA, McKinney AB, Lee S et al. (2014) Intramuscular injection of α-synuclein induces CNS α-synuclein pathology and a rapid-onset motor phenotype in transgenic mice. Proc Natl Acad Sci U S A 201321785
Saijo K, Winner B, Carson CT, Collier JG, Boyer L et al (2009) A Nurr1/CoREST transrepression pathway attenuates neurotoxic inflammation in activated microglia and astrocytes. Cell 137:47
Sarnico I, Lanzillotta A, Benarese M, Alghisi M, Baiguera C et al (2009a) NF-kappaB dimers in the regulation of neuronal survival. International Review of Neurobiology 85:351–362
Sarnico I, Lanzillotta A, Boroni F, Benarese M, Alghisi M, Schwaninger M, Inta I, Battistin L, Spano P, Pizzi M (2009b) NF-κB p50/RelA and c-Rel-containing dimers: opposite regulators of neuron vulnerability to ischaemia. J Neurochem 108:475–485
Schaffer S, Halliwell B (2012) Do polyphenols enter the brain and does it matter? Some theoretical and practical considerations. Genes & Nutrition 7:99–109
Schapira AH (2008) Mitochondrial dysfunction in neurodegenerative diseases. Neurochem Res 33:2502–2509
Schapira A, Cooper J, Dexter D, Clark J, Jenner P et al (1990) Mitochondrial complex I deficiency in Parkinson’s disease. Journal of Neurochemistry 54:823–827
Schmidt-Ullrich R, Mémet S, Lilienbaum A, Feuillard J, Raphaël M et al (1996) NF-kappaB activity in transgenic mice: developmental regulation and tissue specificity. Development 122:2117–2128
Schmitz ML, Baeuerle PA (1991) The p65 subunit is responsible for the strong transcription activating potential of NF-kappa B. EMBO J 10:3805–3817
Schneider A, Martin-Villalba A, Weih F, Vogel J, Wirth T et al (1999) NF-κB is activated and promotes cell death in focal cerebral ischemia. Nature Medicine 5:554
Segura-Aguilar J, Paris I, Muñoz P, Ferrari E, Zecca L et al (2014) Protective and toxic roles of dopamine in Parkinson’s disease. Journal of Neurochemistry 129:898–915
Sen R, Baltimore D (1986a) Inducibility of κ immunoglobulin enhancer-binding protein NF-κB by a posttranslational mechanism. Cell 47:921–928
Sen R, Baltimore D (1986b) Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 46:705–716
Shen W, Qi R, Zhang J, Wang Z, Wang H et al (2012) Chlorogenic acid inhibits LPS-induced microglial activation and improves survival of dopaminergic neurons. Brain Research Bulletin 88:487–494
Singh SS, Rai SN, Birla H, Zahra W, Kumar G et al. (2018) Effect of chlorogenic acid supplementation in MPTP-intoxicated mouse. Front Pharmacol 9
Sivandzade F, Prasad S, Bhalerao A, Cucullo L (2018) NRF2 and NF-κB interplay in cerebrovascular and neurodegenerative disorders: molecular mechanisms and possible therapeutic approaches. Redox Biol
Song J, Cheon SY, Jung W, Lee WT, Lee JE (2014) Resveratrol induces the expression of interleukin-10 and brain-derived neurotrophic factor in BV2 microglia under hypoxia. International Journal of Molecular Sciences 15:15512–15529
Song GJ, Rahman MH, Jha M, Gupta D, Kim J et al (2019) A Bcr-Abl inhibitor GNF-2 Attenuates inflammatory activation of glia and chronic pain. Frontiers in Pharmacology 10:543
Stott SR, Barker RA (2014) Time course of dopamine neuron loss and glial response in the 6-OHDA striatal mouse model of P arkinson’s disease. Eur J Neurosci 39:1042–1056
Suganuma M, Okabe S, Oniyama M, Tada Y, Ito H et al (1998) Wide distribution of [3H](-)-epigallocatechin gallate, a cancer preventive tea polyphenol, in mouse tissue. Carcinogenesis 19:1771–1776
Sulzer D, Cassidy C, Horga G, Kang UJ, Fahn S et al (2018) Neuromelanin detection by magnetic resonance imaging (MRI) and its promise as a biomarker for Parkinson’s disease. NPJ Parkinson’s Disease 4:11
Sun S-C (2011) Non-canonical NF-κB signaling pathway. Cell Research 21:71
Suresh D, Srinivasan K (2010) Tissue distribution & elimination of capsaicin, piperine & curcumin following oral intake in rats. Indian Journal of Medical Research 131
Tai Y, Qiu Y, Bao Z (2018) Magnesium lithospermate B suppresses lipopolysaccharide-induced neuroinflammation in BV2 microglial cells and attenuates neurodegeneration in lipopolysaccharide-injected mice. Journal of Molecular Neuroscience 64:80–92
Tak PP, Firestein GS (2001) NF-κB: a key role in inflammatory diseases. The Journal of Clinical Investigation 107:7–11
Tak PP, Gerlag DM, Aupperle KR, Van De Geest DA, Overbeek M et al (2001) Inhibitor of nuclear factor κB kinase β is a key regulator of synovial inflammation. Arthritis & Rheumatism 44:1897–1907
Tansey MG, Goldberg MS (2010) Neuroinflammation in Parkinson’s disease: its role in neuronal death and implications for therapeutic intervention. Neurobiology of Disease 37:510–518
Taylor D, Krige D, Barnes P, Kemp G, Carroll M, Mann VM, Cooper JM, Marsden CD, Schapira AH (1994) A 31P magnetic resonance spectroscopy study of mitochondrial function in skeletal muscle of patients with Parkinson’s disease. J Neurol Sci 125:77–81
Thanos D, Maniatis T (1995) NF-κB: a lesson in family values. Cell 80:529–532
Thirumangalakudi L, Yin L, Rao HV, Grammas P (2007) IL-8 induces expression of matrix metalloproteinases, cell cycle and pro-apoptotic proteins, and cell death in cultured neurons. Journal of Alzheimer’s Disease 11:305–311
Tripanichkul W, Jaroensuppaperch E (2013) Ameliorating effects of curcumin on 6-OHDA-induced dopaminergic denervation, glial response, and SOD1 reduction in the striatum of hemiparkinsonian mice. Eur Rev Med Pharmacol Sci 17:1360–1368
Tsao R (2010) Chemistry and biochemistry of dietary polyphenols. Nutrients 2:1231–1246
Tsoulfas G, Geller DA (2001) NF-κB in transplantation: friend or foe? Transplant Infectious Disease: Basic Science 3:212–219
Vallabhapurapu S, Karin M (2009) Regulation and function of NF-κB transcription factors in the immune system. Annual Review of Immunology 27:693–733
van der Merwe C, Jalali Sefid Dashti Z, Christoffels A, Loos B, Bardien S (2015) Evidence for a common biological pathway linking three Parkinson’s disease-causing genes: parkin, PINK1 and DJ-1. European Journal of Neuroscience 41:1113–1125
Vauzour D (2012) Dietary polyphenols as modulators of brain functions: biological actions and molecular mechanisms underpinning their beneficial effects. Oxidative Med Cellular Longevity 2012
Venkateshappa C, Harish G, Mythri RB, Mahadevan A, Bharath MS et al (2012) Increased oxidative damage and decreased antioxidant function in aging human substantia nigra compared to striatum: implications for Parkinson’s disease. Neurochemical Research 37:358–369
Verma IM, Stevenson JK, Schwarz EM, Van Antwerp D, Miyamoto S (1995) Rel/NF-kappa B/I kappa B family: intimate tales of association and dissociation. Genes & Development 9:2723–2735
Vitale N, Kisslinger A, Paladino S, Procaccini C, Matarese G et al (2013) Resveratrol couples apoptosis with autophagy in UVB-irradiated HaCaT cells. PLoS One 8:e80728
Waak J, Weber SS, Waldenmaier A, Görner K, Alunni-Fabbroni M et al (2009) Regulation of astrocyte inflammatory responses by the Parkinson’s disease-associated gene DJ-1. The FASEB Journal 23:2478–2489
Wakabayashi K, Hayashi S, Yoshimoto M, Kudo H, Takahashi H (2000) NACP/α-synuclein-positive filamentous inclusions in astrocytes and oligodendrocytes of Parkinson’s disease brains. Acta Neuropathologica 99:14–20
Wang J-Y, Xu L-Z (2009) The research of the effect of curcumin on dopaminergic neurons in mouse model of parkinson's disease. Journal of Taishan Medical College 8:001
Wang Q, Sun AY, Simonyi A, Jensen MD, Shelat PB et al (2005) Neuroprotective mechanisms of curcumin against cerebral ischemia-induced neuronal apoptosis and behavioral deficits. Journal of Neuroscience Research 82:138–148
Wang Y-H, Yu H-T, Pu X-P, Du G-H (2013) Baicalein prevents 6-hydroxydopamine-induced mitochondrial dysfunction in SH-SY5Y cells via inhibition of mitochondrial oxidation and up-regulation of DJ-1 protein expression. Molecules 18:14726–14738
Wang X-S, Zhang Z-R, Zhang M-M, Sun M-X, Wang W-W et al (2017) Neuroprotective properties of curcumin in toxin-base animal models of Parkinson’s disease: a systematic experiment literatures review. BMC Complementary and Alternative Medicine 17:412
Whiteside ST, Epinat JC, Rice NR, Israël A (1997) I kappa B epsilon, a novel member of the IκB family, controls RelA and cRel NF-κB activity. EMBO J 16:1413–1426
Williams CM, El Mohsen MA, Vauzour D, Rendeiro C, Butler LT et al (2008) Blueberry-induced changes in spatial working memory correlate with changes in hippocampal CREB phosphorylation and brain-derived neurotrophic factor (BDNF) levels. Free Radical Biology and Medicine 45:295–305
Wilms H, Rosenstiel P, Sievers J, Deuschl GN, Zecca L et al (2003) Activation of microglia by human neuromelanin is NF-κB dependent and involves p38 mitogen-activated protein kinase: implications for Parkinson’s disease. The FASEB Journal 17:500–502
Xu W, Zheng D, Liu Y, Li J, Yang L et al (2017) Glaucocalyxin B alleviates lipopolysaccharide-induced Parkinson’s disease by inhibiting TLR/NF-κB and activating Nrf2/HO-1 pathway. Cellular Physiology and Biochemistry 44:2091–2104
Yamamoto Y, Verma UN, Prajapati S, Kwak Y-T, Gaynor RB (2003) Histone H3 phosphorylation by IKK-α is critical for cytokine-induced gene expression. Nature 423:655
Yang Y-j, Hu L, Xia Y-P, Jiang C-Y, Miao C et al (2016) Resveratrol suppresses glial activation and alleviates trigeminal neuralgia via activation of AMPK. J Neuroinflammation 13:84
Yang X, Xu S, Qian Y, Xiao Q (2017) Resveratrol regulates microglia M1/M2 polarization via PGC-1α in conditions of neuroinflammatory injury. Brain Behav Immun 64:162–172
Yang J, Jia M, Zhang X, Wang P (2019) Calycosin attenuates MPTP-induced Parkinson’s disease by suppressing the activation of TLR/NF-κB and MAPK pathways. Phytotherapy Research 33:309–318
Yoshino H, Nakagawa-Hattori Y, Kondo T, Mizuno Y (1992) Mitochondrial complex I and II activities of lymphocytes and platelets in Parkinson’s disease. Journal of Neural Transmission-Parkinson’s Disease and Dementia Section 4:27–34
Youdim KA, Dobbie MS, Kuhnle G, Proteggente AR, Abbott NJ et al (2003) Interaction between flavonoids and the blood–brain barrier: in vitro studies. Journal of Neurochemistry 85:180–192
Yu S, Zheng W, Xin N, Chi Z-H, Wang N-Q et al (2010) Curcumin prevents dopaminergic neuronal death through inhibition of the c-Jun N-terminal kinase pathway. Rejuvenation Research 13:55–64
Yu Y, Shen Q, Lai Y, Ou X, Lin D et al (2018) Anti-inflammatory effects of curcumin in microglial cells. Frontiers in Pharmacology 9:386
Zaitone SA, Ahmed E, Elsherbiny NM, Mehanna ET, El-Kherbetawy MK et al (2019) Caffeic acid improves locomotor activity and lessens inflammatory burden in a mouse model of rotenone-induced nigral neurodegeneration: relevance to Parkinson’s disease therapy. Pharmacological Reports 71:32–41
Zecca L, Guadagni L, Bareggi S (1982) Determination of sodium flavodate in body fluids by high-performance liquid chromatography: application to clinical pharmacokinetic studies. Journal of Chromatography B: Biomedical Sciences and Applications 230:168–174
Zecca L, Wilms H, Geick S, Claasen J-H, Brandenburg L-O et al (2008) Human neuromelanin induces neuroinflammation and neurodegeneration in the rat substantia nigra: implications for Parkinson’s disease. Acta Neuropathologica 116:47–55
Zhang Y, Barres BA (2010) Astrocyte heterogeneity: an underappreciated topic in neurobiology. Current Opinion in Neurobiology 20:588–594
Zhang F-X, Xu R-S (2018) Juglanin ameliorates LPS-induced neuroinflammation in animal models of Parkinson’s disease and cell culture via inactivating TLR4/NF-κB pathway. Biomedicine & Pharmacotherapy 97:1011–1019
Zhang W, Phillips K, Wielgus AR, Liu J, Albertini A et al (2011) Neuromelanin activates microglia and induces degeneration of dopaminergic neurons: implications for progression of Parkinson’s disease. Neurotoxicity Research 19:63–72
Zhang F, Zhou H, Wilson BC, Shi J-S, Hong J-S et al (2012a) Fluoxetine protects neurons against microglial activation-mediated neurotoxicity. Parkinsonism & Related Disorders 18:S213–S217
Zhang Z, Cui W, Li G, Yuan S, Xu D et al (2012b) Baicalein protects against 6-OHDA-induced neurotoxicity through activation of Keap1/Nrf2/HO-1 and involving PKCα and PI3K/AKT signaling pathways. Journal of Agricultural and Food Chemistry 60:8171–8182
Zhang G, Li J, Purkayastha S, Tang Y, Zhang H et al (2013a) Hypothalamic programming of systemic ageing involving IKK-β, NF-κB and GnRH. Nature 497:211
Zhang J, Chen J, Yang J, Xu C-W, Pu P et al (2013b) Resveratrol attenuates oxidative stress induced by balloon injury in the rat carotid artery through actions on the ERK1/2 and NF-kappa B pathway. Cellular Physiology and Biochemistry 31:230–241
Zhang H, Hilton MJ, Anolik JH, Welle SL, Zhao C et al (2014) NOTCH inhibits osteoblast formation in inflammatory arthritis via noncanonical NF-κB. The Journal of Clinical Investigation 124:3200–3214
Zhang X, Yang Y, Du L, Zhang W, Du G (2017) “Baicalein exerts anti-neuroinflammatory effects to protect against rotenone-induced brain injury in rats.” Int Immunopharmacol 50:38–47
Zhou Y, Zhang T, Wang X, Wei X, Chen Y, Guo L, Zhang J, Wang C (2015) Curcumin modulates macrophage polarization through the inhibition of the toll-like receptor 4 expression and its signaling pathways. Cell Physiol Biochem 36:631–641
Zhou J, Deng Y, Li F, Yin C, Shi J et al (2019) Icariside II attenuates lipopolysaccharide-induced neuroinflammation through inhibiting TLR4/MyD88/NF-κB pathway in rats. Biomedicine & Pharmacotherapy 111:315–324
Zucca FA, Basso E, Cupaioli FA, Ferrari E, Sulzer D et al (2014) Neuromelanin of the human substantia nigra: an update. Neurotoxicity Research 25:13–23
Zuo L, Motherwell MS (2013) The impact of reactive oxygen species and genetic mitochondrial mutations in Parkinson’s disease. Gene 532:18–23
Acknowledgments
SSS, SNR, HB, WZ, and ASR are sincerely thankful to ICMR, DBT, and BHU, India, for their respective fellowship. The present study was not supported by any funding agencies.
Availability of Data and Material
NA
Author information
Authors and Affiliations
Contributions
SSS planed the work and drafted the manuscript; SNR, WZ, and ASR helped in the manuscript preparation; HB designed and has drawn the figures and pathway; and SPS guided throughout the manuscript.
Corresponding author
Ethics declarations
Ethics Approval and Consent to Participate
NA
Consent for Publication
NA
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Singh, S.S., Rai, S.N., Birla, H. et al. NF-κB-Mediated Neuroinflammation in Parkinson’s Disease and Potential Therapeutic Effect of Polyphenols. Neurotox Res 37, 491–507 (2020). https://doi.org/10.1007/s12640-019-00147-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-019-00147-2