
Abstract
Neurodegenerative diseases are part of the central nervous system (CNS) disorders that indicate their presence with neuronal loss, neuroinflammation, and increased oxidative stress. Several pathophysiological factors and biomarkers are involved in this inflammatory process causing these neurological disorders. The nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) is an inflammation element, which induced transcription and appears to be one of the important players in physiological procedures, especially nervous disorders. NF-κB can impact upon series of intracellular actions and induce or inhibit many inflammation-related pathways. Multiple reports have focused on the modification of NF-κB activity, controlling its expression, translocation, and signaling pathway in neurodegenerative disorders and injuries like Alzheimer’s disease (AD), spinal cord injuries (SCI), and Parkinson’s disease (PD). Curcumin has been noted to be a popular anti-oxidant and anti-inflammatory substance and is the foremost natural compound produced by turmeric. According to various studies, when playing an anti-inflammatory role, it interacts with several modulating proteins of long-standing disease signaling pathways and has an unprovocative consequence on pro-inflammatory cytokines. This review article determined to figure out curcumin’s role in limiting the promotion of neurodegenerative disease via influencing the NF-κB signaling route. Preclinical studies were gathered from plenty of scientific platforms including PubMed, Scopus, Cochrane, and Google Scholar to evaluate this hypothesis. Extracted findings from the literature review explained the repressing impact of Curcumin on the NF-κB signaling pathway and, occasionally down-regulating the cytokine expression. Yet, there is an essential need for further analysis and specific clinical experiments to fully understand this subject.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Neurodegenerative diseases (ND), or continuous loss of neurons’ function or structure, are a set of features that gradually lead to neural dysfunction or neural loss. Huntington’s disease (HD), spinal cord injury (SCI), Parkinson's disease (PD), and Alzheimer’s disease (AD) are among the prevalent central nervous system (CNS) diseases. Neuro-inflammation is considered a common driver of several neurological impairments, which may be induced by trauma, toxic metabolites, and infections, for instance (Kumar et al. 2015b). Furthermore, reports have shown that inflammation cascades affect CNS-exerted functions, i.e., movement, memory and learning, judgment, cognition, and coordination, as well as neuron vitality and structures. In this regard, selective neurons have undergone experiments to obtain more information about the molecular pathogenesis of neurodegenerative diseases (Ransohoff 2016). Multiple studies have demonstrated the capacity of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) production in the development of ND, such as amyotrophic lateral sclerosis (ALS), Huntington’s, ischemia, and AD (Fridmacher et al. 2003).
NF-κB is one of the pathways that is involved in neurodegeneration processes. NF-κB is an inflammatory transcription element that is composed of five family members entitled RelA (p65), RelB, c-Rel, NF-κB1 (p105/p50), and NF-κB2 (p100/p52). Stimulation of NF-κB pathway results in the nuclear translocation of the NF-κB p52/RelB or p50/p65 dimers, as well as anti-apoptotic gene expression regulation via these dimers (Sun et al. 2022). NF-κB can be actuated by three distinct pathways: atypical, canonical, and non-canonical. The apparent contradictory effects of these pathways are due to factors for instance the pathway provocations, the cellular position, and the cell sort. During the canonical pathway, pro-inflammatory genes undergo temporary and immediate transcription. In contrast, non-canonical pathway involves protein synthesis through tumor necrosis factor receptors (TNFR) in response to activated stimuli (Mincheva-Tasheva and Soler 2013). RelA/p50 heterodimers Observation in astrocytes, Schwann cells, microglia, also neurons in many portions of promoting and mature nervous system elucidated the participation of NF-κB, hence examining the role of NF-κB in ND was initiated (Mincheva et al. 2011). According to these analysis, cytokines and chemokines from microglia are released by NF-κB expression, contributing to neural inflammations (Thawkar and Kaur 2019). According to NF-κB’s broad presence in reacting to cellular inflammation, it has turned to an appealing intention to study (Chiarini et al. 2020). Various research endeavors have been accomplished to fully understand the underlying procedures by which NF-κB plays a part in development of NDs. Moreover, multiple chemical and phytochemicals have been tested for their possible impact regulation of NF-κB pathways. One of the phytochemicals that has been vastly investigated due to its anti-inflammatory features is curcumin. The effect of curcumin in the reduction of undesired activation of NF-κB pathway during inflammation has been demonstrated via many evaluations. Curcumin is known as an active component of a plant called Curcuma longa L. This plant, also recognized as turmeric, is an eternal herb from the Zingiberaceae family with more than 100 discovered species around the world. Upon the investigation of the rhizomes of turmeric, a yellow pigment known as curcumin was isolated. Curcumin is not only a food-coloring agent, spice, or dietary supplement, but also has been categorized as an herbal medicament (Aggarwal and Sung 2009). Curcumin has a polyphenolic structure and has given the IUPAC ID of (1E,6E)-1,7-bis(4-hydroxy-3-methoxyphenyl)-1,6-heptadiene-3,5-dione)11 by chemical (Fig. 1) (Priyadarsini 2014).

Curcumin chemical structure and active functional groups (blue: methoxy phenolic groups, red: β-diketone)
The antimicrobial, antineoplastic, anti-inflammatory, and antioxidant features of curcumin in different organs have made it a considerable option for the prohibition or deferment of neurodegenerative diseases (Bhat et al. 2019). However, the low bioavailability and the absorption rate of oral intake of curcumin, along with its high biotransformation and elimination rate, have been of great concern (Rachmawati et al. 2013). Therefore, a wide range of surveys have focused on improving the pharmacokinetics (PK) and delivery systems of curcumin (Anand et al. 2008b).
Recently, extensive surveys have been carried out to discover the pharmacological aim of curcumin to define the molecular processes stimulated through this composite (Basnet and Skalko-Basnet 2011). Evidence shows that curcumin can interact with various factors involved in the inflammation process, including p38 mitogen-activated protein kinase (MAPK), vascular endothelial growth factor (VEGF), glutathione (GSH), p-Tau (p-τ), reactive oxygen species (ROS), cyclin D1, cyclooxygenase-2 (COX-2), tumor necrosis factor- (TNF-) α, inducible nitric oxide synthase (iNOS), NF-κB, DNA (cytosine-5)-methyltransferase-1, nuclear factor E2-related factor 2 (Nrf2), β-catenin, cytosolic PLA2, FOXO3,5-lipoxygenase (5-LOX), and prostaglandin E2 (PGE2), which can activate signaling pathways relevant to chronic diseases (Anand et al. 2008a). Curcumin diminishes the expression of many proinflammatory intracellular inflammatory systems like hypoxia-inducible factor-1 (HIF-1), proinflammatory cytokines in particular TNF-α, interleukin-(IL-)6, and IL-1β, iNOS, and NF-κB, at the molecular level. It also exerts an antiapoptotic impression via a decrease of the bax/Bcl-2 ratio and overexpressing B cell lymphoma 2 (Bcl-2). As an antioxidant, curcumin provokes the nuclear factor erythroid 2-related factor 2/antioxidant responsive element (Nrf2/ARE) pathway, suppresses ROS and mitochondrial cell death pathway, elevates Cu–Zn superoxide dismutase (SOD), and reinstates deficiency of the Glutathione (GSH) levels (Fu et al. 2015).
Unlike the known interaction with functional proteins, the benefits of curcumin at the system levels are still unclear (Grynkiewicz and Ślifirski 2012). Several investigations have declared a lower occurrence of neurological disorders in people consuming curcumin (Sharma et al. 2017). Considering this information, curcumin could be assumed as a prospective candidate for precluding neurodegenerative disease (Garcia-Alloza et al. 2007).
To explicate the defensive influence of curcumin signaling pathways in neurodegenerative diseases, multiple studies have been designed. Recently, curcumin has been exhibited to impede the NF-κB ways as it was a trigger for neuro-inflammation occurrence (Schmidt-Ullrich et al. 1996; Aggarwal and Harikumar 2009). Therefore, this review aims at discovering the mechanisms by which curcumin prevents different kinds of NDs by NF-κB pathway modulation.
Search strategy
Research findings were assembled from scientific databases involving Google Scholar, PubMed, Scopus, and Cochrane Library for, in vitro and in vivo research disclosed in English language from 1997 to 2023 Search words of titles include “Curcumin longa” OR “Curcuma” AND “Neurodegenerative diseases” AND “NF-κB pathway” in order to collect related articles. In the next step, the full-text of each publication was read as well as investigated to find the proper information, which resulted in the elimination of some articles in this stage. Finally, based on available papers, the process of data extraction started.
Results
Curcumin: an overview of the pharmacological actions
Curcumin, with a diferuloylmethane structure, is a natural compound with a variety of functional groups including ketone, phenol, enone, and aromatic ether. It has been proved that methoxy phenolic groups and β-diketone are the active functional groups of this phytochemical that may engage in oxidation (Fig. 1). It is also a diarylheptanoid isolated from C. longa, which is native to South Asia (Kumar et al. 2015a).
Curcumin is a well-known spice for coloring and flavoring foods. It is also used in the cosmetic industry. Numerous studies have evaluated curcumin’s therapeutic and biological activities. Results have shown that curcumin can ameliorate liver ailments, cardiovascular diseases, urinary tract infections, rheumatoid arthritis, eye diseases namely, conjunctivitis and chronic anterior uveitis, different malignancies including lung, skin, breast, prostate, gastrointestinal, and many neurological diseases including PD, epilepsy, AD, Multiple sclerosis (MS). It also has wound-healing properties (Sharifi-Rad et al. 2020).
The common routes of administration of curcumin for many in vivo and clinical studies are gavage, intra-peritoneal (i.p.), and intravenous (Pivari et al.). However, it has been declared that curcumin’s serum level was higher when it was utilized IV or IP. This may be due to its hydrophobicity which is one of the reasons for its low bioavailability. Curcumin is poorly absorbed in the gastrointestinal (GI) tract, rapidly metabolized in the liver and intestine by sulfation and glucuronidation, and is mainly defecated when it is administrated orally (Anand et al. 2007). Different tactics have been assessed to ameliorate curcumin’s bioavailability, from installing curcumin in nanoparticles, phospholipid complexes, liposomes, and micelles, to searching for analogs with higher bioavailability i.e., curcuminoids. For example, nanoparticle formulations of curcumin can enhance water solubility and secure intracellular delivery. Micelles can increase GI absorption, and conjugation with piperine, an alkaloid from black pepper, can slow down its metabolism by suppressing of glucuronidation in the intestine and liver (Nocito et al. 2021).
Most of the curative impacts of curcumin are due to its activity against inflammation and oxidative stress. It is also beneficial in autoimmune diseases, such as MS, regulating cytokines like interleukin-12 (IL-12) and IL-6. Curcumin can affect oxidative stress via scavenging ROS, suppressing enzymes that can generate ROS, such as COX, lipoxygenase (LOX), and xanthine oxidase, up-regulating antioxidant enzymes like SOD, glutathione peroxidase (GPx), catalase (CAT), and heme oxygenase 1 (HO-1), which consequently reduce lipid peroxidation (LPO) (23). These activities can be related to the Nrf2 pathway excitement that can increase antioxidant enzymes and decrease oxidative stress (Ashrafizadeh et al. 2020; Khayatan et al. 2022). Moreover, curcumin can down-regulate the COX pathway which is involved in oncogenesis and inflammation. It can also inhibit the NF-κB pathway so that pro-inflammatory cytokines including IL-8, IL-1β, IL-6, IL-2, TNF-α, PGE2, macrophage inflammatory protein-1alpha (MIP-1α), monocyte chemoattractant protein 1 (MCP-1) and C-reactive protein (CRP) decrease (Sharifi-Rad et al. 2020; Menon and Sudheer 2007). Furthermore, curcumin can down-regulate toll-like receptor 4 (TLR4)/tumor necrosis factor receptor (TNFR)-associated factor 6 (TRAF6)/MAPK pathway, which causes the reduction of pro-inflammatory mediators (Yang et al. 2017).
Several experiments manifested that curcumin ameliorates inflammation and oxidative stress in type 2 diabetes mellitus (T2DM) and it can inhibit the NF-κB signaling pathway that is one of the pathways involved in inflammation. Also, it can regulate lipid metabolism. Additionally, curcumin attenuates T2DM-related diseases including diabetic neuropathy and retinopathy (Pivari et al. 2019).
Various studies on several kinds of cancers like lung, liver, and breast declared that curcumin can suppress the NF-κB pathway. In cancers, curcumin reduces vascular endothelial growth factor receptor (VEGFR) by down-regulation of the phosphoinositide-3-kinase (PI3K)/protein kinase B (Akt) pathway. Thus, angiogenesis is suppressed. Therefore, by the management of NF-κB and activator protein 1 (AP-1), the VEGF level declines (Shanmugam et al. 2015).
Curcumin can act as a neuroprotective compound by scavenging ROS, inducing the Nrf2 pathway, suppressing the NF-κB pathway, decreasing inflammatory cytokines, and affecting other related pathways in the CNS. For instance, an in vivo experiment by Huang et al. in 2018 on Sprague Dawley (SD) male rats with middle cerebral artery occlusion (MCAO) elucidated that curcumin can regulate TLR4/p38/MAPK pathway and inhibit inflammation. They also declared that curcumin can modulate the PI3K/Akt/mammalian target of the rapamycin (mTOR) pathway and lower autophagy in cerebral ischemia–reperfusion (Huang et al. 2018). Another experiment on AD revealed that curcumin decreases microglia and astrocyte activation, suppresses the NF-κB pathway, and elevates peroxisome proliferators-activated receptor γ (PPARγ) expression. These data suggest that curcumin is a potential bioactive molecule against neurodegenerative disorders (Sharifi-Rad et al. 2020).
NF-κB signaling pathways in neurodegenerative disorders
The NF-κB family and its activation pathways
NF-κB contains a group of five transcription factors engaged in different cellular processes, with a notable function in inflammation. This family includes NF-κB1 (p105/p50), NF-κB2 (p100/p52), RelA (p65), RelB, and c-Rel. NF-κB stimulation prompts the transcription of specific genes the majority of which are pro-inflammatory genes. The canonical pathway is well-studied and crucial in inflammatory reactions, a key feature in the development of AD. Initially inert, the p65/p50 dimers in the canonical pathway are confined in the cytoplasm by IκB. Just after encountering pro-inflammatory signals like cytokines, danger-associated molecular patterns, and pathogens, the p65/p50 dimers are liberated from IκB owing to a phosphorylation process that leads to IκB's degradation. Following this, p65/p50 translocates to the nucleus, binding to its target motif (κB motif), and activating NF-κB target genes. Conversely, the noncanonical pathway is induced by certain tumor necrosis factor Receptor (TNFR) superfamily members, triggering NF-κB-inducing kinase (NIK). NIK phosphorylates IκB kinase alpha (IKKα), which then phosphorylates p100’s C-terminal, generating p52. During the post-phosphorylation cascade, p52/RelB moves to the nucleus, initiating NF-κB target gene expression that influences immune cell development (Sun 2011). In the nervous system, several methods can trigger stimulation of the NF-κB pathway. The well-known IKK-dependent processes encompass both non-canonical and canonical pathways. However, a novel IKK-independent mechanism, the atypical pathway, has also been identified (Bender et al. 1998). Typically, the non-canonical and the canonical pathways exhibit two key distinctions: the NF-κB dimer translocation to the nucleus (RelA/p50 for canonical, RelB/p52 for non-canonical) and the involvement of IκB in their activation (IκB-dependent for canonical, IκB-independent for non-canonical). NF-κB activation in the cells through the canonical pathway is the most common pathway of its activation (Heissmeyer et al. 1999).
The “atypical pathway” operates independently of IKK but relies on IκBα for its functioning, leading to the movement of RelA/p50 dimers to the nucleus. In the nervous system, the initiation of this pathway has been linked to stimuli like neurotrophic factors, erythropoietin, and hydrogen peroxide, the non-canonical pathway is also referred to as the IκB-independent pathway (Bonizzi et al. 2004).
NF-κB in neurodegenerative disorders
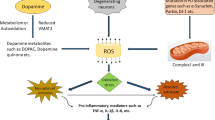
There are numerous kinds of neurons in the mammalian CNS, which can be classified as sensory, motor, and interneuron or excitatory and inhibitory neurons. NF-κB function has been studied in diverse types of neurons, such as excitatory (glutamatergic) and inhibitory (GABAergic) neurons, as well as at the synapse where neuron connections are formed. Neuronal NF-κB can be activated by several stimuli, like inflammatory mediators and growth factors like nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), and excitatory neurotransmitters including glutamate, epinephrine, and norepinephrine (Snow and Albensi 2016). In addition to distinct pathways for activation, there are multiple specific ways to measure NF-κB activation at both behavioral and cellular levels. Within the NF-κB family, members like c-Rel, p50, and p65/RelA as well as IKK, have been associated with the regulation of memory and learning in mice (Dresselhaus and Meffert 2019). However, many studies investigating the specific effects of NF-κB subunits on behavior have not considered cell type specificity. Instead, they have used mouse models lacking certain subunits throughout the body, which result in effects beyond the nervous system. By disrupting NF-κB signaling pathway, especially in specific types of neurons, such as forebrain excitatory neurons or GABAergic interneurons (O'Mahony et al. 2006), researchers have confirmed NF-κB's importance in synaptic plasticity and cognitive behavior. Still, this manipulation does not offer precise subunit-specific insights. Multiple reviews have covered the cognitive impacts of NF-κB signaling (Kaltschmidt and Kaltschmidt 2015). Impairments in NF-κB signaling can to lead to deficits in evaluations of long-term plasticity, long-term potentiation (LTP) (Kyrargyri et al. 2015), and long-term depression (LTD) (O'Riordan et al. 2006). Activating the NF-κB pathway in the excitatory glutamatergic neurons of murine stimulates dendritic spines and excitatory synapse development, whereas reduced NF-κB activity (due to loss of RelA/p65) diminishes dendritic spine size, density, and miniature excitatory post-synaptic currents (mEPSCs). This occurs during the developmental stages of synapse formation and in mature neurons that respond to greater synaptic demand (Boersma et al. 2011). Research from various laboratories suggests that in normal physiological conditions, neuronal NF-κB serves to enhance synapse growth, boost the activity of synapses, and support lasting forms of plasticity. Beyond the gene targets studied before in cancer and immune, NF-κB has also been found to modulate specific downstream targets that significantly contribute to synaptic plasticity. These include Postsynaptic density protein 95 (PSD-95), Spine-associated Rap GTPase-activating protein (SPAR), Protein kinase A (PKA), neuronal Nitric Oxide synthase (nNOS), and growth factors like insulin-like growth factor 2 (IGF-2) and BDNF. The functions related to neuronal plasticity might explain why NF-κB is necessary for the assessment of behavioral indicators of cognition, as observed in numerous investigations. However, it is worth noting that certain behavioral experiments have not exclusively manipulated the NF-κB pathway in neurons, potentially allowing NF-κB in other cell types to contribute to the observed effects. The presence of NF-κB at stimulatory synapses and its triggering via excitatory synaptic activity are also pivotal factors in defining the distinct functions of the NF-κB pathway in mammalian neurons (38). Studies reveal that astrocytes, macrophages, and microglia release radicals, proinflammatory cytokines, and excitotoxins upon activation of NF-κB triggered by TNF-α, promoting neuronal death. Furthermore, inhibiting the NF-κB pathway, only in astrocytes, protects neurons from cell death (Green et al. 1997). Comparable outcomes were seen in hippocampal neurons affected by kainic acid excitotoxicity, where microglial IKKβ gene deletion yielded similar outcomes (Cho et al. 2008). Thus, inhibitors targeting the NF-κB pathway specifically in glial cells are suggested for prospective treatment in neurodegenerative disorders like ALS and AD (Fig. 2) (Mattson and Camandola 2001).

Divergent impacts of NF-κB activation on neurons and glial cells in pathological contexts. The same trigger that activates NF-κB can yield opposing outcomes in neurons
The role of curcumin in regulation of NF-κB in Alzheimer's disease
Alzheimer's disease, a neurodegenerative condition linked to aging, leads to gradual memory loss, cognitive decline, behavior changes, and difficulties with daily tasks. It is characterized by amyloid-β (Ab) plaques, neurofibrillary tangles, inflammation, and vascular changes in the brain. The precise connection between these plaques and tangles and the disease process remains uncertain but is being actively studied. The decline in cognitive function along with nerve cell degeneration is closely tied to abnormal activity within neural networks, synaptic dysfunction, and loss of synapses. Yet, the exact molecular mechanisms driving these effects aren’t fully comprehended, with some being challenging to reproduce in mouse models of the disease (De Strooper and Karran 2016). Numerous genes have been linked to the progression of AD through genetic investigations. These genes include amyloid precursor protein (APP), Presenilin1 (PSEN1), Presenilin2 (PSEN2), apolipoprotein E (ApoE), Triggering Receptor Expression on Myeloid cells 2 (TREM2), and others (Kumar and Thakur 2016). Analyzing promoters and conducting examinations have revealed that the expression of each of these genes is regulated by NF-κB (Chami et al. 2012). In certain instances, products originating from genes linked to AD, such as presenilin1, have demonstrated the ability to reciprocally activate NF-κB, specifically, the RelA/p65 containing dimers, within potential pro-inflammatory cascades (Tanaka et al. 2018).
The TREM family of proteins plays a pivotal role in neuroinflammatory cascades, and emerging evidence points to NF-κB as a primary regulator influencing the expression of both TREM1 and TREM2 (Owens et al. 2017). Within microglia, TREM2 acts as a receptor on the surface essential for activation-related responses, such as survival, proliferation, and phagocytosis. Variants causing reduced TREM2 function have been related to elevated AD with late-onset risk in humans, and the absence of TREM2, as well as TREM2 deficiency in mice, lead to AD-related pathological changes (Guerreiro et al. 2013). It’s also suggested that NF-κB’s control over microglial TREM2 expression might extend to unexplored effects on synaptic plasticity, which is integral to memory and learning. The comprehensive role of NF-κB in AD is extensively addressed in a specialized review (Snow and Albensi 2016), discussing its regulation by Ab and presenting a broad spectrum of potential NF-κB targets implicated in AD progression or cognitive symptoms, encompassing cAMP response element-binding protein (CREB), manganese superoxide dismutase (MnSOD), Postsynaptic density protein 95 (PSD95), and Calcium–calmodulin (CaM)-dependent protein kinase II (CAMKII). Moreover, NF-κB has been related to the control of ApoE (Fig. 3), wherein the ApoE4 variant is a significant genetic risk factor for AD with late-onset. Analysis of gene promoters has recognized NF-κB-binding sites upstream of the ApoE transcription initiation start site, and functional control of ApoE4 expression through NF-κB signaling has been confirmed through experiments using glial cells (Lahiri 2004). Given NF-κB’s multifaceted engagement in cognitive functions and inflammatory processes, it has garnered attention as a potential treatment focus for early intervention in AD. Notably, it’s not just genetic factors but additionally, environmental factors contributing to AD risk, along with protective elements like dietary habits, exercise, and anti-inflammatory medications, exhibit correlations with NF-κB. Furthermore, since aging, the primary AD risk factor is linked to heightened brain activation of NF-κB and inflammation specific to certain tissues, there are implications for AD and other neurodegenerative conditions (Jones and Kounatidis 2017). Figure 3 illustrates the pivotal role of NF-κB in AD. Notably, NF-κB activates Beta-Secretase 1 (BACE1), a factor that encourages the establishment of Aβ fibrils. This, in turn, triggers a feedback loop where Aβ fibrils initiate a loop where they directly activate NF-κB, causing the expression of APOE4 and mGluR5. In microglia and astrocytes, Aβ42 also activates NF-κB, leading to the expression of inflammatory molecules that contribute to myelin injury. Moreover, NF-κB's activation initiates the production of microRNAs that suppress the production of diverse neuroprotective substances. Likewise, the accumulation of hyperphosphorylated tau in the AD brain is heightened through NF-κB-driven initiation of SET Nuclear Proto-Oncogene (SET), which inhibits the dephosphorylation of tubulin-associated unit (tau). Conversely, the presence of glycated tau triggers the production of reactive oxygen species (ROS), consequently activating NF-κB. The intricate network of pathways that collectively lead to neurodegeneration in AD demonstrates a strong interconnection, perpetuated by continuous NF-κB activity in both glial cells and neurons (Sun et al. 2022). An in vivo study by Zhanget al. on Apolipoprotein E4 transgenic mice (ApoE4-Tg) revealed that delivery of curcumin, 40 mg/kg, i.p., impeded the translocation of the NF-κB subunit p65. This study elucidated that the incidence of Endoplasmic reticulum (ER) stress occurrence and the NF-κB signaling pathway activation is evident in ApoE4- Tg mice was inhibited by curcumin (40 mg/kg, i.p.) therapy. Therefore, neuroinflammation and subsequently leading and memory capability in ApoE4-Tg mice got better (Kou et al. 2021). In a study performed in 2023 by Kumar et al., the effect of different treatments against Alzheimer’s disease was measured. The treatments included Scopolamine hydrobromide (2.5 mg/kg i.p.), Curcumin (100, 200 mg/kg p.o.), Coenzyme Q10 (Co-Q10) (200 mg/kg p.o.), Memantine (MEM) (10 mg/kg i.p.), and their combination. The results showed that administering a high dose of curcumin for 4 weeks, either alone or in combination with Co-Q10, significantly enhanced the function of mature female Wistar rats with Alzheimer’s disease and reduced the expression of NF-κB (Kumar et al. 2023). An in vitro study on SH-SY5Y cells elucidated that curcumin (10 μM) upregulates PPARγ expression, which regulates inflammation in transcription level and inhibits NF-κB signaling pathway in APOE4-induced neurological damage. It was also manifested that this agent could decrease neuro-inflammation by reducing TNF-α, IL-1β, and NO creation while simultaneously reducing COX-2 and iNOS protein production (Wang et al. 2020). Another in vitro study exerted by Wang et al. confirmed that upregulation of PPARγ by curcumin (0–30 μmol/l) can inhibit NF-κB signaling in SH-SY5Y cells, which led to alleviation of injuries induced by ApoE4 (Wang et al. 2020). In addition, Song Yu et al. attempted to evaluate the beneficial impact of curcumin on the pathophysiology of PD in an in vitro rat pups’ astrocyte cell. An administration of 1, 2, 4, and 8 Μμ of curcumin reported a reduction in quantities of TLR4 and its subsequent components including NF-κB, MyD88, interferon regulatory factor 3 (IRF3), and transmucosal immediate-release fentanyl (TIRF), which are stimulated by 1-methyl-4-phenylpyridinium ion (MPP+). Additionally, curcumin can inactivate the immune response of morphological and TLR4 stimulation in MPP+-induced astrocytes (Yu et al. 2016). Curcumin’s efficacy in 2 μM concertation has been assessed in oxidative stress and hypoxia in hippocampal cells by Chhunchha et al. in 2013. Results have shown that curcumin can reduce ROS, inhibit NF-κB stimulation, decrease Bax and caspases, and prevent apoptosis via peroxiredoxin6 (Prdx6) up-regulation after hypoxia (Chhunchha et al. 2013). To investigate their claim, Wang et al. experimented in vitro study on MES23.5 cells. The result showed, 2 × 104 µmol per L curcumin in dimethyl sulfoxide (DMSO) for 24 h reduced ROS generation, which led to inhibition and regulation of 6-OHDA-inudced NF-κB nuclear translocation (42). Alzheimer’s pathology is related to toxic plaque accumulation that is composed of Aβ. Rate-limiting protease in Aβ generation is BACE1 accordingly. BACE1 inhibitors are a potent treatment for AD. In 2020, Huang et al. study demonstrated enhanced BACE1 promotor activity and Aβ production correlated to NF-κB activation and indicated that curcumin’s role in inhibiting NF-κB activity is at promotor level (Huang et al. 2020). Due to the possible impacts of high levels of PGs on neurodegeneration, Kang et al. performed a study that indicated the therapeutic consequences of curcumin on BV2 microglial cells stimulated by LPS in a concentration-dependent manner, 16 μM curcumin reduced more than 50% of NF-κB and AP-1 DNA bindings and also had a great impact on reduction of COX-2 mRNA, protein, and enzyme activity (Kang et al. 2004).

NF-κB plays a central role in initiating a cascade of factors that are involved in Alzheimer disease-related changes
The role of curcumin in the regulation of NF-κB in Parkinson’s disease
Parkinson’s disease (PD) is a significant neurodegenerative condition that influences around 1% of individuals over 60 years old. It causes unintended or uncontrollable movements, such as tremors, rigidity, and bradykinesia (DeMaagd and Philip 2015). Oxidative stress is thought to prompt the apoptosis of the substantia nigra’s dopaminergic neurons, leading to the observed neurodegenerative symptoms in PD patients, PD was initially reported by James Parkinson in 1817 (Parkinson 2002). Like AD, PD entails alterations in the neuronal cytoskeleton, albeit affecting specific nerve cell types that are particularly susceptible. Cases are mostly sporadic and many genes have been identified to relate to several gene’s mutations. The main genes implicated in PD are a-synuclein (SNCA), parkin, PTEN-induced putative kinase 1 (PINK1), leucine-rich repeat kinase 2 (LRRK2), and DJ-1 (Wood-Kaczmar et al. 2006). SNCA is a protein pivotal in synaptic dopamine release process. It can accumulate in Lewy bodies in Lewy body dementia (LBD), which can be an infection associated with abnormal accumulation of SNCA in the brain. These deposits, called Lewy bodies, affect chemicals in the brain, changes in which can lead to problems with development, considering, temperament, and behavior (Prasad et al. 2023). This SNCA accumulation in Lewy bodies is in a similar manner to prions, and is considered a key component of PD (Steiner et al. 2018). PD biological staging system describes PD according to neuronal pathologic SNCA and dopaminergic neuron dysfunction/degeneration (Chahine et al. 2023). It is also believed that the characteristic SNCA pathology in PD can expand from the gut to the brain through the vagus nerve (Braak et al. 2003). Furthermore, evidence has showed transmission of gut-to-brain pathology and dopaminergic neuron loss after injecting prion-like alpha-synuclein fibrils into gut muscles in mice (Braak et al. 2003).
The low function of the PARKIN E3 ubiquitin ligase has long been genetically linked to PD (Panicker et al. 2021). Recent in vitro analysis demonstrated that overexpressing PARKIN enhances TNF-α-mediated NF-κB activation, suggesting a function for this E3 ubiquitin ligase in TNFα-mediated NF-κB signaling by stabilizing linear ubiquitin chain assembly complex (LUBAC) (Meschede et al. 2020). TNFα activated NF-κB and caused neuroprotection of cell death from oxidative stress-induced in both sexes. The gene expression behind this neuroprotection is sexually dimorphic, with females upregulating superoxide dismutase 2 (SOD2) and IGF2 and males showing elevated protein kinase A (PKA) cat alpha expression. Inhibition of cRel, a key regulator of neuronal expansion from neural crest cells, shifts the fate of these stem cells from neuronal to oligodendrocytic lineage (Ruiz-Perera et al. 2018). From neurons, it is recognized as damage-associated molecular patterns (DAMP) or pathogen-associated molecular patterns (PAMP) by microglial toll-like receptor2 (TLR2). Following TLR2 stimulation, myeloid differentiation factor 88 (MyD88) and NF-κB pathways are activated, leading to the production of IL-1β and TNF-α (Fig. 4) (Béraud and Maguire-Zeiss 2012). Accordingly, it has been revealed that stromal cell-derived factor (CXCL12)/CXCR4/Focal adhesion kinase (FAK)/Sarcoma (Src)/Rac1 signaling pathway leads to SNCA-induced accumulation of microglia and thereby promoting neuroinflammation (Li et al. 2019). Figure 4 shows aggregated SNCA binds to CD36 on microglia, leading to the recruitment of Fyn kinase, which in turn triggers protein kinase C-delta (PKC-δ) activation. This sequence facilitates the migration of the p65 subunit of NF-κB to the nucleus (Panicker et al. 2019). Within the nucleus, the p65 subunit enhances the expression of IL-1β and the NLR family pyrin domain containing 3 (NLRP3) inflamed (Dolatshahi et al. 2021). According to another in vivo study on APP/PS1 double-transgenic mice-induced AD, improvement of memory deficits could be through inhibition of NF-κB and stimulation of PPARγ pathway, which can occur by curcumin (150 mg/kg, i.p.) administration. The valuable impacts of curcumin on AD were because of neuroinflammation inhibition, as exhibited by the declined cytokine expression and stimulation of glia, as much as suppression of the NF-κB pathway (Liu et al. 2016). In a 2017 study on 10- to 12-week-old male Sprague Dawley rats as shown in reverse transcription polymerase chain reaction (RT-PCR) and electrophoretic mobility shift assay (EMSA), 40 mg/kg of birthweight of Curcumin, i.p., impeded NF-κB, microglia, and astrocytes activation. The activity of brain’s DNA-binding to NF-κB lessened in EMSA assay and NF-κB expression down-regulated, reducing pro-inflammatory cytokines TNF-α and IL-1β. This process caused dopaminergic neuron protection in opposition to LPS-induced PD and inflammatory action (Sharma et al. 2017). Zhang et al. in 2019 evaluated the effects of curcumin, 5 and 10 μM, on BV2 microglial cell lines after LPS-induced neurotoxicity. It was manifested that curcumin could suppress pro-inflammatory pathways like NF-κB/ TLR4 and decrease IL-6, IL-1β, iNOS, and CD16/32 expression. Moreover, it was declared that curcumin could exert its beneficial effects by regulating the triggering receptor expressed on the TREM2 pathway and increasing IL-10, IL-4, arginase 1, and CD206 (Zhang et al. 2019). Yang et al. investigated curcumin as a microglia-dependent neuroprotector that affects midbrain dopaminergic (DA) neurons against LPS-induced neurotoxicity. The administration of curcumin (10 µM) along with LPS-induced neurotoxicity in mesencephalic neuron-glia cultures showed a considerable decrease in NF-κB DNA binding ability, and expression of NO, iNOS mRNA, IL-1β, and its mRNA, and COX-2 mRNA, TNF-α and its mRNA, as well as, decreased neurotoxic factors and inhibited NF-κB and AP-1 (Yang et al. 2008). Scientists have declared that the NF-κB signaling pathway may be effective in curcumin's impact on PD. Xu et al. induced PD by 3 mg/kg/day rotenone subcutaneous injection in mice's substantia nigra and concluded that treatment with intraperitoneal injection of 50 mg/kg/day curcumin resulted in NF-κB signaling pathway inhibition. To analyze in detail, they performed western blots to examine NF-κB and p-NF-κB levels, which were down-regulated in the curcumin-treated group (Xu et al. 2023).

Clumped α-synuclein, when it attaches to CD36 on microglia and enlists Fyn kinase, prompts the activation of protein kinase C-delta (PKC-δ)
The role of curcumin in the regulation of NF-κB in Huntington’s disease
Huntington’s disease (HD) is an inheritable dominant autosomal neurodegenerative disease characterized by mood changes, decreased neuromuscular coordination, and cognitive decline, ultimately leading to fatality (Gatto et al. 2020). The disease is caused by mutations that expand the repeating region of the nucleobase cytosine–adenine–guanine (CAG) triplet of the Huntington gene, which encodes a polyglutamine tract (polyQ) within the amino terminus of the Huntingtin protein (HTT). A normal HTT has less than 26 repetitions, while in the disease-associated mutant version, typically more than 35 repeats exist. A higher count is associated with a greater severity and an earlier onset, the expansion of polyglutamine in HTT appears to alter its conformation (Bates et al. 2015). Although HTT is expressed in every part of the body, the precise role of normal HTT and why the mutated form disrupts neurons to a greater extent remains unclear. Notably, normal HTT interacts with various neuronal proteins, such as NF-κB subunits p50 and p65 (Takano and Gusella 2002). HTT is notably concentrated at the post-synaptic density of neuronal synapses alongside these NF-κB subunits. Notably, HTT tends to bind selectively to activated NF-κB, facilitating the movement of p65-containing NF-κB dimers away from dendritic spines. However, the polyQ expansion found in HD-associated HTT disrupts this localization at the post-synaptic density, hindering its movement out of dendritic spines and the subsequent nuclear aggregation of NF-κB (Marcora and Kennedy 2010). This indicates that the atypical transport of NF-κB from synapses to the nucleus in neurons could potentially play a role in the development of HD. Although most experiments have focused on the links among NF-κB and HD within types of neuronal cells, mutant HTT seems to influence neuroinflammation, potentially indicating its role in glial cells. In HD patients and an HD mouse model, astrocytes from the caudate nucleus brain region and the cortex display heightened NF-κB activation (Hsiao et al. 2013). This increase in activation primarily occurs in astrocytes rather than neurons or microglia. Elevated astrocyte IKK activity seems to drive this NF-κB activation, consistent with a study showing excessive IKK activities in an HD mouse model's brain (Khoshnan et al. 2004). HD astrocytes ameliorate HD symptoms and block IKK, which mitigates neurotoxicity (Hsiao et al. 2013). NF-κB also regulates HTT at the transcription level. An NF-κB-binding site and a single-nucleotide polymorphism (SNP) within it were identified as the HTT promoter. This SNP impairs NF-κB binding and reduces HTT transcription. Notably, this SNP's influence on the HTT mutant allele delays HD onset, while its existence on the wild-type HTT allele is related to early onset. Though primarily studied at the genomic level, the effects of the NF-κB-binding site and SNP in the HTT promoter were verified in ST14A cells, derived from the striatal brain region that coordinates several facets of cognition, including both motor and action planning, decision-making, motivation, reward perception, and reinforcement and also displaying medium spiny neuron features (Bečanović et al. 2015). In conclusion, the NF-κB pathway and its role in regulating HTT gene expression are in the advancement and progression of HD (Fig. 5).

Effect of curcumin on NF-ĸB signaling pathway. Inducible nitric oxide synthase (iNOS), Cyclooxygenase 2 (COX2), Nitric oxide (NO), Tumor necrosis factor alpha (TNF-α), Interleukin 6 (IL-6), Interleukin 1 Beta (IL1B), Inhibitor of nuclear factor kappa-B kinase subunit beta (IKK-β), hypoxamiR (miR-199a-5p), Reactive oxygen species (ROS), Peroxisome proliferators–activated receptor γ (PPARγ), Monocyte chemoattractant protein 1 (MCP-1), Toll-like receptor 4 (TLR 4), Myeloid differentiation primary response 88 (MYD88), Nucleotide-binding domain, leucine-rich containing family, pyrin domain containing 3 (NLRP3), Glial fibrillary acidic protein (GFAP)
The role of curcumin in modulation of NF-κB in ALS and other neuronal injuries
Amyotrophic lateral sclerosis (ALS) involves the degradation in motor neurons, which leads to increasing loss of voluntary muscle control, speech difficulties, and breathing problems. Despite almost all ALS patients are periodical, about 10% have a hereditary basis (Masrori and Van Damme 2020). Several genes are associated with familial ALS, and NF-κB’s involvement in their regulation or interaction is notable (Mead et al. 2023). The human SOD1 promoter’s NF-κB-binding site responds to phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt) pathway, leading to increased SOD1 levels (Rojo et al. 2004). The NF-κB’s p65 subunit links with TAR DNA-binding protein 43 (TDP-43), a connection heightened in ALS. TDP-43 is believed to act as NF-κB’s co-activator. Mutations of Optineurin (OPTN) have shown its relation to ALS (Ouali Alami et al. 2018). Optineurin is one of the four genes linked to glaucoma that interacts with proteins including myosin VI, Rab8, TANK-binding kinase 1, huntingtin, and transferrin receptor. It also involves in intracellular processes, such as transportation of proteins, Golgi's apparatus maintenance, as well as NF-κB pathway’s antiviral, and antibacterial signaling (Ying and Yue 2012). While normal OPTN inhibits TNF-α-induced NF-κB activation, ALS-associated OPTN mutations abolish this inhibitory effect. Neuroinflammation and activated microglia are distinct features of ALS (Swarup et al. 2011). Both inherited and sporadic ALS forms exhibit activation of NF-κB located in glial cells. Recent research indicates that astrocyte NF-κB activation might contribute to ALS by influencing microglial proliferation and immune responses (Ouali Alami et al. 2018).
To wrap up the whole evidence, besides its effects on motor neurons, NF-κB operation in non-neuronal cells significantly participates in the development of ALS (Dresselhaus and Meffert 2019).
NF-κB activation, triggered by traumatic as well as ischemic circumstances of brain and spinal cord, has emerged as a pivotal player in modulating neuronal responses. Experimental stroke and injury models have indicated that the NF-κB path exerts diverse effects on neuronal degeneration and protection (Kaltschmidt et al. 1999). Opposing outcomes are obtained according to the findings that mice with p50 subunit deficiency exhibit reduced infarct size (Nurmi et al. 2004), while IKK/NF-κB signaling has been exposed in ischemic brain damage (Schwaninger et al. 2006). Conversely, in the hippocampus and the striatum, NF-κB contributes to survival signaling post-temporary focal ischemia (Duckworth et al. 2006). Recent research even suggests that inhibiting glial’s NF-κB can mitigate discomfort and inflammation responses in chronic sciatic nerve constriction (Fu et al. 2010). Collectively, these findings underscore the contentious position of the NF-κB pathway in neuron's related harm and illnesses. In order to address these seeming contradictions, two conjectures have been suggested. The more recent one, proposed by Kaltschmidt et al. (2005), is the NF-κB homeostasis model (Kaltschmidt et al. 2005). This model states that sustained different stages of NF-κB operation over long-term periods may induce neuronal loss, in physiological situations, nuclear RelA activates replications of apoptosis-inhibitor genes, contingent upon IκB protein’s role in localizing RelA to the cytoplasm. Pathological conditions can disrupt this balance, leading to NF-κB shifting from a critical promoter to a supreme blocker of apoptosis-inhibitor genes, thereby triggering neuronal cell death. An alternate hypothesis, advocated by Mattson and Camandola (2001), suggests that NF-κB operation in neuronal cells stimulates anti-apoptotic genes promoting their survival. Conversely, NF-κB operation in glial cells stimulates the creation of proinflammatory cytokines, culminating in neuronal loss. This divergence in responses to the same stimulus results from NF-κB activation in distinct cell units. TNF-α, which glial cells produce, exemplifies this duality. TNF-α and TNFR1 connection in glial cells incites NO production and may cause loss of neurons, while TNFR2 neuronal activation prompts anti-apoptotic gene expression. This theory gains support from studies revealing that neuronal toxicity of TNF-α hinges on the presence of glial cells (Reich and Hölscher 2022). Notably, microglia, macrophages, and astrocytes release proinflammatory cytokines, excitotoxins, as well as free radicals due to TNF-α-stimulated activation of NF-κB, driving neuronal loss, in the models of neuron harm, inhibiting the route exclusively in astrocytes which indicated neuroprotection (Meunier et al. 2007). Consequently, glial-specific NF-κB pathway inhibition emerges as a potential therapy perspective for neurodegenerative circumstances such as AD, as well as ALS (Thomsen et al. 2009). An in vivo study on SD rats with SCI conducted by Yu et al. showed that curcumin injection in the dose of 150 mg/kg i.p. can reduce NF-κB and TNF-α expression. It also can amend motor dysfunction caused by SCI. Moreover, they manifested that curcumin increases heme oxygenase 1 (HO-1), tight junction protein (TJ), zonula occludens 1 (ZO-1), and occludin (Yu et al. 2014). Besides, another study on SD rats with TBI conducted by Sun et al., demonstrated that the use of Curcumin, 30 mg/kg i.p., can improve neurogenesis, as well as reduce pro-inflammatory regulators like TNF-α, IL-18, IL-6, as well as IL-1β expression. It was presented that blockage of TLR4-MAPK/NF-κB route might represent a possible mechanism involved in the reduction of these pro-inflammatory mediators (Sun et al. 2020). Yuan et al. in 2015 evaluated the curcumin's impacts (300, 100, and 30 mg per kg, i.p.) on the Female SD rat model of SCI. By assessing behavior, western blotting, immunohistochemistry, real-time polymerase chain reaction, and enzyme-linked immunosorbent assay, it has been observed that curcumin given to rats showed an anti-inflammatory property through NF-κB signaling pathway, decreased glial scar, improved functional recovery, and impeded proinflammatory cytokines expression like TNF-α, interleukin-1β (IL-1β), as well as NF-κB (Yuan et al. 2015). According to an in vivo study, SCI was induced by the method of Allen's and western blot. Enzyme-linked immunosorbent assay (ELISA), Basso mouse scale (BMS), immunohistochemical, and Griess assay are other methods of experiment. 100 adult female Kun-Ming (KM) mice conducted by Zhang et al. in 2017, curcumin administration (50,100, or 200 mg per kg) can inhibit the TGF-b-activated kinase 1 (TAK1)/mitogen-activated protein kinase kinase 6(MKK6)/p38 mitogen-activated protein kinases (p38MAPK) via the TAK1 and NF-κB routes and inflammation. Curcumin decreased the phosphorylation levels of IκB Kinase β (IKKβ) and IkB. IKKβ and IkB are the main upstream regulators for NF-κB activity. The overall protein transcription of IKKβ and IkB was reduced too. It has been discovered that curcumin reacts as inhibitor of SCI-stimulated IKK and IkB (Zhang et al. 2017). In a study on adult KunMing mice (male), it has been examined the effects of curcumin (50 mg/kg, i.p.) on attenuated lipopolysaccharide (LPS)-stimulated microglial activity, as well as overproduction of proinflammatory cytokine IL-1, TNF-α, the levels of iNOS and COX2 mRNA in the hippocampus and prefrontal cortex (PFC). Furthermore, curcumin reduced LPS-induced NF-κB activity in the hippocampus and PFC. Curcumin showed antidepressant-like effects that might be regulated by decreasing the levels of stress-stimulated pro-inflammatory cytokines, iNOS and COX-2 mRNA by NF-κB signaling mechanism. There have been observed alterations in the stages of IL-1β, TNF-α, iNOS, and COX-2, as well as alterations of NF-κB activities in special brain sectors like the PFC and hippocampus (Wang et al. 2014). According to an in vivo experiment on female SD rats (curcumin 100 mg per kg, i.p.) and an in vitro experiment on cortical astrocytes (curcumin 1 μmol/l) in SCI by Yuan et al. in 2017, curcumin can reduce monocyte chemoattractant protein 1 (MCP-1), Regulated upon Activation, Normal T Cell Expressed, and Secreted (RANTES), C-X-C motif chemokine ligand 10 (CXCL10) and T cell infiltration by blockage of NF-κB pathway, as well as astrocyte activity. In addition, curcumin can down-regulate SRY-Box Transcription Factor 9 (SOX9) pathway, reduce α-smooth muscle actin (α-SMA) and glial fibrillary acidic protein (GFAP) expression and decrease chondroitin sulfate proteoglycan (CSPG) deposition (Yuan et al. 2017). In another study conducted in 2015, density depletion of NF-κB-positive nuclei and lessened NF-κB activity occurred in SCI rats when they were on i.p. injection containing 6 mg/ kg/ day curcumin diluted in olive oil during and after 28 days of treatment duration (Machova Urdzikova et al. 2015). According to a 2013 investigation on the inflammatory impacts of curcumin in experimental white matter harms in rats, administration of 100 mg per kg curcumin in phosphate-buffered saline (PBS), which contains 1% dimethyl sulfoxide (DMSO) by i.p. injection, led to reduced binding activity of NF-κB (Ni et al. 2015). Daverey & colleagues performed an in vitro trial on male Wistar rats’ spinal cord dorsal columns. In their model, they observed 50 µM curcumin in 95% N2 & 5% CO2 lessened NF-κB expression induced by hypoxia in the cytosol, which secured curcumin-mediated protection of neuron cells. This group also tried NF-κB-specific inhibition experiment that resulted in a noticeable regulatory weight of the NF-κB signaling route in preventing pro-inflammatory cytokines (Daverey and Agrawal 2020). Curcumin, as a NF-κB/TLR inhibitor, was administrated in the Giacomeli et al. study in which lipid core nanocapsules (LNC) were used to overcome the low bioavailability of curcumin. Three drugs containing curcumin were investigated and all three caused a reduction of mRNA expression of NF-κB. Meanwhile, lipid nanocapsules of curcumin 10 mg per kg (LNC10) treatment presented a noticeable reduction in mRNA transcription of NF-κB in comparison with the two following drugs: lipid nanocapsules of curcumin 1, and 10 mg/kg (Giacomeli et al. 2019). Ni H et al. investigation examined the influence of curcumin on the expression of TLR4 and NF-κB inflammatory signaling pathway in SCI rats. The effect of curcumin (100 mg/kg, i.p) on suppressing the growth of TLR4 levels was proved. Moreover, curcumin down-regulated NF-κB DNA-binding activity and reduced TNF-α, IL-1β, and IL-6 along with BBB locomotion score, spinal cord edema, and apoptotic index in contrast to those in the sham group (Ni et al. 2015). In 2019, Gao et al. conducted an in vitro study on BV2 microglial cells. The study results suggested that curcumin at a concentration of 8 μM can down-regulate the IKKβ/ NF-κB pathway by up-regulating miR-199b-5p. Additionally, curcumin at this concentration can also inhibit the replication of TNF-α, IL-1β, iNOS, and phosphorylated-p65 (Gao et al. 2019). Xie et al. researched the role of Curcumin (2, 4, & 8 µM) impact in LPS-influenced inflammatory injury on BV2 cells. In this process, they found out the preventive position of Curcumin on NF-κB signaling using miR3623p/TLR4 axis (Xie et al. 2020).
Curcumin’s beneficial impacts on neurodegenerative diseases based on clinical evidence
There are no clinical studies about the effects of curcumin and its derivatives on ND via regulating NF-kB; however, there are a few clinical studies about the beneficial effects of curcumin on ND improvement, which could be due to inflammation reduction that can be mediated via NF-kB signaling pathway regulation. In a clinical study published in 2008, curcumin at the dose of 1 and 4 g/day was given orally to 34 patients who have been involved in the experiment. There were no significant changes in MMSE scores between placebo and curcumin groups. Moreover, there was a rise in amyloid-beta 40 levels in the blood that could be due to the curcumin disaggregation effect of amyloid-beta 40 in the brain (Baum et al. 2008). In another clinical study by Ringman et al. (2012), the effects and adverse effects of curcumin in AD patients were evaluated. Curcumin was given to 36 persons in a 24- to 48-week period at a dose of 2 and 4 g/day orally. There were no significant changes in amyloid-beta 40 and 42 levels in blood or CSF between the two groups. Furthermore, curcumin did not affect Alzheimer’s Disease Assessment Scale–cognitive subscale (ADAS-Cog) and Alzheimer’s Disease Cooperative Society Study-Activities of Daily Living (ADCS-ADL) scale. Positively, curcumin showed no important adverse effect and it was well tolerated in patients (Ringman et al. 2012).
Dolati et al. reported that daily 80 mg of nano-curcumin has advantageous impacts on MS patients by affecting Treg cells function and circulation period (Dolati et al. 2019). 7 days of treatment with 500 mg curcuminoids can improve conditions in hospitalized TBI patients compared to placebo. Curcuminoid supplementation attenuated levels of CRP, MCP-1, IL-6, and TNF-α levels while it didn’t alter enzymatic activities of SOD and GPx significantly. Moreover, it reduced the mortality risk rate based on the acute physiology and chronic health evaluation (APACHE) II test. It could be possible that curcuminoid exert its anti-inflammatory effects via regulating NF-kB pathway (Zahedi et al. 2021). Moreover, addition of curcuminoids (500 mg), besides 5 mg/day piperine to routine therapeutic approaches compared to placebo for 7 days, diminished levels of leptin, but it didn’t change adiponectin in TBI patients (Shadnoush et al. 2020). Curcumin (180 mg/day) prevents development of T2DM and AD. It was observed that curcumin treatment for 12 weeks led to glycogen synthase kinase-3-β and islet amyloid polypeptide levels reduction. It also decreased insulin resistance compared to placebo (Thota et al. 2020). Ahmadi et al. considered the addition of nano-curcumin (containing 80 mg curcuminoids) addition to riluzole in ALS patients. Compared to a placebo, it enhanced the survival rate without exerting any major side effects (Ahmadi et al. 2018).
Discussion, concluding remarks and future perspectives
The development of neurodegenerative diseases is highly dependent on the neuroinflammation and neural cell loss. Hence, regulating these two factors can prevent further progression of the ND. Scientists have tested and analyzed different natural and chemical compounds to manage the sign and symptoms of NDs, i.e., ALS, PD, SCI, HD, and AD. The noteworthy remark is that how inflammatory cascades in cellular levels can directly affect the patients' health-related circumstances. Hence, the ultimate goal is to investigate involved pathophysiologic mechanism and their affected pathways for better understanding the process of the incidents, discover suitable compounds and agent, hinder the progression of the disease, and raise the well-being of the patients.
NF-κB expression and its following signaling pathway have gained noticeable attention due to enormous impact on production of inflammatory proteins. Understanding the NF-κB mechanism and targeting it, seems to be an effective strategy for ameliorating NDs.
Curcumin is approved as a promising anti-oxidant, and anti-inflammatory compound in previous experiments.
Other review articles have pointed out curcumin's therapeutic role in AD, MS, and other neurological disorders due to its antioxidant, anti-inflammatory, and anti-apoptotic properties (Ghanaatian et al. 2019; Farkhondeh et al. 2019; Bland et al. 2023; Bhat et al. 2019). However, our study is the first review highlighting the NF-κB signaling pathway as curcumin's main target in all forms of NDs. Also, different studies have shown the curcumin’s helpful anti-inflammatory effect in neurodegenerative diseases, mainly induced by modulation of NF-κB signaling route. The current review highlighted curcumin’s remarkable impacts on restricting neuronal cell loss and inflammatory-related conditions by down-regulation of involved chemicals, cytokines, and signaling pathways.
Particularly, results from in vitro, as well as in vivo studies manifest the inhibitory influence of curcumin which happens on NF-κB signaling pathway as a way of reducing neuroinflammation, which is the primary cause of NDs.
The number of clinical trials on this matter is not satisfying yet. Thus, it seems more profound and concentrated experiments are needed to determine curcumin's exact administration dose, pharmacokinetics, pharmacodynamics, safety, and adverse consequences for reducing NF-κB activity in neurodegenerative disorders. Additional studies are required to examine not only the short-term effects but also the long-term outcomes of using curcumin to impede the NF-κB signaling mechanism in the referred disease (Tables 1, 2).
Data availability
Not applicable.
Abbreviations
- MPP+:
-
1-Methyl-4-phenylpyridinium ion
- SNCA:
-
A-Synuclein
- AD:
-
Alzheimer disease
- ALS:
-
Amyotrophic lateral sclerosis
- ApoE4-Tg:
-
ApoE4 transgenic
- APOE4:
-
Apolipoprotein E4
- BMS:
-
Basso mouse scale
- BACE1:
-
Beta-Secretase 1
- BDNF:
-
Brain-derived neurotrophic factor
- CAMKII:
-
Calcium–calmodulin (CaM)-dependent protein kinase II
- CREB:
-
CAMP response element-binding protein
- CAT:
-
Catalase
- CNS:
-
Central nervous system
- ChAT:
-
Choline acetyltransferase
- CSPG:
-
Chondroitin sulfate proteoglycan
- Co-Q10:
-
Coenzyme Q10
- CRP:
-
C-reactive protein
- CXCL10:
-
C-X-C motif chemokine ligand 10
- COX-2:
-
Cyclooxygenase 2
- DAMP:
-
Damage-associated molecular patterns
- DCM:
-
Diabetic cardiomyopathy
- DR:
-
Diabetic retinopathy
- DMSO:
-
Dimethyl sulfoxide
- EMSA:
-
Electrophoretic mobility shift assay
- ER:
-
Endoplasmic reticulum
- ELISA:
-
Enzyme-linked immunosorbent assay
- GI:
-
Gastrointestinal
- GFAP:
-
Glial fibrillary acidic protein
- GSH:
-
Glutathione
- GPx:
-
Glutathione peroxidase
- HO-1:
-
Heme oxygenase 1
- HTT:
-
Huntingtin protein
- HD:
-
Huntington’s disease
- iNOS:
-
Inducible nitric oxide synthase
- IRF3:
-
Interferon regulatory factor 3
- IL:
-
Interleukin
- i.p.:
-
Intraperitoneal
- IP:
-
Intra-peritoneal
- IV:
-
Intravenous
- Iba1:
-
Ionized calcium-binding adapter molecule 1
- IKKβ:
-
IκB Kinase β
- KM:
-
Kun-Ming
- LRRK2:
-
Leucine-rich repeat kinase 2
- LBD:
-
Lewy body dementia
- LTP:
-
Like long-term potentiation
- LUBAC:
-
Linear ubiquitin chain assembly complex
- LPO:
-
Lipid peroxidation
- LPS:
-
Lipopolysaccharides
- LOX:
-
Lipoxygenase
- LTD:
-
Long-term depression
- LUBAC:
-
Linear ubiquitin chain assembly complex
- MIP-1α:
-
Macrophage inflammatory protein-1alpha
- mTOR:
-
Mammalian target of rapamycin
- MnSOD:
-
Manganese superoxide dismutase
- MEM:
-
Memantine
- MCAO:
-
Middle cerebral artery occlusion
- mEPSCs:
-
Miniature excitatory post-synaptic currents
- MAPK:
-
Mitogen-activated protein kinase
- MKK6:
-
Mitogen-activated protein kinase kinase 6
- MCP-1:
-
Monocyte chemoattractant protein 1
- MS:
-
Multiple sclerosis
- MyD88:
-
Myeloid differentiation primary response 88
- NGF:
-
Nerve growth factor
- ND:
-
Neurodegenerative disease
- NO:
-
Nitric oxide
- Nrf2:
-
Nuclear factor erythroid 2-related factor 2/antioxidant responsive element
- NF-κB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- OPTN:
-
Optineurin
- p38MAPK:
-
P38 mitogen-activated protein kinases
- PAMP:
-
Pathogen-associated molecular patterns
- PO:
-
Per os
- Prdx6:
-
Peroxiredoxin6
- PPARγ:
-
Peroxisome proliferator-activated receptor γ
- PBS:
-
Phosphate-buffered saline
- PI3K:
-
Phosphoinositide-3-kinase
- PSD-95:
-
Postsynaptic density protein 95
- PFC:
-
Prefrontal cortex
- PSEN:
-
Presenilin
- PGE2:
-
Prostaglandin E2
- Akt:
-
Protein kinase B
- AP-1:
-
Activator protein 1
- PKC-δ:
-
Protein kinase C-delta
- PINK1:
-
PTEN-induced putative kinase 1
- ROS:
-
Reactive oxygen species
- RANTES:
-
Regulated upon Activation Normal T Cell Expressed, and Secreted
- RT-PCR:
-
Reverse transcription polymerase chain reaction
- SCO:
-
Scopolamine hydrobromide
- SNP:
-
Single-nucleotide polymorphisms
- SCI:
-
Spinal cord injury
- SPAR:
-
Spine-associated Rap GTPase-activating protein
- SOX9:
-
SRY-Box Transcription Factor 9
- SN:
-
Substantia nigra
- SOD:
-
Superoxide dismutase
- TDP-43:
-
TAR DNA-binding protein 43
- TAK1:
-
TGF-b-activated kinase 1
- TJ:
-
Tight junction protein
- TLR4:
-
Toll-like receptor 4
- TGF:
-
Transforming growth factor
- TIRF:
-
Transmucosal immediate-release fentanyl
- TBI:
-
Traumatic brain injury
- TREM2:
-
Triggering receptor expressed on myeloid cells 2
- Tau:
-
Tubulin-associated unit
- TRAF6:
-
Tumor necrosis factor receptor (TNFR)-associated factor 6
- TNF-α:
-
Tumor necrosis factor-α
- T2DM:
-
Type 2 diabetes mellitus
- VEGF:
-
Vascular endothelial growth factor
- VEGFR:
-
Vascular endothelial growth factor receptor
- ZO-1:
-
Zonula occludens 1
- α-SMA:
-
α-Smooth muscle actin
- Aβ:
-
β-Amyloid peptides
- APACHE:
-
Acute physiology and chronic health evaluation
- ADAS-Cog:
-
Alzheimer’s disease Assessment Scale–cognitive subscale
- ADCS-ADL:
-
Alzheimer’s disease Cooperative society Study-Activities of Daily Living
References
Aggarwal BB, Harikumar KB (2009) Potential therapeutic effects of curcumin, the anti-inflammatory agent, against neurodegenerative, cardiovascular, pulmonary, metabolic, autoimmune and neoplastic diseases. Int J Biochem Cell Biol 41:40–59
Aggarwal BB, Sung B (2009) Pharmacological basis for the role of curcumin in chronic diseases: an age-old spice with modern targets. Trends Pharmacol Sci 30:85–94
Ahmadi M, Agah E, Nafissi S, Jaafari MR, Harirchian MH, Sarraf P, Faghihi-Kashani S, Hosseini SJ, Ghoreishi A, Aghamollaii V, Hosseini M, Tafakhori A (2018) Safety and efficacy of nanocurcumin as add-on therapy to riluzole in patients with amyotrophic lateral sclerosis: a pilot randomized clinical trial. Neurotherapeutics 15:430–438
Anand P, Kunnumakkara AB, Newman RA, Aggarwal BB (2007) Bioavailability of curcumin: problems and promises. Mol Pharm 4:807–818
Anand P, Sundaram C, Jhurani S, Kunnumakkara AB, Aggarwal BB (2008a) Curcumin and cancer: an “old-age” disease with an “age-old” solution. Cancer Lett 267:133–164
Anand P, Thomas SG, Kunnumakkara AB, Sundaram C, Harikumar KB, Sung B, Tharakan ST, Misra K, Priyadarsini IK, Rajasekharan KN, Aggarwal BB (2008b) Biological activities of curcumin and its analogues (Congeners) made by man and Mother Nature. Biochem Pharmacol 76:1590–1611
Ashrafizadeh M, Ahmadi Z, Mohammadinejad R, Farkhondeh T, Samarghandian S (2020) Curcumin activates the Nrf2 pathway and induces cellular protection against oxidative injury. Curr Mol Med 20:116–133
Basnet P, Skalko-Basnet N (2011) Curcumin: an anti-inflammatory molecule from a curry spice on the path to cancer treatment. Molecules 16:4567–4598
Bates GP, Dorsey R, Gusella JF, Hayden MR, Kay C, Leavitt BR, Nance M, Ross CA, Scahill RI, Wetzel R (2015) Huntington disease. Nat Rev Dis Primers 1:1–21
Baum L, Lam CW, Cheung SK, Kwok T, Lui V, Tsoh J, Lam L, Leung V, Hui E, Ng C, Woo J, Chiu HF, Goggins WB, Zee BC, Cheng KF, Fong CY, Wong A, Mok H, Chow MS, Ho PC, Ip SP, Ho CS, Yu XW, Lai CY, Chan MH, Szeto S, Chan IH, Mok V (2008) Six-month randomized, placebo-controlled, double-blind, pilot clinical trial of curcumin in patients with Alzheimer disease. J Clin Psychopharmacol 28:110–113
Bečanović K, Nørremølle A, Neal SJ, Kay C, Collins JA, Arenillas D, Lilja T, Gaudenzi G, Manoharan S, Doty CN, Beck J, Lahiri N, Portales-Casamar E, Warby SC, Connolly C, de Souza RA, Tabrizi SJ, Hermanson O, Langbehn DR, Hayden MR, Wasserman WW, Leavitt BR (2015) A SNP in the HTT promoter alters NF-κB binding and is a bidirectional genetic modifier of Huntington disease. Nat Neurosci 18:807–816
Bender K, Göttlicher M, Whiteside S, Rahmsdorf HJ, Herrlich P (1998) Sequential DNA damage-independent and -dependent activation of NF-kappaB by UV. Embo j 17:5170–5181
Béraud D, Maguire-Zeiss KA (2012) Misfolded α-synuclein and Toll-like receptors: therapeutic targets for Parkinson’s disease. Parkinsonism Relat Disord 18(Suppl 1):S17-20
Bhat A, Mahalakshmi AM, Ray B, Tuladhar S, Hediyal TA, Manthiannem E, Padamati J, Chandra R, Chidambaram SB, Sakharkar MK (2019) Benefits of curcumin in brain disorders. BioFactors 45:666–689
Bland AR, Ashton JC, Kamal MA, Sahebkar A (2023) The current evidence for the therapeutic role of curcumin in Alzheimer’s disease. CNS Neurol Disord Drug Targets 22:318–320
Boersma MC, Dresselhaus EC, de Biase LM, Mihalas AB, Bergles DE, Meffert MK (2011) A requirement for nuclear factor-kappaB in developmental and plasticity-associated synaptogenesis. J Neurosci 31:5414–5425
Bonizzi G, Bebien M, Otero DC, Johnson-Vroom KE, Cao Y, Vu D, Jegga AG, Aronow BJ, Ghosh G, Rickert RC, Karin M (2004) Activation of IKKalpha target genes depends on recognition of specific kappaB binding sites by RelB:p52 dimers. Embo j 23:4202–4210
Braak H, Rüb U, Gai WP, del Tredici K (2003) Idiopathic Parkinson’s disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm (vienna) 110:517–536
Chahine LM, Merchant K, Siderowf A, Sherer T, Tanner C, Marek K, Simuni T (2023) Proposal for a biologic staging system of Parkinson’s disease. J Parkinsons Dis 13:297–309
Chami L, Buggia-Prévot V, Duplan E, del Prete D, Chami M, Peyron JF, Checler F (2012) Nuclear factor-κB regulates βAPP and β- and γ-secretases differently at physiological and supraphysiological Aβ concentrations. J Biol Chem 287:24573–24584
Chhunchha B, Fatma N, Kubo E, Rai P, Singh SP, Singh DP (2013) Curcumin abates hypoxia-induced oxidative stress based-ER stress-mediated cell death in mouse hippocampal cells (HT22) by controlling Prdx6 and NF-κB regulation. Am J Physiol Cell Physiol 304:C636–C655
Chiarini A, Armato U, Hu P, Dal Prà I (2020) Danger-sensing/patten recognition receptors and neuroinflammation in Alzheimer’s disease. Int J Mol Sci 21:9036
Cho IH, Hong J, Suh EC, Kim JH, Lee H, Lee JE, Lee S, Kim CH, Kim DW, Jo EK, Lee KE, Karin M, Lee SJ (2008) Role of microglial IKKbeta in kainic acid-induced hippocampal neuronal cell death. Brain 131:3019–3033
Daverey A, Agrawal SK (2020) Curcumin protects against white matter injury through NF-κB and Nrf2 cross talk. J Neurotrauma 37:1255–1265
de Strooper B, Karran E (2016) The cellular phase of Alzheimer’s disease. Cell 164:603–615
Demaagd G, Philip A (2015) Parkinson’s disease and its management: part 1: disease entity, risk factors, pathophysiology, clinical presentation, and diagnosis. P t 40:504–532
Dolati S, Babaloo Z, Ayromlou H, Ahmadi M, Rikhtegar R, Rostamzadeh D, Roshangar L, Nouri M, Mehdizadeh A, Younesi V, Yousefi M (2019) Nanocurcumin improves regulatory T-cell frequency and function in patients with multiple sclerosis. J Neuroimmunol 327:15–21
Dolatshahi M, Ranjbar Hameghavandi MH, Sabahi M, Rostamkhani S (2021) Nuclear factor-kappa B (NF-κB) in pathophysiology of Parkinson disease: diverse patterns and mechanisms contributing to neurodegeneration. Eur J Neurosci 54:4101–4123
Dresselhaus EC, Meffert MK (2019) Cellular specificity of NF-κB function in the nervous system. Front Immunol 10:1043
Duckworth EA, Butler T, Collier L, Collier S, Pennypacker KR (2006) NF-kappaB protects neurons from ischemic injury after middle cerebral artery occlusion in mice. Brain Res 1088:167–175
Farkhondeh T, Samarghandian S, Pourbagher-Shahri AM, Sedaghat M (2019) The impact of curcumin and its modified formulations on Alzheimer’s disease. J Cell Physiol 234:16953–16965
Fridmacher V, Kaltschmidt B, Goudeau B, Ndiaye D, Rossi FM, Pfeiffer J, Kaltschmidt C, Israël A, Mémet S (2003) Forebrain-specific neuronal inhibition of nuclear factor-kappaB activity leads to loss of neuroprotection. J Neurosci 23:9403–9408
Fu ES, Zhang YP, Sagen J, Candiotti KA, Morton PD, Liebl DJ, Bethea JR, Brambilla R (2010) Transgenic inhibition of glial NF-kappa B reduces pain behavior and inflammation after peripheral nerve injury. Pain 148:509–518
Fu W, Zhuang W, Zhou S, Wang X (2015) Plant-derived neuroprotective agents in Parkinson’s disease. Am J Transl Res 7:1189–1202
Gao F, Shen J, Zhao L, Hao Q, Yang Y (2019) Curcumin alleviates lipopolysaccharide (LPS)-activated neuroinflammation via modulation of miR-199b-5p/IκB Kinase β (IKKβ)/Nuclear Factor Kappa B (NF-κB) pathway in microglia. Med Sci Monit 25:9801–9810
Garcia-Alloza M, Borrelli LA, Rozkalne A, Hyman BT, Bacskai BJ (2007) Curcumin labels amyloid pathology in vivo, disrupts existing plaques, and partially restores distorted neurites in an Alzheimer mouse model. J Neurochem 102:1095–1104
Gatto EM, Rojas NG, Persi G, Etcheverry JL, Cesarini ME, Perandones C (2020) Huntington disease: advances in the understanding of its mechanisms. Clin Park Relat Disord 3:100056
Ghanaatian N, Lashgari NA, Abdolghaffari AH, Rajaee SM, Panahi Y, Barreto GE, Butler AE, Sahebkar A (2019) Curcumin as a therapeutic candidate for multiple sclerosis: molecular mechanisms and targets. J Cell Physiol 234:12237–12248
Giacomeli R, Izoton JC, dos Santos RB, Boeira SP, Jesse CR, Haas SE (2019) Neuroprotective effects of curcumin lipid-core nanocapsules in a model Alzheimer’s disease induced by β-amyloid 1–42 peptide in aged female mice. Brain Res 1721:146325
Green KL, Gatto GJ, Grant KA (1997) The nitric oxide synthase inhibitor L-NAME (N omega-nitro-L-arginine methyl ester) does not produce discriminative stimulus effects similar to ethanol. Alcohol Clin Exp Res 21:483–488
Grynkiewicz G, Ślifirski P (2012) Curcumin and curcuminoids in quest for medicinal status. Acta Biochim Pol 59:201–212
Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S, Hazrati L, Collinge J, Pocock J, Lashley T, Williams J, Lambert JC, Amouyel P, Goate A, Rademakers R, Morgan K, Powell J, St George-HyslopSingletonHardy PPJ (2013) TREM2 variants in Alzheimer’s disease. N Engl J Med 368:117–127
Heissmeyer V, Krappmann D, Wulczyn FG, Scheidereit C (1999) NF-kappaB p105 is a target of IkappaB kinases and controls signal induction of Bcl-3-p50 complexes. Embo j 18:4766–4778
Hsiao HY, Chen YC, Chen HM, Tu PH, Chern Y (2013) A critical role of astrocyte-mediated nuclear factor-κB-dependent inflammation in Huntington’s disease. Hum Mol Genet 22:1826–1842
Huang L, Chen C, Zhang X, Li X, Chen Z, Yang C, Liang X, Zhu G, Xu Z (2018) Neuroprotective effect of curcumin against cerebral ischemia-reperfusion via mediating autophagy and inflammation. J Mol Neurosci 64:129–139
Huang P, Zheng N, Zhou HB, Huang J (2020) Curcumin inhibits BACE1 expression through the interaction between ERβ and NFκB signaling pathway in SH-SY5Y cells. Mol Cell Biochem 463:161–173
Jones SV, Kounatidis I (2017) Nuclear factor-kappa B and Alzheimer disease, unifying genetic and environmental risk factors from cell to humans. Front Immunol 8:1805
Kaltschmidt B, Kaltschmidt C (2015) NF-KappaB in long-term memory and structural plasticity in the adult mammalian brain. Front Mol Neurosci 8:69
Kaltschmidt B, Uherek M, Wellmann H, Volk B, Kaltschmidt C (1999) Inhibition of NF-kappaB potentiates amyloid beta-mediated neuronal apoptosis. Proc Natl Acad Sci U S A 96:9409–9414
Kaltschmidt B, Widera D, Kaltschmidt C (2005) Signaling via NF-kappaB in the nervous system. Biochim Biophys Acta 1745:287–299
Kang G, Kong PJ, Yuh YJ, Lim SY, Yim SV, Chun W, Kim SS (2004) Curcumin suppresses lipopolysaccharide-induced cyclooxygenase-2 expression by inhibiting activator protein 1 and nuclear factor kappab bindings in BV2 microglial cells. J Pharmacol Sci 94:325–328
Khayatan D, Razavi SM, Arab ZN, Niknejad AH, Nouri K, Momtaz S, Gumpricht E, Jamialahmadi T, Abdolghaffari AH, Barreto GE, Sahebkar A (2022) Protective effects of curcumin against traumatic brain injury. Biomed Pharmacother 154:113621
Khoshnan A, Ko J, Watkin EE, Paige LA, Reinhart PH, Patterson PH (2004) Activation of the IkappaB kinase complex and nuclear factor-kappaB contributes to mutant huntingtin neurotoxicity. J Neurosci 24:7999–8008
Kou J, Wang M, Shi J, Zhang H, Pu X, Song S, Yang C, Yan Y, Döring Y, Xie X, Pang X (2021) Curcumin reduces cognitive deficits by inhibiting neuroinflammation through the endoplasmic reticulum stress pathway in apolipoprotein E4 transgenic mice. ACS Omega 6:6654–6662
Kumar A, Thakur MK (2016) Binding of transcription factors to Presenilin 1 and 2 promoter cis-acting elements varies during the development of mouse cerebral cortex. Neurosci Lett 628:98–104
Kumar A, Chetia H, Sharma S, Kabiraj D, Talukdar NC, Bora U (2015a) Curcumin resource database. Database (oxford). https://doi.org/10.1093/database/bav070
Kumar A, Sharma S, Prashar A, Deshmukh R (2015b) Effect of licofelone–a dual COX/5-LOX inhibitor in intracerebroventricular streptozotocin-induced behavioral and biochemical abnormalities in rats. J Mol Neurosci 55:749–759
Kumar P, Singh A, Kumar A, Kumar R, Pal R, Sachan AK, Dixit RK, Nath R (2023) Effect of curcumin and coenzyme Q10 alone and in combination on learning and memory in an animal model of Alzheimer’s disease. Biomedicines 11:1422
Kyrargyri V, Vega-Flores G, Gruart A, Delgado-García JM, Probert L (2015) Differential contributions of microglial and neuronal IKKβ to synaptic plasticity and associative learning in alert behaving mice. Glia 63:549–566
Lahiri DK (2004) Apolipoprotein E as a target for developing new therapeutics for Alzheimer’s disease based on studies from protein, RNA, and regulatory region of the gene. J Mol Neurosci 23:225–233
Li Y, Niu M, Zhao A, Kang W, Chen Z, Luo N, Zhou L, Zhu X, Lu L, Liu J (2019) CXCL12 is involved in α-synuclein-triggered neuroinflammation of Parkinson’s disease. J Neuroinflammation 16:263
Liu ZJ, Li ZH, Liu L, Tang WX, Wang Y, Dong MR, Xiao C (2016) Curcumin attenuates beta-amyloid-induced neuroinflammation via activation of peroxisome proliferator-activated receptor-gamma function in a rat model of Alzheimer’s disease. Front Pharmacol 7:261
Machova Urdzikova L, Karova K, Ruzicka J, Kloudova A, Shannon C, Dubisova J, Murali R, Kubinova S, Sykova E, Jhanwar-Uniyal M, Jendelova P (2015) The anti-inflammatory compound curcumin enhances locomotor and sensory recovery after spinal cord injury in rats by immunomodulation. Int J Mol Sci 17:49
Marcora E, Kennedy MB (2010) The Huntington’s disease mutation impairs Huntingtin’s role in the transport of NF-κB from the synapse to the nucleus. Hum Mol Genet 19:4373–4384
Masrori P, van Damme P (2020) Amyotrophic lateral sclerosis: a clinical review. Eur J Neurol 27:1918–1929
Mattson MP, Camandola S (2001) NF-kappaB in neuronal plasticity and neurodegenerative disorders. J Clin Invest 107:247–254
Mead RJ, Shan N, Reiser HJ, Marshall F, Shaw PJ (2023) Amyotrophic lateral sclerosis: a neurodegenerative disorder poised for successful therapeutic translation. Nat Rev Drug Discov 22:185–212
Menon VP, Sudheer AR (2007) Antioxidant and anti-inflammatory properties of curcumin. Adv Exp Med Biol 595:105–125
Meschede J, Šadić M, Furthmann N, Miedema T, Sehr DA, Müller-Rischart AK, Bader V, Berlemann LA, Pilsl A, Schlierf A, Barkovits K, Kachholz B, Rittinger K, Ikeda F, Marcus K, Schaefer L, Tatzelt J, Winklhofer KF (2020) The parkin-coregulated gene product PACRG promotes TNF signaling by stabilizing LUBAC. Sci Signal 13:1256
Meunier A, Latrémolière A, Dominguez E, Mauborgne A, Philippe S, Hamon M, Mallet J, Benoliel JJ, Pohl M (2007) Lentiviral-mediated targeted NF-κB blockade in dorsal spinal cord glia attenuates sciatic nerve injury-induced neuropathic pain in the rat. Mol Ther 15:687–697
Mincheva S, Garcera A, Gou-Fabregas M, Encinas M, Dolcet X, Soler RM (2011) The canonical nuclear factor-κB pathway regulates cell survival in a developmental model of spinal cord motoneurons. J Neurosci 31:6493–6503
Mincheva-Tasheva S, Soler RM (2013) NF-κB signaling pathways: role in nervous system physiology and pathology. Neuroscientist 19:175–194
Ni H, Jin W, Zhu T, Wang J, Yuan B, Jiang J, Liang W, Ma Z (2015) Curcumin modulates TLR4/NF-κB inflammatory signaling pathway following traumatic spinal cord injury in rats. J Spinal Cord Med 38:199–206
Nocito MC, de Luca A, Prestia F, Avena P, la Padula D, Zavaglia L, Sirianni R, Casaburi I, Puoci F, Chimento A, Pezzi V (2021) Antitumoral activities of curcumin and recent advances to improve its oral bioavailability. Biomedicines 9:1476
Nurmi A, Lindsberg PJ, Koistinaho M, Zhang W, Juettler E, Karjalainen-Lindsberg ML, Weih F, Frank N, Schwaninger M, Koistinaho J (2004) Nuclear factor-kappaB contributes to infarction after permanent focal ischemia. Stroke 35:987–991
O’Mahony A, Raber J, Montano M, Foehr E, Han V, Lu SM, Kwon H, Lefevour A, Chakraborty-Sett S, Greene WC (2006) NF-kappaB/Rel regulates inhibitory and excitatory neuronal function and synaptic plasticity. Mol Cell Biol 26:7283–7298
O’Riordan KJ, Huang IC, Pizzi M, Spano P, Boroni F, Egli R, Desai P, Fitch O, Malone L, Ahn HJ, Liou HC, Sweatt JD, Levenson JM (2006) Regulation of nuclear factor kappaB in the hippocampus by group I metabotropic glutamate receptors. J Neurosci 26:4870–4879
Ouali Alami N, Schurr C, Olde Heuvel F, Tang L, Li Q, Tasdogan A, Kimbara A, Nettekoven M, Ottaviani G, Raposo C, Röver S, Rogers-Evans M, Rothenhäusler B, Ullmer C, Fingerle J, Grether U, Knuesel I, Boeckers TM, Ludolph A, Wirth T, Roselli F, Baumann B (2018) NF-κB activation in astrocytes drives a stage-specific beneficial neuroimmunological response in ALS. EMBO J 37:e98697
Owens R, Grabert K, Davies CL, Alfieri A, Antel JP, Healy LM, McColl BW (2017) Divergent neuroinflammatory regulation of microglial TREM expression and involvement of NF-κB. Front Cell Neurosci 11:56
Panicker N, Sarkar S, Harischandra DS, Neal M, Kam TI, Jin H, Saminathan H, Langley M, Charli A, Samidurai M, Rokad D, Ghaisas S, Pletnikova O, Dawson VL, Dawson TM, Anantharam V, Kanthasamy AG, Kanthasamy A (2019) Fyn kinase regulates misfolded α-synuclein uptake and NLRP3 inflammasome activation in microglia. J Exp Med 216:1411–1430
Panicker N, Ge P, Dawson VL, Dawson TM (2021) The cell biology of Parkinson’s disease. J Cell Biol 220:12095
Parkinson J (2002) An essay on the shaking palsy. J Neuropsychiatry Clin Neurosci 14:223–236
Pivari F, Mingione A, Brasacchio C, Soldati L (2019) Curcumin and type 2 diabetes mellitus: prevention and treatment. Nutrients 11:1837
Prasad S, Katta MR, Abhishek S, Sridhar R, Valisekka SS, Hameed M, Kaur J, Walia N (2023) Recent advances in Lewy body dementia: a comprehensive review. Dis Mon 69:101441
Priyadarsini KI (2014) The chemistry of curcumin: from extraction to therapeutic agent. Molecules 19:20091–20112
Rachmawati H, Al Shaal L, Müller RH, Keck CM (2013) Development of curcumin nanocrystal: physical aspects. J Pharm Sci 102:204–214
Ransohoff RM (2016) How neuroinflammation contributes to neurodegeneration. Science 353:777–783
Reich N, Hölscher C (2022) The neuroprotective effects of glucagon-like peptide 1 in Alzheimer’s and Parkinson’s disease: an in-depth review. Front Neurosci 16:970925
Ringman JM, Frautschy SA, Teng E, Begum AN, Bardens J, Beigi M, Gylys KH, Badmaev V, Heath DD, Apostolova LG, Porter V, Vanek Z, Marshall GA, Hellemann G, Sugar C, Masterman DL, Montine TJ, Cummings JL, Cole GM (2012) Oral curcumin for Alzheimer’s disease: tolerability and efficacy in a 24-week randomized, double blind, placebo-controlled study. Alzheimers Res Ther 4:43
Rojo AI, Salinas M, Martín D, Perona R, Cuadrado A (2004) Regulation of Cu/Zn-superoxide dismutase expression via the phosphatidylinositol 3 kinase/Akt pathway and nuclear factor-kappaB. J Neurosci 24:7324–7334
Ruiz-Perera LM, Schneider L, Windmöller BA, Müller J, Greiner JFW, Kaltschmidt C, Kaltschmidt B (2018) NF-κB p65 directs sex-specific neuroprotection in human neurons. Sci Rep 8:16012
Schmidt-Ullrich R, Mémet S, Lilienbaum A, Feuillard J, Raphaël M, Israel A (1996) NF-kappaB activity in transgenic mice: developmental regulation and tissue specificity. Development 122:2117–2128
Schwaninger M, Inta I, Herrmann O (2006) NF-kappaB signalling in cerebral ischaemia. Biochem Soc Trans 34:1291–1294
Shadnoush M, Zahedi H, Norouzy A, Sahebkar A, Sadeghi O, Najafi A, Hosseini S, Qorbani M, Ahmadi A, Ardehali SH, Hosseinzadeh-Attar MJ (2020) Effects of supplementation with curcuminoids on serum adipokines in critically ill patients: a randomized double-blind placebo-controlled trial. Phytother Res 34:3180–3188
Shanmugam MK, Rane G, Kanchi MM, Arfuso F, Chinnathambi A, Zayed ME, Alharbi SA, Tan BK, Kumar AP, Sethi G (2015) The multifaceted role of curcumin in cancer prevention and treatment. Molecules 20:2728–2769
Sharifi-Rad J, Rayess YE, Rizk AA, Sadaka C, Zgheib R, Zam W, Sestito S, Rapposelli S, Neffe-Skocińska K, Zielińska D, Salehi B, Setzer WN, Dosoky NS, Taheri Y, el Beyrouthy M, Martorell M, Ostrander EA, Suleria HAR, Cho WC, Maroyi A, Martins N (2020) Turmeric and Its Major Compound Curcumin on Health: Bioactive Effects and Safety Profiles for Food, Pharmaceutical. Biotechnol Med Appl Front Pharmacol 11:01021
Sharma N, Sharma S, Nehru B (2017) Curcumin protects dopaminergic neurons against inflammation-mediated damage and improves motor dysfunction induced by single intranigral lipopolysaccharide injection. Inflammopharmacology 25:351–368
Snow WM, Albensi BC (2016) Neuronal gene targets of NF-κB and their dysregulation in Alzheimer’s disease. Front Mol Neurosci 9:118
Steiner JA, Quansah E, Brundin P (2018) The concept of alpha-synuclein as a prion-like protein: ten years after. Cell Tissue Res 373:161–173
Sun SC (2011) Non-canonical NF-κB signaling pathway. Cell Res 21:71–85
Sun G, Miao Z, Ye Y, Zhao P, Fan L, Bao Z, Tu Y, Li C, Chao H, Xu X, Ji J (2020) Curcumin alleviates neuroinflammation, enhances hippocampal neurogenesis, and improves spatial memory after traumatic brain injury. Brain Res Bull 162:84–93
Sun E, Motolani A, Campos L, Lu T (2022) The pivotal role of NF-kB in the pathogenesis and therapeutics of Alzheimer’s disease. Int J Mol Sci 23:8972
Swarup V, Phaneuf D, Dupré N, Petri S, Strong M, Kriz J, Julien JP (2011) Deregulation of TDP-43 in amyotrophic lateral sclerosis triggers nuclear factor κB-mediated pathogenic pathways. J Exp Med 208:2429–2447
Takano H, Gusella JF (2002) The predominantly HEAT-like motif structure of huntingtin and its association and coincident nuclear entry with dorsal, an NF-kB/Rel/dorsal family transcription factor. BMC Neurosci 3:15
Tanaka Y, Sabharwal L, Ota M, Nakagawa I, Jiang JJ, Arima Y, Ogura H, Okochi M, Ishii M, Kamimura D, Murakami M (2018) Presenilin 1 regulates NF-κB activation via association with breakpoint cluster region and casein kinase II. J Immunol 201:2256–2263
Thawkar BS, Kaur G (2019) Inhibitors of NF-κB and P2X7/NLRP3/Caspase 1 pathway in microglia: novel therapeutic opportunities in neuroinflammation induced early-stage Alzheimer’s disease. J Neuroimmunol 326:62–74
Thomsen MB, Foster E, Nguyen KH, Sosunov EA (2009) Transcriptional and electrophysiological consequences of KChIP2-mediated regulation of CaV1.2. Channels (austin) 3:308–310
Thota RN, Rosato JI, Dias CB, Burrows TL, Martins RN, Garg ML (2020) Dietary supplementation with curcumin reduce circulating levels of glycogen synthase kinase-3β and islet amyloid polypeptide in adults with high risk of type 2 diabetes and Alzheimer’s disease. Nutrients 12:1032
Wang Z, Zhang Q, Yuan L, Wang S, Liu L, Yang X, Li G, Liu D (2014) The effects of curcumin on depressive-like behavior in mice after lipopolysaccharide administration. Behav Brain Res 274:282–290
Wang M, Kou J, Wang C, Yu X, Xie X, Pang X (2020) Curcumin inhibits APOE4-induced injury by activating peroxisome proliferator-activated receptor-γ (PPARγ) in SH-SY5Y cells. Iran J Basic Med Sci 23:1576–1583
Wood-Kaczmar A, Gandhi S, Wood NW (2006) Understanding the molecular causes of Parkinson’s disease. Trends Mol Med 12:521–528
Xie P, Deng M, Sun Q, Jiang B, Xu H, Liu J, Zhou Y, Ma Y, Chen Z (2020) Curcumin protects BV2 cells against lipopolysaccharide-induced injury via adjusting the miR-362-3p/TLR4 axis. Mol Biol Rep 47:4199–4208
Xu L, Hao LP, Yu J, Cheng SY, Li F, Ding SM, Zhang R (2023) Curcumin protects against rotenone-induced Parkinson’s disease in mice by inhibiting microglial NLRP3 inflammasome activation and alleviating mitochondrial dysfunction. Heliyon 9:e16195
Yang S, Zhang D, Yang Z, Hu X, Qian S, Liu J, Wilson B, Block M, Hong JS (2008) Curcumin protects dopaminergic neuron against LPS induced neurotoxicity in primary rat neuron/glia culture. Neurochem Res 33:2044–2053
Yang H, Huang S, Wei Y, Cao S, Pi C, Feng T, Liang J, Zhao L, Ren G (2017) Curcumin enhances the anticancer effect Of 5-fluorouracil against gastric cancer through down-regulation of COX-2 and NF-κB signaling pathways. J Cancer 8:3697–3706
Ying H, Yue BY (2012) Cellular and molecular biology of optineurin. Int Rev Cell Mol Biol 294:223–258
Yu DS, Cao Y, Mei XF, Wang YF, Fan ZK, Wang YS, Lv G (2014) Curcumin improves the integrity of blood-spinal cord barrier after compressive spinal cord injury in rats. J Neurol Sci 346:51–59
Yu S, Wang X, He X, Wang Y, Gao S, Ren L, Shi Y (2016) Curcumin exerts anti-inflammatory and antioxidative properties in 1-methyl-4-phenylpyridinium ion (MPP(+))-stimulated mesencephalic astrocytes by interference with TLR4 and downstream signaling pathway. Cell Stress Chaperones 21:697–705
Yuan J, Zou M, Xiang X, Zhu H, Chu W, Liu W, Chen F, Lin J (2015) Curcumin improves neural function after spinal cord injury by the joint inhibition of the intracellular and extracellular components of glial scar. J Surg Res 195:235–245
Yuan J, Liu W, Zhu H, Chen Y, Zhang X, Li L, Chu W, Wen Z, Feng H, Lin J (2017) Curcumin inhibits glial scar formation by suppressing astrocyte-induced inflammation and fibrosis in vitro and in vivo. Brain Res 1655:90–103
Zahedi H, Hosseinzadeh-Attar MJ, Shadnoush M, Sahebkar A, Barkhidarian B, Sadeghi O, Najafi A, Hosseini S, Qorbani M, Ahmadi A, Ardehali SH, Norouzy A (2021) Effects of curcuminoids on inflammatory and oxidative stress biomarkers and clinical outcomes in critically ill patients: A randomized double-blind placebo-controlled trial. Phytother Res 35:4605–4615
Zhang N, Wei G, Ye J, Yang L, Hong Y, Liu G, Zhong H, Cai X (2017) Effect of curcumin on acute spinal cord injury in mice via inhibition of inflammation and TAK1 pathway. Pharmacol Rep 69:1001–1006
Zhang J, Zheng Y, Luo Y, Du Y, Zhang X, Fu J (2019) Curcumin inhibits LPS-induced neuroinflammation by promoting microglial M2 polarization via TREM2/ TLR4/ NF-κB pathways in BV2 cells. Mol Immunol 116:29–37
Acknowledgements
None.
Funding
The authors declare that no funds, grants, or other support was received during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
Niusha Esmaealzadeh: data curation, writing- original draft preparation. Mahdis Sadat Miri: data curation, writing—original draft preparation. Helia Mavvadat: data curation, writing—original draft preparation. Amirreza Peyrovinasab: data curation, writing—original draft preparation. Sara Ghasemi Zargar: data curation, writing—original draft preparation. Shirin Sirous Kabiri: data curation, writing—original draft preparation. Seyed Mehrad Razavi: conceptualization, methodology, reviewing and editing, supervision. Amir Hossein Abdolghaffari: conceptualization, reviewing and editing, supervision.
Corresponding authors
Ethics declarations
Conflict of interests
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Esmaealzadeh, N., Miri, M., Mavaddat, H. et al. The regulating effect of curcumin on NF-κB pathway in neurodegenerative diseases: a review of the underlying mechanisms. Inflammopharmacol 32, 2125–2151 (2024). https://doi.org/10.1007/s10787-024-01492-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10787-024-01492-1