
Abstract
The brain of patients affected by Alzheimer’s disease (AD) develops progressive neurodegeneration linked to the formation of proteins aggregates. However, their single actions cannot explain the extent of brain damage observed in this disorder, and the characterization of co-adjuvant involved in the early toxic processes evoked in AD is essential. In this line, quinolinic acid (QUIN) and homocysteine (Hcy) appear to be involved in the AD neuropathogenesis. Herein, we investigate the effects of QUIN and Hcy on early toxic events in cortical neurons and astrocytes. Exposure of primary cortical cultures to these neurometabolites for 24 h induced concentration-dependent neurotoxicity. In addition, QUIN (25 μM) and Hcy (30 μM) triggered ROS production, lipid peroxidation, diminished of Na+,K+-ATPase activity, and morphologic alterations, culminating in reduced neuronal viability by necrotic cell death. In astrocytes, QUIN (100 μM) and Hcy (30 μM) induced caspase-3-dependent apoptosis and morphologic alterations through oxidative status imbalance. To establish specific mechanisms, we preincubated cell cultures with different protective agents. The combined toxicity of QUIN and Hcy was attenuated by melatonin and Trolox in neurons and by NMDA antagonists and glutathione in astrocytes. Cellular death and morphologic alterations were prevented when co-culture was treated with metabolites, suggesting the activation of protector mechanisms dependent on soluble factors and astrocyte and neuron communication through gap junctions. These findings suggest that early damaging events involved in AD can be magnified by synergistic toxicity of the QUIN and Hcy. Therefore, this study opens new possibilities to elucidate the molecular mechanisms of neuron-astrocyte interactions and their role in neuroprotection against QUIN and Hcy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Neurodegeneration is characterized by progressive loss of neuronal integrity, in both structure and function. Among neurodegenerative diseases, Alzheimer’s disease (AD) is the most common worldwide, affecting approximately 10% of individuals over the age of 60. According to data revealed by the World Health Organization, the number of cases of dementia among the elderly is expected to increase to over 115 million people by 2050 (www.Alz.co.uk/research/statics). Thus, million people will be suffering from dementia and will require significant medical, monetary, and human resources for their care, leading AD to epidemic proportions.
AD is characterized by progressive memory loss and cognitive dysfunction, resulting in severe dementia (Goedert and Spillantini 2006a). The key feature of the disease is the formation of amyloid plaques and neurofibrillary tangles, leading to neuronal death mostly in the neocortex and hippocampus (Mudher and Lovestone 2002). However, cortical and hippocampal neuronal degeneration could be the consequence of a combination of several mechanisms, including perturbations in protein metabolism, excitotoxicity, oxidative stress, mitochondrial perturbations, and inflammation (Goedert and Spillantini 2006b; Mudher and Lovestone 2002). Although the biochemical and cellular basis of AD symptoms have received great attention in scientific researches, the understanding of the pathologic mechanisms underlying the disease remains unknown. Furthermore, the study and discovery of therapeutic strategies to prevent or delay neurodegeneration neural tissue is a high priority, since no cure is currently available.
Along with genetic factors that are the prime cause of homocysteine (Hcy) imbalance, the nutritional, aging, and hormonal factors are also contributing to high Hcy levels in the body (Sharma et al. 2015). Several studies have reported that elevated levels of serum Hcy are correlated with AD (Gottfries et al. 1998; Seshadri 2012). Individuals with serum Hcy concentration above 14 μM had a 4.6 higher risk of having AD than those subjects with plasma Hcy below 11 μM (Clarke et al. 1998). Hcy has been associated with the degree of cognitive impairment or stage of dementia and could act as a biomarker for the progression of AD. Accordingly, hyperhomocisteinemia is associated with poor performance on objective tests of cognitive function in studies with AD patients (Bell et al. 1992; Williams et al. 2002). Also, results from Heijer and collaborators (den Heijer et al. 2003) showed that high plasma Hcy levels are associated with atrophy of the hippocampus and cortical regions in elderly at risk of AD. Based in all this, it is logical to assume that homocysteine plays a role in neurodegeneration seen in the disease. Oxidative/nitrative stress and neuroinflammation has been associated to the deleterious effect of Hcy (Sharma et al. 2015). However, to our knowledge, the effects of lower doses of Hcy (30 μM) in neural cells have not fully understood.
Kynurenine pathway is the major route of tryptophan metabolism leading to production of a number of biologically active molecules. Among them, quinolinic acid (QUIN) is considered a more important neurotoxin since it is involved in the pathogenesis of a number of neurological diseases, including AD (Guillemin and Brew 2002; Tan et al. 2012). A cleavage product of amyloid precursor protein, Aβ 1-42, induces production of QUIN in neurotoxic concentrations (Guillemin et al. 2003). High QUIN concentrations can lead to inflammation and excitotoxicity around the senile plaques (Guillemin and Brew 2002). Thus, it can cause glutamate accumulation, oxidative stress, and neural death by cytotoxicity (Kincses et al. 2010). These findings imply that QUIN may be one of the critical factors in the pathogenesis of neural damage in AD.
Both QUIN and Hcy exert their effects through glutamatergic receptors overactivation, leading to excitotoxicity, oxidative stress, and cell death. Furthermore, high Hcy levels can exacerbate QUIN effects through decrease of kynurenic acid (KYNA), another active molecule of kynurenine pathway with antagonistic effects on QUIN. In addition, it has been shown that QUIN and Hcy are involved in energy and redox status changes (Lugo-Huitron et al. 2013; Staehelin 2005), and it is well-known that those alterations could lead to activation of cell death pathways (Cobb and Cole 2015; Prentice et al. 2015).
One problem with most studies involving the role of potential metabolites in neurodegenerative diseases is that they have typically been studied separately, whereas the actual situation clearly involves interaction between multiple toxic agents. Also, the limited understanding of the mechanisms underlying the onset of symptoms is responsible, at least in part, by the lack of effective treatment for AD cases. Since AD is characterized by a long prodromal period, its etiology may be better investigated in relation to early preclinical pathophysiology of the disease.
Considering that both QUIN and Hcy are increased in the early stages of the disease and are involved in the AD pathophysiology, the present study aimed to investigate the death mechanisms triggered by QUIN and Hcy in primary cortical astrocytes and neurons, and evaluate some mechanisms underlying these actions. We focused on pro-apoptotic proteins, mitochondrial potential, redox imbalance, Na+,K+-ATPase activity, morphologic alterations, and astrocyte-neuron communication on QUIN and Hcy effects. In addition, glutamate antagonists and antioxidants were investigated in the paradigm of cell death and protective mechanisms through GJ.
Materials and Methods
Materials
Phalloidin-fluorescein isothiocynate, bis-acrylamide, benzamidine, leupeptin, antipain, pepstatin, chymostatin, acrylamide, the antibodies anti-β III-tubulin, anti-mouse IgG (whole molecule), anti-mouse IgG (whole molecule)-fluorescein isothiocyante (FITC), F(ab)2 fragment-Cy3, peroxidase-conjugated anti-mouse IgG, anti-active caspase-3, melatonin, Trolox, L-glutathione reduced, ibuprofen, diclofenac sodium salt, acetylsalicylic acid, MK-801, quinolinic acid, DL-homocysteine, and material for cell culture were obtained from Sigma (St. Louis, MO). Polyclonal anti-GFAP antibody was from Dako (Glostrup, Denmark). 4,6-Diamidino-2-phenylindole (DAPI) was from Calbiochem (La Jolla, CA). An enhanced chemiluminescence (ECL) detection kit was from Millipore (Billerica, MA). Anti-actin, anti-phospho Akt, anti-GSK-3β, anti-phosphoGSK-3β, and anti-MAP2 antibodies were from Cell Signaling Technology (Boston, MA). Fetal bovine serum (FBS), Dulbecco’s modified Eagle’s medium nutrient mixture F-12 (DMEM/F12), Neurobasal medium, B-27 supplement, fungizone, and penicillin/streptomycin were purchased from Gibco BRL (Carlsbad, CA). The potent competitive non-NMDA iGluR antagonist 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) and the non-selective group I/group II mGluR antagonist (R,S)-a-methyl-4-carboxyphenylglycine (MCPG) were purchased from Tocris Neuramin (Bristol, UK). FITC Annexin V Apoptosis Detection Kit was from BD Pharmigen (San Diego, CA). Mitotracker red and green were purchased from Thermofisher Scientific (Waltham, MA).
Animals
Pregnant Wistar rats (200–250 g) were obtained from our breeding stock. Rats were maintained under a 12-h light-dark cycle in a constant temperature (22 °C) colony room, with food and water ad libitum. Animals were observed on gestational day 22 and as soon as delivery had finished, mothers and pups were sacrificed by decapitation without anesthesia, and pups were used for astrocyte culture. For neuron culture, mothers were sacrificed on gestational day 18 by decapitation and the embryos were used for neuronal cultures. The experimental protocol followed the official governmental guidelines in issued by the Brazilian Federation of Societies for Experimental Biology, following the Guide for the Care and Use of Laboratory Animals (No. 80-23, revised 1996) and Arouca Law (Law n° 11.794/2008). In addition, all experimental protocols of this study were approved by the Ethics Committee of Federal University of Rio Grande do Sul (UFRGS, RS, Brazil) under license (#26995). We further attest that all efforts were made to minimize the number of animals used and their suffering.
Astrocyte and Neuron Primary Culture
Astrocytes were assessed from the cerebral cortex of newborn (0–1 day old; P0) Wistar rats as previously described (Pierozan et al. 2014). Briefly, rats were decapitated and the cerebral cortex was removed. Mechanically dissociated cells were plated in DMEM/F12/10% fetal bovine serum (FBS) (pH 7.4) supplemented with glucose (33 mM), glutamine (2 mM), and sodium bicarbonate (3 mM) in 15.6- and 34.8-mm-diameter wells (six- and 24-well plates; Corning, Corning, NY) that had previously been coated with polyornithine (1.5 mg/mL; Sigma). These cells were grown in a humid incubator (37 °C; 5% CO2), with the media replaced every 3 days. After astrocytes had reached semiconfluence (15 days in vitro [DIV]), the culture medium was removed by suction, and the cells were incubated for 24 h at 37 °C in an atmosphere of 5% CO2 in DMEM/F12 without FBS and treated with the metabolites (QUIN plus DL-Hcy). Some cultures were treated with the same incubation medium but without metabolites (control cultures). Morphological studies were performed with cells fixed for immunocytochemistry.
Primary neuronal cell cultures were prepared from embryonic day 18 Wistar rat striatum as previously described (Pierozan et al. 2015). Briefly, single-cell suspensions were obtained by dissociating cerebral cortical cells in DMEM/F12 medium supplemented with 33 mM glucose, 2 mM glutamine, and 3 mM sodium bicarbonate. Approximately 5 × 104 neuronal cells were plated on polylysine-treated coverslips placed on a 24-well plate or 1.5 × 106 neuronal cells for a six-well plate. The neuronal cultures were kept in Neurobasal medium supplemented with 2 mM glutamine and B27 for up to 24 h. After this, the culture medium was removed by suction, and the cells were incubated for 7 days in a humid incubator (37 °C; 5% CO2). When neuronal cultures were 8 DIV, the culture medium was removed and cells were treated for an additional 24 h in Neurobasal medium in the presence of metabolites. Some cultures were treated with the same incubation medium but without metabolites (control cultures). Morphological studies were performed with cells fixed for immunocytochemistry.
Co-culture consisted of recently isolated striatal neurons plated on top of nonrelated astrocyte monolayers (15 DIV). Co-cultures was kept for 7 days at 37 °C in a humidified 5% CO2 air atmosphere. When neurons were at 8 DIV, the cultures were used for subsequent treatment.
3-(4,5-Dimethyl-2-yl)22,5-Diphenyl-2H-Tetrazolium Bromide Assay
Neurons and astrocytes were seeded at a density of 2 × 104 cells/well in 96-well plates (8-DIV neurons and 15-DIV astrocytes). They were treated with 25–100 μM QUIN and/or 30 μM Hcy for 24 h, with three replicates of each treatment. Cell viability was measured by the 3-(4,5-dimethyl-2-yl)22,5-diphenyl-2H-tetrazolium bromide (MTT) assay, which is based on the conversion of the tetrazolium salt to the colored product formazan. In brief, 0.5 mg MTT was added to each well of the 96-well plates (containing 100 μL medium and cells) 4 h before the end of incubation with metabolites. The supernatant was then separated, and 100 μL dimethylsulfoxide (DMSO) was added to each well, followed by incubation and shaking for 10 min. The formazan product generated during the incubation was solubilized in DMSO and measured at 540 and 620 nm. Only viable cells are able to reduce MTT.
In the experiments designed to study signaling mechanisms of cell death, cells were preincubated for 30 min in the presence or absence of 100 μM MK-801 (Jiang et al. 2010), 50 μM DL-AP5, 25 μM CNQX, 50 μM MCPG, 30 μM MPEP, (Loureiro et al. 2015), 1 mM GSH (de Lima Pelaez et al. 2007), 10 μM melatonin and 50 μM Trolox (Loureiro et al. 2013), 100 μM LaCl3 (Rana and Dringen 2007) (Santos-Carvalho et al. 2013), 10 μM AAS, 1 μg/mL sodium diclofenac and 100 μg/mL ibuprofen, and 1.5 μg/mL nimesulide (Loureiro et al. 2016). After the preincubation, the culture medium was removed and cells were treated for an additional 24 h in the presence or absence of metabolites plus antagonists or antioxidants.
In experiments designed to evaluate the role of the conditioned medium of astrocytes and neuron on cell survival, the astrocytes (15 DIV), neurons (8DIV), and co-cultures were pretreated with QUIN and Hcy for 24 h in serum-free DMEM/F12 medium, and after 24 h the medium was collected. The cultures were treated with the astrocyte or neuron-conditioned medium plus QUIN and Hcy for 24 h.
In the experiments designed to evaluate the role of GJ on cell survival, the co-culture were pretreated with 50 μM La3Cl2 for 30 min (Moore et al. 2014), followed by treatment with QUIN, Hcy or both for 24 h. In the experiments designed to investigate the mechanisms by which GJ are involved in the protective mechanisms triggered by co-culture, the cells were pretreated for 30 min in the presence or absence of 100 μM MK-801 (Jiang et al. 2010), 50 μM DL-AP5, 25 μM CNQX, 50 μM MCPG, 30 μM MPEP, (Loureiro et al. 2015), 1 mM GSH (de Lima Pelaez et al. 2007), 10 μM melatonin and 50 μM Trolox (Loureiro et al. 2013), 100 μM LaCl3 (Rana and Dringen 2007) (Santos-Carvalho et al. 2013), 10 μM AAS, 1 μg/mL sodium diclofenac and 100 μg/mL ibuprofen, and 1.5 μg/mL nimesulide (Loureiro et al. 2016). After the preincubation, the culture medium was removed and cells were treated for an additional 24 h in the presence or absence of metabolites plus antagonists, antioxidants, and La3Cl2.
Annexin V-Pi Labeling
The apoptotic/necrotic detection assay was carried out by surface labeling with the Ca2+-dependent phosphatidylserine-binding protein annexin V and propidium iodide. After they had been incubated with QUIN (25–100 μM) and/or 30 μM Hcy for 24 h, cells were recovered from the culture plates by 0.25% trypsin-EDTA treatment, centrifuged (1000 g for 5 min), and washed once with phosphate-buffered saline (PBS). Cells were labeled by incubation with annexin V-FITC and propidium iodide (Pi) in a binding buffer (apoptosis detection kit I-556547; BD Pharmingen) for 15 min at room temperature in the dark, according to the manufacturer’s instruction. Stained cells were acquired (10,000 for gated astrocytes and neurons) on an FACS Calibur flow cytometer. Analysis was performed in the FlowJo Software.
Mitochondrial Mass and Membrane Potential Measurements
MitoTracker Red (MTR) and MitoTracker Green (MTG) dyes were employed to assess mitochondrial function. MTR is a lipophilic cationic fluorescent dye that is concentrated inside mitochondria because of the negative mitochondrial membrane potential (Pendergrass et al. 2004). The loss of membrane potential results in release of MTR from mitochondria and a subsequent decrease in fluorescence (Khanal et al. 2011). MTG is a green-fluorescing fluorophore which is taken up electrophoretically into mitochondria and has been used as a measure of mitochondrial mass independent of mitochondrial membrane potential. MTR and MTG were dissolved in dimethylsulfoxide (DMSO) to a 1-mM stock concentration. Dissociated cells, previously filtered, were stained with 100 nm MTR and 100 nm MTG for 45 min at 37 °C in a water bath in a dark room according to method described by Keij et al. (2000) and Pendergrass et al. (2004) with some modifications. Immediately after staining, cell suspensions were removed from the water bath and analyzed by flow cytometry on a FACSCalibur flow cytometer (Becton Dickinson, San Jose, CA). The emission of fluorochromes was recorded through specific band-pass fluorescence filters: red (FL-3; 670 nm long pass) and green (FL-1; 530 nm/30). Data from 10,000 events (intact cells) were acquired and mean relative fluorescence intensity was determined after exclusion of debris events from the data set. All flow cytometric acquisitions and analyses were performed using CELLQuest Pro data acquisition (BD Biosciences) and the FlowJo analysis software.
2′7′-Dichlorofluorescein Fluorescence Assay
Reactive oxygen species production was measured according to the method of LeBel et al. (1990) and based on the oxidation of 2′7′-dichlorofluorescein (H2DCF). The samples were incubated in a medium containing 100 μM 2′7′-dichlorofluorescein diacetate (H2DCF-DA) solution. Reaction produces the fluorescent compound dichlorofluorescein (DCF) which is measured at λem = 525 nm and λex = 488 nm.
Sulfhydryl Content
This assay was performed as described by Aksenov and Markesbery (2001), which is based on the reduction of 5,5′-dithio-bis(2-nitrobenzoic acid) (DTNB) by sulfhydryl groups, which in turn become oxidized (disulfide), generating a yellow derivative (TNB) whose absorption is measured spectrophotometrically at 412 nm. Briefly, 15 μL of homogenate were added to 275 μL of phosphate-buffered saline pH 7.4 containing 1 mM EDTA. The reaction was started by the addition of 10 μL of 10 mM DTNB and incubated for 30 min at room temperature in a dark room. The sulfhydryl content is inversely correlated to oxidative damage to the protein.
Thiobarbituric Acid-Reactive Substances (TBARS) Assay
TBA-RS levels were measured according to the method described by Yagi (1998) with slight modifications. Briefly, 50 μL of 10% trichloroacetic acid and 50 μL of cell homogenate were incubated for 1 h in a boiling water bath in the presence of 400 μL of 0.67% thiobarbituric acid in 7.1% sodium sulfate. The mixture was allowed to cool on running tap water for 5 min. The resulting pink-stained complex was extracted with 400 μL of butanol. Fluorescence of the organic phase was read at 515 and 553 nm as excitation and emission wavelengths, respectively. Calibration curve was performed using 1,1,3,3-tetramethoxypropane and subjected to the same treatment as supernatants. TBA-RS levels were calculated as percentage of controls.
Na+,K+-ATPase Activity Assay
The reaction mixture for the Na+,K+-ATPase assay contained 5 mM MgCl2, 80 mM NaCl, 20 mM KCl, and 40 mM Tris HCl, pH 7.4, to give a final volume of 200 μL. The reaction was initiated by adding ATP to a final 3 mM concentration. Controls were carried out under the same conditions with the addition of 1 mM ouabain. Na+K+-ATPase activity was calculated from the difference between the two assays, according to the method of Wyse et al. (1999). Released inorganic phosphate (Pi) was measured by the method of Chan et al. (1986). The enzyme-specific activity was expressed as nanomoles of Pi released per minute per milligram protein.
Western Blot Analysis
After 24 h of metabolites treatment, cells were disrupted by lysis solution containing 2 mM EDTA, 50 mMTris-HCl, pH 6.8, and 4% SDS. For electrophoretic analysis, samples were dissolved in 25% (v/v) of a solution containing 40% glycerol, 5% mercaptoethanol, and 50 mM TrisHCl, pH 6.8, and boiled for 3 min. Total protein homogenate was analyzed by 10% SDS-PAGE (30 mg total protein/lane) and transferred (Trans-blot SD semidry transfer cell; Bio-Rad, Hercules, CA) to nitrocellulose membranes for 1 h at 15 V in transfer buffer (48 mM Trizma, 39 mM glycine, 20% methanol, and 0.25% SDS). The blot was then washed for 20 min in Tris-buffered saline (TBS; 500 mM NaCl, 20 mM Trizma, pH 7.5), followed by 2 h incubation in blocking solution (TBS plus 5% defatted dry milk). After the incubation was finished, the blot was washed twice for 5 min with blocking solution plus 0.05% Tween-20 (T-TBS) and then incubated overnight at 4 °C in blocking solution containing monoclonal antibodies diluted 1:1000. The blot was then washed twice for 5 min with T-TBS and incubated for 2 h in a solution containing peroxidase-conjugated rabbit anti-mouse IgG diluted 1:2000 or peroxidase-conjugated anti-rabbit IgG diluted 1:2000. The blot was washed twice with T-TBS for 5 min and twice with TBS for 5 min. The blot was developed with the chemiluminescence ECL kit.
Immunocytochemistry
When neuronal cultures had reached 8 DIV and astrocytes had reached 15 DIV, the culture medium was removed and cells were treated for an additional 24 h with the metabolites. Immunocytochemistry was performed as previously described (Pierozan et al. 2015). Briefly, cultured cells that had been plated on glass coverslips were fixed with 4% paraformaldehyde for 30 min and permeabilized with 0.1% Triton X-100 in PBS for 5 min at room temperature. After having been blocked, neurons were incubated overnight with anti-β III-tubulin (1:200) and anti-MAP2 (1:200), and astrocytes were incubated with anti-actin (1:200) and anti-GFAP (1:500) at room temperature, followed by PBS washes and incubation with specific secondary antibody conjugated with Cy3 (sheep anti-rabbit, 1:1000) or with FITC (sheep anti-mouse, 1:400) for 1 h. Co-cultures were preincubated with La3Cl2 and antioxidants or glutamate antagonists, before treatment with the metabolites. To analyze the morphology of the cells, the co-culture were incubated with anti-β III-tubulin and anti-GFAP antibodies. In all immunostaining, negative controls reactions were performed by omitting the primary antibody. No reactivity was observed when the primary antibody was excluded. The nucleus was stained with DAPI (0.25 mg/mL). Cells were viewed with a Nikon inverted microscope, and images were transferred to a computer with a digital camera (Sound Vision).
Morphometric Analysis
Neurons stained with anti-βIII-tubulin and anti-MAP2 antibodies were captured by a system coupled to a Nikon Eclipse T E300 inverted fluorescence microscope (Nikon, Tokyo, Japan). The neurite/neuron ratio and the neurite length were analyzed in ImageJ (NIH) as previously described (Baranes et al. 2012). The number of processes originating from the soma was measured manually, and all neurites emerging from neuronal soma were considered. To measure the neurite length, we used an ImageJ plugin that allows semiautomatic tracing of neuritis for measurements of neurite length. Neurite length was analyzed by considering only the major process of each neuron. At least four independent experiments were performed in triplicate, encompassing 10 fields randomly chosen in each group. The data were stored and morphometric analyses were carried out in GraphPad Prism 5 (GraphPad Software, La Jolla, CA).
Astrocytes stained for actin and GFAP cytoskeleton were measured by using labeling for phalloidin-fluorescein, GFAP-Cy3 labeling and DAPI, respectively. Measurements of cytoplasm area was obtained in ImageJ and used as a criterion for morphological alteration. An ImageJ plugin that allows semiautomatic tracing of the area of interest was used to measure the cytoplasmic area. Four independent experiments were performed in triplicate, encompassing 10 fields randomly chosen in each group. The data were stored and morphometric analyses were carried out in GraphPad Prism 5.
Statistical Analysis
Data from the experiments were analyzed statistically by two-way ANOVA, followed by the Tukey-Kramer test when the F test was significant. P < 0.05 was considered significant. All analyses were carried out with an IBM-compatible PC in SPSS.
Results
First, we searched for cellular viability of cortical neurons and astrocytes treated with 25–100 μM QUIN concentrations and/or 30 μM Hcy. This Hcy concentration is considered mild homocysteinemia, and it is well-established risk factor for Alzheimer’s disease (Sharma et al. 2015). Cells were incubated with 25–100 μM QUIN and/or 30 μM Hcy for 24 h and cell viability was determinate by MTT assay. No significant differences were found when metabolites were incubated alone. However, we found significantly decrease in neuronal viability in concentration from 25 μM QUIN + 30 μM Hcy (Fig. 1a). In astrocytes, cell viability was statistically decreased only from 100 μM QUIN + 30 μM Hcy (Fig. 1b), suggesting a different susceptibility for these two cell types to these compounds.

The profile of cell death and mitochondrial function induced by QUIN and Hcy on neuron and astrocyte. Cortical neuron (a) and astrocyte (b) were incubated in the presence or absence of 25–100 μM QUIN and/or 30 μM Hcy for 24 h. One hour before completing treatment with metabolites, 0.5 mg/mL of MTT was added to the incubation medium, and the formazan product generated was measured at 540 and 620 nm. Apoptotic and necrotic cortical neuron (c) and astrocytes (d) were assessed with annexin V-FITC and Pi assay. Apoptotic cell death was detected with FITC-labeled annexin V; necrotic cells were detected with Pi; cells that underwent apoptosis followed by necrosis were detected with annexin plus Pi; cells without labeling were live cells. Evaluation of mitochondrial mass (MTG), permeability (MTR), and function (ratio) on neurons (e) and astrocytes (f). Data are reported as mean ± SD of four independent experiments performed in triplicate. Results were statistically analyzed by two-way ANOVA followed by the Tukey-Kramer test. Statistically significant differences from control are indicated as follows: ***p < 0.001, **p < 0.01, and *p < 0.05
Next, we evaluated the type of cell death triggered by QUIN and Hcy in the primary neurons and astrocytes using the Annexin-Pi assay. Interestingly, we found significant diminished live neurons in all treated groups (Fig. 1c). However, the type of cell death triggered by metabolites alone or in combination it was shown to be different. Neurons treated with 25 μM of QUIN dye by apoptotic mechanisms, while 30 μM Hcy triggered both apoptotic and necrotic mechanisms of cell death. In neurons treated with 25 μM QUIN plus 30 μM Hcy, the predominant mechanism of neuronal death is necrosis. On the other hand, in astrocytes, the 100 μM QUIN plus 30 μM Hcy caused apoptotic cell death (Fig. 1d), demonstrating that the metabolites triggered different death mechanisms depending on cell type and concentration.
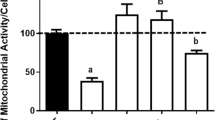
Decrease in mitochondrial membrane potential occurs during apoptosis (Gottlieb et al. 2003). In order to analyze mitochondrial mass and membrane potential, we used MitoTracker Green and MitoTracker Red (MTG and MTR) probes, respectively. The subsequent experiments were performed using the minimal QUIN concentration that combined with 30 μM Hcy was able to cause cell death in each cell type. Results showed that, in neurons, no significant differences were observed in mitochondrial mass and mitochondrial membrane potential (Fig. 1e). However, 100 μM QUIN plus 30 μM Hcy induce a decrease in mitochondrial membrane potential, as judged by decrease on MTR fluorescence and MTR/MTG ratio in astrocytes (Fig. 1f). This is in agreeing with apoptotic death determined by annexin-PI assay in this cell type.
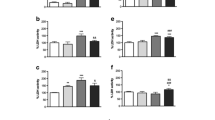
To better clarify the signaling pathways of cell death triggered by QUIN and Hcy in primary neurons and astrocytes, we evaluate some proteins involved in death mechanisms such as caspase-3 cleaved, cytochrome c, PARP cleaved (85 kDa fragment), and Bax immunocontent by western blot analysis. We found that, in neuron, the immunocontent of cytoplasmic Bax and cytochrome c, as well as caspase-3 cleaved was not altered in the QUIN + Hcy group compared with control (Fig. 2a–c, respectively). However, the immunocontent of mitochondrial Bax is increased (Fig. 2a) and the immunocontent of PARP cleaved is diminished (Fig. 2d). Also, treatment with QUIN alone increased cytoplasmic cytochrome c and caspase-3 cleaved immunocontent compared with control group.

Effects of QUIN and Hcy on Bax, cytochrome c, caspase-3, and PARP-1 immunocontent in cortical neurons (a–d) and astrocytes (e–h). Representative western blots of the proteins are shown. Actin was used as loading control. Data are reported as mean ± SD of four independent experiments performed in triplicate. Results were statistically analyzed by two-way ANOVA followed by the Tukey-Kramer test. Statistically significant differences from are indicated as follows: ***p < 0.001, **p < 0.01, and *p < 0.05 different from control and # p < 0.05 different from QUIN group
In astrocytes, the increase of mitochondrial Bax immunocontent (Fig. 2e) is accompanied by the increase of cytoplasmatic cytochrome c (Fig. 2f), caspase-3 cleaved (Fig. 2g) and PARP cleaved immunocontent (Fig. 2h).
Electron transport chain in mitochondria is considered as a major producer of reactive oxygen species (ROS) (Costa et al. 2016). Also, excess cellular levels of ROS can cause damage to proteins, nucleic acids and lipids, contributing to cell death (Halliwell 2011). Based on that, we investigated the involvement of oxidative stress on cell death triggered by QUIN and Hcy in cortical neurons and astrocytes. Figure 3a shows that 25 μM QUIN + Hcy caused increased DCF-DA oxidation in neurons. Otherwise, 100 μM QUIN plus Hcy caused decreased of DCF-DA oxidation in astrocytes (Fig. 3d). To evaluate the consequences of redox imbalance, we evaluated the sulfhydryl content and TBA-RS oxidation to analyze the role of oxidative stress in protein and lipid damage downstream of metabolites. Figure 3b, e shows that sulfhydryl content was significantly decreased by metabolites in astrocytes but not in neurons, indicating the higher vulnerability of astrocytes to protein damage. Moreover, the TBA-RS oxidation showed that, in neurons, 25 μM QUIN + Hcy was able to cause lipid peroxidation (Fig. 3c). Interestingly, in astrocytes, 100 μM QUIN + Hcy caused a decreased in lipid peroxidation (Fig. 3f).

Effect of QUIN and Hcy on DCF production (a, d), sulfhydryl content (b, e), and TBARS production (c, f) in cortical neurons and astrocytes of rats. Results were statistically analyzed by two-way ANOVA followed by the Tukey-Kramer test. Statistically significant differences from are indicated as follows: **p < 0.01 and *p < 0.05 different from control and ### p < 0.001 and # p < 0.05 different from QUIN group
Several studies suggest that oxidative stress can affects Na+,K+-ATPase activity, and inhibition of this enzyme can triggers cell death (Schweinberger et al. 2014). Based on that, we therefore investigate the effects of metabolites on Na+,K+-ATPase activity, and results showed a decrease in Na+,K+-ATPase activity in neurons after 24 h of exposure to metabolites (Fig. 4a), while the enzyme activity was unaltered in astrocytes (Fig. 4b), suggesting that misregulated ion gradient could underlie necrotic death mechanisms in neurons.

Effect of QUIN and Hcy on Na+,K+-ATPase activity in cortical neurons (a) and astrocytes (b). Results were statistically analyzed by two-way ANOVA followed by the Tukey-Kramer test. Data are reported as mean ± SD of four different experiments. Statistically significant differences from are indicated as follows: ***p < 0.001 different from control and # p < 0.05 different from QUIN group
Considering that redox status imbalance and mitochondrial dysfunction could disrupt cytoskeleton, and this can lead to cell dysfunction and death (Buja et al. 1993; Green and Kroemer 2004), we searched for a role for the cytoskeleton in the QUIN and Hcy toxicity. To evaluate the effect of metabolites on glial and neuronal cytoskeleton, we used immunocytochemical analysis of specific filaments present in astrocytes (GFAP) and neurons (MAP2). Disruption of the microtubular cytoskeleton was reflected by changes in the morphology of neurons, in particular by shortening and loss of neurites, since neurons treated with the metabolites presented a reduced neurite length (Fig. 5a, b) and neurite/neuron ratio (Fig. 5a, c) compared with controls. These results suggest the compromised framework of neurites in response to metabolites.

Effect of QUIN and Hcy on cytoskeletal organization in cortical neurons and astrocyte cultures. Representative images of cells immunostained with anti-MAP2 (a) for neurons and with phalloidin-fluorescein and anti-GFAP (d) for astrocytes. Morphometric analysis from control and treated cells with the ImageJ software: b neurite length, c neurites/neuron, and e cytoplasmic area of astrocytes. Data are reported as mean ± SD of four different experiments and results were statistically analyzed by one-way ANOVA followed by the Tukey-Kramer test, ***p < 0.001, **p < 0.01, and *p < 0.05 compared with control group and ### p < 0.001 compared with the QUIN group. Scale bar = 30 μm
Immunocytochemical results from astrocytes showed that cells treated with metabolites changed the morphology from polygonal to an elongated shape, presented reorganization of actin and GFAP network (Fig. 5d) and decreased cytoplasmic area (Fig. 5e), which is associated with altered cell morphology and cytoskeletal network.
Since cultured neurons at DIV8 express NMDA receptors (Kovacs et al. 2001; Sinor et al. 2000; Zhang and Wong-Riley 1999), in an attempt to investigate the mechanisms responsible for cell death caused by metabolites, we tested different glutamate antagonists in preventing the QUIN and Hcy effects.
Surprisingly, none of the glutamate antagonists was able to prevent cell death in cortical neurons, indicating that glutamate excitotoxicity was not involved in QUIN and Hcy neuronal death (Fig. 6a). On the other hand, in astrocytes, the NMDA antagonists MK-801 and DL-AP5 were able to prevent astrocytic death (Fig. 6b), indicating that the cell death triggered by metabolites on cortical astrocytes are dependent on NMDA receptors.

Protection against QUIN and Hcy-induced cell death in cortical neurons and astrocyte cultures. Cortical neurons (a, b) and astrocytes (c, d) were preincubated with glutamate antagonists (MK-801, DL-AP5, CNQX, MCPG, and MPEP) or antioxidants (melatonin (Mlt), Trolox (Trx), and GSH) for 30 min before treatment with metabolites for 24 h. One hour before completing treatment, 0.5 mg/mL of MTT was added to the incubation medium, and the formazan product was measured at 540 and 620 nm. Data are reported as mean ± SD of four independent experiments performed in triplicate. Results were statistically analyzed by two-way ANOVA followed by the Tukey-Kramer test: ***p < 0.001 and *p < 0.05 compared with the control group; ### p < 0.001 and ## p < 0.01 compared with the QUIN group
Since redox imbalance was involved in QUIN and Hcy actions, we treated cells with antioxidants (melatonin, Trolox, and GSH), and the results showed that melatonin and Trolox were able to prevent cell death in cortical neurons (Fig. 6c) while in astrocytes only GSH could prevent cell death triggered by QUIN and Hcy (Fig. 6d).
Next, in order to evaluate the role of astrocyte-neuron interaction on the effects caused by metabolites, after primary astrocytes had reached confluence, neurons were plated onto the astrocytic monolayers and the co-cultures proceeded for 7 days. After this, QUIN (25, 50, or 100 μM) and Hcy (30 μM) were added for 24 h and MTT assay was performed to test cell viability. Interestingly, when co-cultures were treated with the metabolites, none of the concentrations were able to decrease cell viability (Fig. 7a).

Role of astrocyte-neuron interaction on QUIN and Hcy effects. a Cortical co-cultures viability was measured by incubation with/without 100 μM QUIN and/or 30 μM Hcy for 24 h. Effect of the conditioned medium of astrocytes (MCA), neurons (MCN) and co-cultures (MCC) on neurons (b) and astrocytes (c) viability. d Effect of GJs blocker LaCl3 on co-culture viability. One hour before completing treatment with metabolites, 0.5 mg/mL of MTT was added to the incubation medium, and the formazan product generated was measured at 540 and 620 nm. Data are reported as mean ± SD of four independent experiments performed in triplicate. Results were statistically analyzed by two-way ANOVA followed by the Tukey-Kramer test: ***p < 0.001, **p < 0.01, and *p < 0.05 compared with the control group
Given that, we are attempted to investigate the mechanisms by which the two cells types are protecting each other against the insult caused by QUIN and Hcy. For this, we used the conditioned medium secreted by astrocytes (CMA) or co-culture (CMC) in the response of neurons to metabolites. The CMA or CMC treated with 25, 50, and 100 μM QUIN and 30 μM Hcy for 24 h was added to primary neurons for an additional 24 h and the results showed unaltered MTT assay in neurons treated with CMC, but not CMA (Fig. 7b), suggesting that the astrocyte-neuron interaction secrete protective factors that preserve neuron viability against QUIN + Hcy-induced cell death. On the other hand, the conditioned medium of astrocytes failed to prevent neurons against the insult.
Moreover, we tested the conditioned medium secreted by neurons (CMN) and CMC in the response of astrocyte to metabolites, and the results showed that neither CMN nor CMC was able to prevent cell death caused by metabolites on astrocytes (Fig. 7c). This suggests that the protection observed in astrocytes is not due to soluble factors secreted by these cells, but maybe due to cell-cell interactions.
In line with this, we investigate the role of cell-cell interaction in the protection of co-culture against QUIN and Hcy-insult. For this, we used lanthanum chloride (LaCl3), a gap junction (GJ) blocker (Eskandari et al. 2002), to test the participation of these channels on the co-culture protection. Results showed that co-cultures exposed to metabolites and pretreated with LaCl3 presented a decrease in cell viability in a concentration of 100 μM QUIN + 30 μM Hcy (Fig. 7d). This suggests that GJs is involved in the mechanisms of protection triggered by co-culture against the metabolites. Moreover, to better clarify by which mechanisms the GJs are involved in these protection, we pretreated the cells with the same antagonists and antioxidants. The results showed that just the NMDA antagonists MK-801 and DL-AP5 were able to prevent cell death in the co-culture treated with La3Cl2 (Fig. 8a), indicating that the protection triggered by GJs on co-culture involves glutamate mechanisms dependent on NMDA receptors. We also observed that none of the antioxidants were able to prevent cell death caused by La3Cl2 (Fig. 8b), suggesting that the redox homeostasis is not involved in the protective mechanisms triggered by cell-cell interactions.

Mechanisms by which GJs protect the co-cultures against QUIN and Hcy toxicity. a Effect of the glutamate antagonists and antioxidants (b) on co-cultures treated with La3Cl2. One hour before completing treatment with metabolites, 0.5 mg/mL of MTT was added to the incubation medium, and the formazan product generated was measured at 540 and 620 nm. Data are reported as mean ± SD of four independent experiments performed in triplicate. Results were statistically analyzed by two-way ANOVA followed by the Tukey-Kramer test: ***p < 0.001, **p < 0.01, and *p < 0.05 compared with the control group and ## p < 0.01 and # p < 0.05 compared with the QUIN + Hcy + La3Cl2 group
Finally, immunocytochemical studies with the astrocyte and neurons markers showed that, in treated co-cultures, GFAP presented a typical IF array, and this distribution was not altered compared with astrocytes from controls (Fig. 9a). The morphometric analysis showed that the neurite/neuron ratio (Fig. 9b), the neurite length (Fig. 9c), and the cytoplasmic area of astrocytes (Fig. 9d) were not altered in none of the treated groups. Moreover, when co-cultures were treated with La3Cl2, the morphology of the cells was altered, showing a diminished neurite/neuron ratio (Fig. 9b), neurite per neuron (Fig. 9c), and cytoplasmic area of astrocytes (Fig. 9d). These findings corroborate the protective role of GJs against QUIN and Hcy toxicity.

Protection against QUIN and Hcy on morphologic alterations involves GJs participation. Co-cultures were preincubated with 50 μM La3Cl2 for 30 min before QUIN and/or Hcy for 24 h as described in the “Material and Methods” section. a Immunostaining for β III-tubulin (green), GFAP (red), and DAPI (blue) in co-cultures. Morphometric analysis showing neurite length (b), neurite/neuron ratio (c), and cytoplasmic area of astrocytes (d). Data are reported as mean ± SD of four different experiments and results were statistically analyzed by one-way ANOVA followed by the Tukey-Kramer test, ***p < 0.001, **p < 0.01 compared with the control group and ### p < 0.001, ## p < 0.01, and # p < 0.05 compared with the QUIN + Hcy + La3Cl2 group. Scale bar = 30 μm
Discussion
AD pathology is characterized by the formation of extracellular amyloid plaques and intraneuronal neurofibrillary tangles (Blennow et al. 2006) that, although they are the main hallmarks of the disease, there are diverse pathways that provoke the processes of cell death. Despite the significance advancement in our understanding of the mechanisms that contribute to AD progression, the effectiveness of treatment for the disease is still missing. One of the factors responsible for this is that the loss of neurons occurs during many years, which can make the mode of cell death and the initiating factors difficult to determine. In this regard, in vitro and in vivo models have proved invaluable, yielding insight into cell death pathways involved in the disease.
In the present study, we reported that QUIN and Hcy, metabolites that are increased in AD, led to alterations of proteins involved with death signaling pathways in primary cortical cultures. We demonstrated a link among oxidative stress, glutamate dyshomeostasis, and cell death, as well as different susceptibilities and death mechanisms of neurons and astrocytes to metabolites. Also, we found mechanisms of mutual protection of these two cell types to insult.
It is interesting to note that exposure of primary cortical neurons to these compounds independently induced neurotoxicity. However, when treated combined, these two metabolites had synergistic toxicity and different death mechanisms. QUIN and Hcy toxicity seemed mediated by apoptotic pathways, while both metabolites induce cell death by necrotic mechanisms. In this regard, certain neurotoxins can induce both apoptosis and necrosis depending on the conditions, particularly with regard to redox levels and mitochondrial integrity (Bonfoco et al. 1995; Gorman et al. 2005). In addition, the inhibition of specific proteins involved in regulating apoptosis or autophagy can change the type of cell death to necrosis (Fasciani et al. 2013).
We found that QUIN and Hcy diminished PARP-1 cleaved immunocontent, without alter cytochrome c and active caspase-3. Increased in ROS production and lipid peroxidation and a decreased in Na+,K+-ATPase activity were also observed. It has been shown that overactivation of full-length PARP-1 (consequently diminished PARP-1 cleavage) can result in energy depletion and necrosis (D’Amours et al. 2001). Overproduction of ROS also promotes PARP-1 hyperactivation (Virag and Szabo 1999) and is associated with generation of large DNA fragments, characteristic of necrotic cell death (Fujikawa 2015). Moreover, lipid peroxidation could affects membrane permeability and Na+,K+-ATPase activity. In line with this, the activity of Na+,K+-ATPase is significantly lower in the brains of patients with AD. This selective reduction of this enzyme seems to be associated with loss of calcium homeostasis and cell degeneration (Liguri et al. 1990), and evidences suggest that Na+,K+-ATPase may be a potent neuroprotective modulator against AD (Zhang et al. 2013).
Microtubule-associated proteins (MAPs) are responsible for stabilization of the cytoskeleton and protect cells from toxic effects. In the present report, we have demonstrated that QUIN and Hcy disrupt the microtubular cytoskeleton of primary neurons leading to neuritis loss. Also, changes in microtubular cytoskeleton could lead to alterations in organelles transport (Mudher and Lovestone 2002), contributing to necrotic cell death. Since it is well-known that microtubule disruption is a marker of neuronal death in AD, disruption of the cytoskeletal homeostasis of microtubules in QUIN and Hcy-treated neurons could represent a dendritic injury, contributing to necrosis in the disease.
Although it is well-known that toxicity caused by QUIN and Hcy is mediated primarily by activation of glutamate receptors, our present results show that neuronal death in cortical neurons is not directly dependent of NMDA receptors. However, QUIN and Hcy toxicity in neurons is attenuated by the free radical scavenger melatonin and partially by Trolox, but not by GSH, suggesting that the mechanism of toxicity seems to be at the level of high ROS production and not by depletion of cellular GSH. This result indicates a prominent role of oxidative stress in the neurotoxicity of the combined metabolites treatment independently of the NMDA-activation.
In astrocytes, QUIN and Hcy decreased cell viability only when associated, demonstrating a potentiating effect of the two metabolites in this cell type. In contrast with neurons, the metabolites caused decreased ROS production, depletion of sulfhydryl groups, reduction of lipid peroxidation and mitochondrial membrane potential. Also, Bax and cytochrome c was released, caspase-3 was cleaved, and degradation of PARP-1 occurred. These events lead to apoptosis of the cultured astrocytes. Additionally, cytoskeletal rearrangement has been observed.
Mitochondrial dysfunction has been demonstrated to play a critical role in apoptosis (Green and Kroemer 2004), and depolarization of the mitochondrial membrane as a result of opening of the permeability transition pores is an early and irreversible event during apoptosis. The mitochondrial apoptotic pathway is initiated by release of apoptogenic factors, for example, Bax protein, causing the release of cytochrome c, which activates caspase-3. In the cytosol, caspase-3 cleaves PARP-1, releasing the 89 kDa domain and initiates the apoptotic process (Saelens et al. 2004). It is well-known the important role of caspases for the activation of cell death in neurodegenerative diseases, as well as mood disorders (Cui et al. 2012; Dygalo et al. 2012). In this context, improper function of astrocytes may contribute to the onset of neurodegenerative disease, such as AD (Kahlson and Colodner 2015).
Surprisingly, we found a decrease in ROS production in astrocytes treated with the metabolites. Historically, mitochondrial ROS were thought to exclusively cause cellular damage and lack a physiological function. However, they are now appreciated to function as signaling molecules to regulate a wide variety of physiology, like cellular signaling, synaptic plasticity, and cell differentiation (Sena and Chandel 2012), and tight regulation of ROS is critical to their ability to participate in physiological or pathological cell signaling. Therefore, both increases and decreases of ROS production could be harmful to cell proper functions. In line with this, we found a decreased of sulfhydryl and TBARS content, which could be explained, at least in part, by decreasing of ROS production observed in astrocytes treated with QUIN and Hcy. Also, studies indicate that even a slight depolarization of the inner mitochondrial membrane could abolish mitochondrial ROS generation (Kalogeris et al. 2014).
Interestingly, although our results showed a decrease in ROS production, we found depletion in the sulfhydryl content in astrocytes treated with metabolites. However, the method of detection used in this study estimate the total SH groups, including GSH. So, we cannot exclude that the diminished sulfhydryl content could reflect a result of GSH depletion. Several studies have shown a correlation between GSH depletion and the progression of apoptosis, and early and transient rise in intracellular GSSG has been shown to precede cytochrome c release and caspase c activation during apoptosis (Franco and Cidlowski 2009). Moreover, because glia cells synthesize GSH, we could not exclude the possibility that the decrease in GSH concentration was reflective of reduced synthesis, for example as cells were in the process of dying. Importantly, GSH metabolism is reported to be altered in AD patient’s brain. In line with this, we found that treatment with GSH was able to block astrocytic death caused by the metabolites, and replenishment of intracellular GSH pools has been shown to protect cells against apoptosis (Franco and Cidlowski 2009). However, the precise role of GSH in the complex process of cell apoptosis remains unclear.
The cytoskeleton of actin and GFAP were reorganized in response to QUIN and Hcy, leading to altered cell morphology. Alterations on actin and GFAP dynamics would enhance sensitivity to several apoptotic stimuli (Caulin et al. 1997). Also, interactions between actin and mitochondria have been proposed as a general apoptotic phenomenon, occurring before Bax translocation. Accordingly, actin may contribute to the initiation of apoptosis by enabling cytosolic proteins to be carried to mitochondria by the cytoskeleton-driven trafficking system (Tang et al. 2006). Moreover, GFAP is also a putative caspase substrate and can be cleaved by enzyme to generate proapototic fragments that cause filament aggregation and morphologic alterations (Chen et al. 2013). In this regard, caspase-3 activated cleaves GFAP and contributes to the damaged astrocytes in AD (Mouser et al. 2006).
The combination of QUIN and Hcy toxicity in primary astrocytes is mediated primarily by NMDA receptor activation, since the NMDA receptor antagonist MK-801 and DL-AP5 were able to prevent their effect. Also, the mechanism of the synergistic toxicity seems to be at the level of depletion of cellular GSH, since it prevents astrocytic death caused by the compounds. In astrocytes, QUIN and Hcy triggered apoptotic pathways that activates the cysteine protease family of caspases and contributes to apoptotic death, and it is well-known that these mechanisms occur in several neurodegenerative conditions such as AD (Gorman 2008).
In the present data, cell death was prevented when astrocytes and neurons were co-cultured, suggesting the activation of protector mechanisms dependent on astrocyte and neurons communication. Corroborating this has been shown that astrocytes influence the neuronal development, as well as neurons are able to facilitate glial communication and interaction, which play a role in neuroprotection (Rouach et al. 2002). In order to clarify this point, we used conditioned medium of astrocyte, neuron, and co-culture in an attempt to identify if the neuroprotection found in co-culture was caused by soluble factors secreted by these cells. We found that, in neuron, but not in astrocytes, the conditioned medium of co-culture was able to prevent the neuronal death. Thus, it is feasible that, in our experimental conditions, cortical astrocytes secreted factors that promoted neuronal survival. Importantly, since the conditioned medium of astrocytes was not able to prevent this effect, the astrocyte/neuron interaction was important to trigger such kind of response in astrocytes.
Several studies have contributed to demonstrate that neuron and astrocytes actively interact through astrocytic GJs. In this regard, a number of studies showed that compounds that can block GJ could be deleterious after brain injury (Eskandari et al. 2002; Fasciani et al. 2013). Indeed, we observed that, when GJs were blocked by LaCl2, the protection was lost and cell death and morphologic alteration occurred. Moreover, we found that co-cultures treated with MK-801 and DL-AP5 were protected from cell death caused by GJ blockade. This agrees with the fact that neurons in contact with astrocytes are protected against glutamate toxicity (Mattson and Rychlik 1990), and this protection is attributed to glutamate uptake by astrocytes (Rosenberg 1991). GJs are permeable to glutamate, so they can improve glutamate uptake contributing to its dissipation and thus to neuronal protection (Goldberg et al. 1999). In line with this, it has been shown that astrocyte dysfunction may contribute to the earliest neuronal deficit in AD. The astrocytic calcium signaling and gliotransmitter release, processes that are vital for astrocyte-neuron communication, are disrupted in the disease (Vincent et al. 2010).
It is important to note that, in the present data, the morphological alterations in neurons and astrocytes were prevented by astrocyte-neuron co-culture. It is well-known that astrocytes secreted factors that promoted neurite formation and extension (Delgado-Rivera et al. 2009; Hatten et al. 1988; Sorensen et al. 2008). According with this, it is possible that these secreted factors provided a cellular microenvironment in which they protect neurons against QUIN and Hcy insult, preserving the cytoskeletal homeostasis. Moreover, astrocytic GJ represent a target for the interactions occurring between neurons and astrocytes, and it is known that disrupted interaction of GJ proteins with the cytoskeleton decreases cell adhesion and destabilizes the tridimensional cellular network (Toyofuku et al. 1998).
Thus, it is feasible that, under our experimental conditions, striatal astrocytes secreted factors that promoted neurite formation and extension and provided a cellular microenvironment in which astrocyte/neuron interactions reciprocally protected themselves against QUIN insult so that the homeostasis of the cytoskeleton and morphological features were preserved.
In summary, our present results show that the combination of QUIN plus Hcy causes oxidative damage, Na+,K+-ATPase inhibition, and morphologic alterations resulting in necrotic cell death in neurons, while in astrocytes they disrupt redox homeostasis, mitochondrial function, and morphological alterations, triggering apoptotic cell death. In addition, we found a cross talk between neurons and astrocytes mediating a central mechanism of cell homeostasis in QUIN and Hcy-treated co-cultures. Our findings highlight the role of secreted factors and reciprocal interactions between astrocytes and neurons in protecting themselves against an insult. The identification of new toxic mechanisms triggered the deleterious actions produced by neurotoxins involved in AD and the understanding of the mechanisms that trigger cell death can provide new possibilities to design more effective therapeutic approaches. Additional steps are certainly necessary to improve our understanding of the astrocyte-neuron communication on AD pathophysiology.
References
Aksenov MY, Markesbery WR (2001) Changes in thiol content and expression of glutathione redox system genes in the hippocampus and cerebellum in Alzheimer’s disease. Neurosci Lett 302(2–3):141–145
Baranes K, Kollmar D, Chejanovsky N, Sharoni A, Shefi O (2012) Interactions of neurons with topographic nano cues affect branching morphology mimicking neuron-neuron interactions. J Mol Histol 43(4):437–447. https://doi.org/10.1007/s10735-012-9422-2
Bell IR, Edman JS, Selhub J et al (1992) Plasma homocysteine in vascular disease and in nonvascular dementia of depressed elderly people. Acta Psychiatr Scand 86(5):386–390
Blennow K, de Leon MJ, Zetterberg H (2006) Alzheimer’s disease. Lancet 368(9533):387–403. https://doi.org/10.1016/S0140-6736(06)69113-7
Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA (1995) Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-D-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci U S A 92(16):7162–7166
Buja LM, Eigenbrodt ML, Eigenbrodt EH (1993) Apoptosis and necrosis. Basic types and mechanisms of cell death. Arch Pathol Lab Med 117(12):1208–1214
Caulin C, Salvesen GS, Oshima RG (1997) Caspase cleavage of keratin 18 and reorganization of intermediate filaments during epithelial cell apoptosis. J Cell Biol 138(6):1379–1394
Chan KM, Delfert D, Junger KD (1986) A direct colorimetric assay for Ca2+ -stimulated ATPase activity. Anal Biochem 157(2):375–380
Chen MH, Hagemann TL, Quinlan RA, Messing A, Perng MD (2013) Caspase cleavage of GFAP produces an assembly-compromised proteolytic fragment that promotes filament aggregation. ASN Neuro 5(5):e00125. https://doi.org/10.1042/AN20130032
Clarke R, Smith AD, Jobst KA, Refsum H, Sutton L, Ueland PM (1998) Folate, vitamin B12, and serum total homocysteine levels in confirmed Alzheimer disease. Arch Neurol 55(11):1449–1455
Cobb CA, Cole MP (2015) Oxidative and nitrative stress in neurodegeneration. Neurobiol Dis 84:4–21. https://doi.org/10.1016/j.nbd.2015.04.020
Costa LG, Tagliaferri S, Roque PJ, Pellacani C (2016) Role of glutamate receptors in tetrabrominated diphenyl ether (BDE-47) neurotoxicity in mouse cerebellar granule neurons. Toxicol Lett 241:159–166. https://doi.org/10.1016/j.toxlet.2015.11.026
Cui JH, Qiao Q, Guo Y et al (2012) Increased apoptosis and expression of FasL, Bax and caspase-3 in human lupus nephritis class II and IV. J Nephrol 25(2):255–261. https://doi.org/10.5301/JN.2011.8451
D’Amours D, Sallmann FR, Dixit VM, Poirier GG (2001) Gain-of-function of poly(ADP-ribose) polymerase-1 upon cleavage by apoptotic proteases: implications for apoptosis. J Cell Sci 114(Pt 20):3771–3778
de Lima Pelaez P, Funchal C, Loureiro SO et al (2007) Branched-chain amino acids accumulating in maple syrup urine disease induce morphological alterations in C6 glioma cells probably through reactive species. Int J Dev Neurosci 25(3):181–189. https://doi.org/10.1016/j.ijdevneu.2007.01.001
Delgado-Rivera R, Harris SL, Ahmed I et al (2009) Increased FGF-2 secretion and ability to support neurite outgrowth by astrocytes cultured on polyamide nanofibrillar matrices. Matrix Biol 28(3):137–147. https://doi.org/10.1016/j.matbio.2009.02.001
den Heijer T, Vermeer SE, Clarke R et al (2003) Homocysteine and brain atrophy on MRI of non-demented elderly. Brain 126(Pt 1):170–175
Dygalo NN, Kalinina TS, Bulygina VV, Shishkina GT (2012) Increased expression of the anti-apoptotic protein Bcl-xL in the brain is associated with resilience to stress-induced depression-like behavior. Cell Mol Neurobiol 32(5):767–776. https://doi.org/10.1007/s10571-011-9794-y
Eskandari S, Zampighi GA, Leung DW, Wright EM, Loo DD (2002) Inhibition of gap junction hemichannels by chloride channel blockers. J Membr Biol 185(2):93–102. https://doi.org/10.1007/s00232-001-0115-0
Fasciani I, Temperan A, Perez-Atencio LF et al (2013) Regulation of connexin hemichannel activity by membrane potential and the extracellular calcium in health and disease. Neuropharmacology 75:479–490. https://doi.org/10.1016/j.neuropharm.2013.03.040
Franco R, Cidlowski JA (2009) Apoptosis and glutathione: beyond an antioxidant. Cell Death Differ 16(10):1303–1314. https://doi.org/10.1038/cdd.2009.107
Fujikawa DG (2015) The role of excitotoxic programmed necrosis in acute brain injury. Comput Struct Biotechnol J 13:212–221. https://doi.org/10.1016/j.csbj.2015.03.004
Goedert M, Spillantini MG (2006a) A century of Alzheimer’s disease. Science 314(5800):777–781. https://doi.org/10.1126/science.1132814
Goedert M, Spillantini MG (2006b) Frontotemporal lobar degeneration through loss of progranulin function. Brain 129(Pt 11):2808–2810. https://doi.org/10.1093/brain/awl291
Goldberg GS, Lampe PD, Nicholson BJ (1999) Selective transfer of endogenous metabolites through gap junctions composed of different connexins. Nat Cell Biol 1(7):457–459. https://doi.org/10.1038/15693
Gorman AM (2008) Neuronal cell death in neurodegenerative diseases: recurring themes around protein handling. J Cell Mol Med 12(6A):2263–2280. https://doi.org/10.1111/j.1582-4934.2008.00402.x
Gorman AM, Szegezdi E, Quigney DJ, Samali A (2005) Hsp27 inhibits 6-hydroxydopamine-induced cytochrome c release and apoptosis in PC12 cells. Biochem Biophys Res Commun 327(3):801–810. https://doi.org/10.1016/j.bbrc.2004.12.066
Gottfries CG, Lehmann W, Regland B (1998) Early diagnosis of cognitive impairment in the elderly with the focus on Alzheimer’s disease. J Neural Transm (Vienna) 105(8–9):773–786. https://doi.org/10.1007/s007020050094
Gottlieb E, Armour SM, Harris MH, Thompson CB (2003) Mitochondrial membrane potential regulates matrix configuration and cytochrome c release during apoptosis. Cell Death Differ 10(6):709–717. https://doi.org/10.1038/sj.cdd.4401231
Green DR, Kroemer G (2004) The pathophysiology of mitochondrial cell death. Science 305(5684):626–629. https://doi.org/10.1126/science.1099320
Guillemin GJ, Brew BJ (2002) Implications of the kynurenine pathway and quinolinic acid in Alzheimer’s disease. Redox Rep 7(4):199–206. https://doi.org/10.1179/135100002125000550
Guillemin GJ, Smythe GA, Veas LA, Takikawa O, Brew BJ (2003) A beta 1-42 induces production of quinolinic acid by human macrophages and microglia. Neuroreport 14(18):2311–2315. https://doi.org/10.1097/01.wnr.0000097042.56589.ff
Halliwell B (2011) Free radicals and antioxidants—quo vadis? Trends Pharmacol Sci 32(3):125–130. https://doi.org/10.1016/j.tips.2010.12.002
Hatten ME, Lynch M, Rydel RE et al (1988) In vitro neurite extension by granule neurons is dependent upon astroglial-derived fibroblast growth factor. Dev Biol 125(2):280–289
Jiang J, Yan M, Lv Q et al (2010) Inhibition of nitric oxide-induced nuclear localization of CAPON by NMDA receptor antagonist in cultured rat primary astrocytes. Neurochem Int 56(4):561–568. https://doi.org/10.1016/j.neuint.2009.12.019
Kahlson MA, Colodner KJ (2015) Glial tau pathology in tauopathies: functional consequences. J Exp Neurosci 9(Suppl 2):43–50. https://doi.org/10.4137/JEN.S25515
Kalogeris T, Bao Y, Korthuis RJ (2014) Mitochondrial reactive oxygen species: a double edged sword in ischemia/reperfusion vs preconditioning. Redox Biol 2:702–714. https://doi.org/10.1016/j.redox.2014.05.006
Keij JF, Bell-Prince C, Steinkamp JA (2000) Staining of mitochondrial membranes with 10-nonyl acridine orange, MitoFluor Green, and MitoTracker Green is affected by mitochondrial membrane potential altering drugs. Cytometry 39(3):203–210
Khanal G, Chung K, Solis-Wever X, Johnson B, Pappas D (2011) Ischemia/reperfusion injury of primary porcine cardiomyocytes in a low-shear microfluidic culture and analysis device. Analyst 136(17):3519–3526. https://doi.org/10.1039/c0an00845a
Kincses ZT, Toldi J, Vecsei L (2010) Kynurenines, neurodegeneration and Alzheimer’s disease. J Cell Mol Med 14(8):2045–2054. https://doi.org/10.1111/j.1582-4934.2010.01123.x
Kovacs AD, Cebers G, Cebere A, Moreira T, Liljequist S (2001) Cortical and striatal neuronal cultures of the same embryonic origin show intrinsic differences in glutamate receptor expression and vulnerability to excitotoxicity. Exp Neurol 168(1):47–62. https://doi.org/10.1006/exnr.2000.7576
LeBel CP, Ali SF, McKee M, Bondy SC (1990) Organometal-induced increases in oxygen reactive species: the potential of 2′,7′-dichlorofluorescin diacetate as an index of neurotoxic damage. Toxicol Appl Pharmacol 104(1):17–24
Liguri G, Taddei N, Nassi P, Latorraca S, Nediani C, Sorbi S (1990) Changes in Na+,K(+)-ATPase, Ca2(+)-ATPase and some soluble enzymes related to energy metabolism in brains of patients with Alzheimer’s disease. Neurosci Lett 112(2–3):338–342
Loureiro SO, Heimfarth L, Scherer EB et al (2013) Cytoskeleton of cortical astrocytes as a target to proline through oxidative stress mechanisms. Exp Cell Res 319(3):89–104. https://doi.org/10.1016/j.yexcr.2012.11.002
Loureiro SO, Sidegum DS, Biasibetti H et al (2015) Crosstalk among disrupted glutamatergic and cholinergic homeostasis and inflammatory response in mechanisms elicited by proline in astrocytes. Mol Neurobiol. https://doi.org/10.1007/s12035-014-9067-0
Loureiro SO, Sidegum DS, Biasibetti H et al (2016) Crosstalk among disrupted glutamatergic and cholinergic homeostasis and inflammatory response in mechanisms elicited by proline in astrocytes. Mol Neurobiol 53(2):1065–1079. https://doi.org/10.1007/s12035-014-9067-0
Lugo-Huitron R, Ugalde Muniz P, Pineda B, Pedraza-Chaverri J, Rios C, Perez-de la Cruz V (2013) Quinolinic acid: an endogenous neurotoxin with multiple targets. Oxidative Med Cell Longev 2013:104024. https://doi.org/10.1155/2013/104024
Mattson MP, Rychlik B (1990) Glia protect hippocampal neurons against excitatory amino acid-induced degeneration: involvement of fibroblast growth factor. Int J Dev Neurosci 8(4):399–415
Moore AR, Zhou WL, Sirois CL, Belinsky GS, Zecevic N, Antic SD (2014) Connexin hemichannels contribute to spontaneous electrical activity in the human fetal cortex. Proc Natl Acad Sci U S A 111(37):E3919–E3928. https://doi.org/10.1073/pnas.1405253111
Mouser PE, Head E, Ha KH, Rohn TT (2006) Caspase-mediated cleavage of glial fibrillary acidic protein within degenerating astrocytes of the Alzheimer’s disease brain. Am J Pathol 168(3):936–946. https://doi.org/10.2353/ajpath.2006.050798
Mudher A, Lovestone S (2002) Alzheimer’s disease-do tauists and baptists finally shake hands? Trends Neurosci 25(1):22–26
Pendergrass W, Wolf N, Poot M (2004) Efficacy of MitoTracker Green (TM) and CMX Rosamine to measure changes in mitochondrial membrane potentials in living cells and tissues. Cytometry A 61A:162–169
Pierozan P, Ferreira F, Ortiz de Lima B et al (2014) The phosphorylation status and cytoskeletal remodeling of striatal astrocytes treated with quinolinic acid. Exp Cell Res 322(2):313–323. https://doi.org/10.1016/j.yexcr.2014.02.024
Pierozan P, Ferreira F, de Lima BO, Pessoa-Pureur R (2015) Quinolinic acid induces disrupts cytoskeletal homeostasis in striatal neurons. Protective role of astrocyte-neuron interaction. J Neurosci Res 93(2):268–284. https://doi.org/10.1002/jnr.23494
Prentice H, Modi JP, Wu JY (2015) Mechanisms of neuronal protection against excitotoxicity, endoplasmic reticulum stress, and mitochondrial dysfunction in stroke and neurodegenerative diseases. Oxidative Med Cell Longev 2015:964518. https://doi.org/10.1155/2015/964518
Rana S, Dringen R (2007) Gap junction hemichannel-mediated release of glutathione from cultured rat astrocytes. Neurosci Lett 415(1):45-48
Rosenberg PA (1991) Accumulation of extracellular glutamate and neuronal death in astrocyte-poor cortical cultures exposed to glutamine. Glia 4(1):91–100. https://doi.org/10.1002/glia.440040111
Rouach N, Avignone E, Meme W et al (2002) Gap junctions and connexin expression in the normal and pathological central nervous system. Biol Cell 94(7–8):457–475
Saelens X, Festjens N, Vande Walle L, van Gurp M, van Loo G, Vandenabeele P (2004) Toxic proteins released from mitochondria in cell death. Oncogene 23(16):2861–2874. https://doi.org/10.1038/sj.onc.1207523
Santos-Carvalho A, Aveleira CA, Elvas F, Ambrosio AF, Cavadas C (2013) Neuropeptide Y receptors Y1 and Y2 are present in neurons and glial cells in rat retinal cells in culture. Invest Ophthalmol Vis Sci 54(1):429–443. https://doi.org/10.1167/iovs.12-10776
Schweinberger BM, Schwieder L, Scherer E, Sitta A, Vargas CR, Wyse AT (2014) Development of an animal model for gestational hypermethioninemia in rat and its effect on brain Na(+),K(+)-ATPase/Mg(2)(+)-ATPase activity and oxidative status of the offspring. Metab Brain Dis 29(1):153–160. https://doi.org/10.1007/s11011-013-9451-x
Sena LA, Chandel NS (2012) Physiological roles of mitochondrial reactive oxygen species. Mol Cell 48(2):158–167. https://doi.org/10.1016/j.molcel.2012.09.025
Seshadri S (2012) Homocysteine and the risk of dementia. Clin Chem 58(6):1059–1060. https://doi.org/10.1373/clinchem.2011.181099
Sharma M, Tiwari M, Tiwari RK (2015) Hyperhomocysteinemia: impact on neurodegenerative diseases. Basic Clin Pharmacol Toxicol 117(5):287–296. https://doi.org/10.1111/bcpt.12424
Sinor JD, Du S, Venneti S et al (2000) NMDA and glutamate evoke excitotoxicity at distinct cellular locations in rat cortical neurons in vitro. J Neurosci 20(23):8831–8837
Sorensen A, Moffat K, Thomson C, Barnett SC (2008) Astrocytes, but not olfactory ensheathing cells or Schwann cells, promote myelination of CNS axons in vitro. Glia 56(7):750–763. https://doi.org/10.1002/glia.20650
Staehelin HB (2005) Micronutrients and Alzheimer’s disease. Proc Nutr Soc 64(4):565–570
Tan L, Yu J, Tan L (2012) The kynurenine pathway in neurodegenerative diseases: mechanistic and therapeutic considerations. J Neurol Sci 323:1–8
Tang HL, Le AH, Lung HL (2006) The increase in mitochondrial association with actin precedes Bax translocation in apoptosis. Biochem J 396(1):1–5. https://doi.org/10.1042/BJ20060241
Toyofuku T, Yabuki M, Otsu K, Kuzuya T, Hori M, Tada M (1998) Direct association of the gap junction protein connexin-43 with ZO-1 in cardiac myocytes. J Biol Chem 273(21):12725–12731
Vincent AJ, Gasperini R, Foa L, Small DH (2010) Astrocytes in Alzheimer’s disease: emerging roles in calcium dysregulation and synaptic plasticity. J Alzheimers Dis 22(3):699–714. https://doi.org/10.3233/JAD-2010-101089
Virag L, Szabo C (1999) Inhibition of poly(ADP-ribose) synthetase (PARS) and protection against peroxynitrite-induced cytotoxicity by zinc chelation. Br J Pharmacol 126(3):769–777. https://doi.org/10.1038/sj.bjp.0702332
Williams JH, Pereira EA, Budge MM, Bradley KM (2002) Minimal hippocampal width relates to plasma homocysteine in community-dwelling older people. Age Ageing 31(6):440–444
Wyse AT, Noriler ME, Borges LF et al (1999) Alanine prevents the decrease of Na+,K+-ATPase activity in experimental phenylketonuria. Metab Brain Dis 14(2):95–101
Yagi K (1998) Simple assay for the level of total lipid peroxides in serum or plasma. Methods Mol Biol 108:101-6 https://doi.org/10.1385/0-89603-472-0:101
Zhang C, Wong-Riley M (1999) Expression and regulation of NMDA receptor subunit R1 and neuronal nitric oxide synthase in cortical neuronal cultures: correlation with cytochrome oxidase. J Neurocytol 28(7):525–539
Zhang LN, Sun YJ, Pan S et al (2013) Na(+)-K(+)-ATPase, a potent neuroprotective modulator against Alzheimer disease. Fundam Clin Pharmacol 27(1):96–103. https://doi.org/10.1111/fcp.12000
Acknowledgements
This work was supported by grants of the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and INCT (EN 465671/2014-4)/CNPq, Brazil.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Pierozan, P., Biasibetti-Brendler, H., Schmitz, F. et al. Synergistic Toxicity of the Neurometabolites Quinolinic Acid and Homocysteine in Cortical Neurons and Astrocytes: Implications in Alzheimer’s Disease. Neurotox Res 34, 147–163 (2018). https://doi.org/10.1007/s12640-017-9834-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-017-9834-6