
Abstract
Parkinson’s disease (PD) is the second most common neurodegenerative disorder affecting humans. It is characterized by dopaminergic neurodegeneration, mitochondrial impairment, and oxidative stress, enhanced lipid peroxidation, and induction of pro-inflammatory cytokines. We evaluated the neuroprotective efficacy of glycyrrhizic acid (GA), an active component of licorice, against rotenone-induced-oxidative stress and neuroinflammation in a PD rat model. Since PD is progressive and chronic, we investigated the effect of chronic administration of GA for 4 weeks (50 mg/kg/day), 30 min prior to rotenone administration. Rotenone administration significantly reduced the activity of superoxide dismutase and catalase, and caused the depletion of reduced glutathione. A concomitant increase in the levels of the lipid peroxidation product malondialdehyde was observed. It also significantly enhanced the levels of pro-inflammatory cytokines in the midbrain and elevated the levels of inflammatory mediators such as cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS). Immunohistochemical analysis revealed significant increments in ionized calcium-binding adaptor molecule-1 (Iba-1) levels, and in glial fibrillary acidic protein (GFAP) levels, and loss of dopamine neurons in the substantia nigra pars compacta upon rotenone challenge. GA treatment significantly attenuated the dopamine neuron loss and decreased the Iba-1 and GFAP activation induced by the rotenone insult. GA also improved antioxidant enzyme activity, prevented glutathione depletion, inhibited lipid peroxidation, and attenuated induction of pro-inflammatory cytokines. Subsequently, GA attenuated the increased levels of the inflammatory mediators COX-2 and iNOS. In conclusion, GA protects against rotenone-induced-PD. The neuroprotective effects of GA are attributed to its potent antioxidative and anti-inflammatory properties.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is a progressive neurodegenerative disease characterized by the loss of dopaminergic neurons within the substantia nigra pars compacta (SNc). This in turn contributes to the depletion of dopamine, which results in motor behavioral deficits such as slowness of movements, rigidity, tremors, and postural imbalance (Dexter and Jenner 2013; Schapira et al. 2014). The severity of these motor symptoms is associated with the loss of tyrosine hydroxylase-positive dopaminergic neurons in the SNc area (Anderson and Maes 2014). A number of convincing studies have demonstrated that oxidative stress, mitochondrial dysfunction, proteasomal inhibition, and inflammation are the critical players in the pathogenesis of PD that accelerate dopaminergic neurodegeneration (Anderson and Maes 2014; Celardo et al. 2014; Niranjan 2014).
Oxidative stress and inflammatory processes are intimately connected and are linked to the death of dopaminergic neurons in the brain, which is more susceptible to oxidative stress than other organs (Taylor et al. 2013; Schapira et al. 2014; Anderson and Maes 2014). Although many pharmacotherapeutic interventions are available to provide symptomatic treatment, there is still a paucity of disease-modifying or preventive agents for PD (Al Dakheel et al. 2014; Schapira et al. 2014). Therefore, halting the disease progression appears perplexing and current approaches are far from satisfactory. Thus, there is a need for novel agents that have the potential to target these intimately linked cascades of oxidative stress–inflammatory-cytokine signaling and delay the development and progression of PD (Schapira et al. 2014). In recent years, besides the other pharmacotherapeutic approaches, treatment with antioxidants has gradually gained importance as a disease-preventive strategy in the treatment of PD (Albarracin et al. 2012; Song et al. 2012; Al Dakheel et al. 2014).
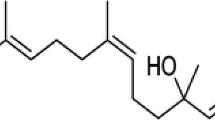
Since oxidative stress and inflammation appear to be major players in brain-aging and neurodegeneration (Taylor et al. 2013; Niranjan 2014; Al Dakheel et al. 2014), it is believed that naturally occurring molecules possessing antioxidant and anti-inflammatory properties along with other pharmacological activities could be effective in preventing or ameliorating these neurodegenerative changes (Albarracin et al. 2012; Song et al. 2012; Fu et al. 2015). In recent years, numerous experimental and epidemiological studies have shown that medicinal herbs or herb-derived antioxidants might be promising drug candidates for PD (Albarracin et al. 2012; Koppula et al. 2012; Song et al. 2012; Takeda et al. 2014; Fu et al. 2015). Thus, in search of better pharmacotherapeutic agents, medicinal herbs have been extensively explored as a major source of novel agents (Fu et al. 2015). Using this approach, licorice, one of the most widely used food additives and herbal medicines, has received wide attention based on current reports of its beneficial effects in several chronic degenerative diseases including age-related neurodegenerative diseases (Mazzio et al. 2013; Ojha et al. 2013). In licorice, glycyrrhizic acid (GA, Fig. 1), a triterpenoid saponin, has been identified as one of the potent active components that contributes to its biological and pharmacological activities (Asl and Hosseinzadeh 2008).

The chemical structure and drug-likeness properties of glycyrrhizic acid
GA has been shown to possess numerous pharmacological properties including anti-inflammatory, antioxidant, antiviral, antitumor, gastroprotective, cardioprotective, hepatoprotective, and neuroprotective properties (Asl and Hosseinzadeh 2008). Recently, GA has garnered attention for its neuroprotective activity in experimental models of brain injury, spinal ischemia, and cerebral ischemia (Kawakami et al. 2010; Luo et al. 2013; Gong et al. 2014). Very recently, in vitro studies have reported its protective role against neurotoxicity induced by β-amyloid (Zhu et al. 2012), glutamate (Wang et al. 2014), and 1-methyl-4-phenylpyridinium (MPP+; Teng et al. 2014). However, to translate the in vitro findings, evidence from in vivo studies is vital in drug development as one of a series of incremental steps leading from bench to bedside. Previous in vitro studies have revealed that anti-inflammatory effects contribute to the neuroprotective properties of GA; however, the precise underlying mechanism is still unclear and evidence of efficacy in in vivo models is lacking.
Therefore, in the present study, we investigated the effects of GA, a naturally derived molecule, in a rotenone (ROT)-induced rat model of PD, an in vivo model that recapitulates human PD pathogenesis and represents a clinically relevant animal model for the screening of novel agents (Cannon et al. 2009; Betarbet et al. 2000; Litteljohn et al. 2011; Johnson and Bobrovskaya 2014). Based on the hypothesis that oxidative injury and inflammation underlie neurodegeneration, investigating the effect of GA in ROT-induced neurodegeneration may provide an alternative and early intervention approach to prevent or halt the progression of neurodegenerative changes in PD.
Materials and Methods
Drugs and Chemicals
Polyclonal rabbit anti-cyclo-oxygenase-2 (COX-2), anti-inducible nitric oxide synthase (iNOS), and anti-glial fibrillary acidic protein (GFAP) were purchased from Abcam, Cambridge, MA. Anti-ionized calcium-binding adaptor molecule-1 (Iba-1) polyclonal rabbit was purchased from Wako Chemicals, Richmond, VA, USA. Anti-tyrosine hydroxylase was obtained from Novus Biologicals, Littleton, CO, USA. Secondary horseradish peroxidase-conjugated goat anti-rabbit antibodies were obtained from Jackson Immunoresearch, West Grove, PA, USA. Alexa Fluor 488 and 594-conjugated secondary goat anti-rabbit antibodies were purchased from Life Technologies, Grand Island, NY, USA. ROT, GA, and the assay kit for reduced glutathione (GSH), and other reagents were purchased from Sigma-Aldrich, St. Louis, MO, USA.
Experimental Animals
Six- to seven-month-old male Wistar rats (280–300 g) bred in the animal research facility of the College of Medicine and Health Sciences, United Arab Emirates University were used. A maximum of four rats were housed per cage and were acclimatized for 1 week to the laboratory conditions prior to the start of the experiment. The animals were housed under standard laboratory conditions of light and dark cycle. The animals had access to commercially available rodent food and water ad libitum. All the experiments were carried out between 09:00 and 15:00 h. The experimental protocol for animal experimentation was approved by the Animal Research Ethics Committee, United Arab Emirates University, UAE.
Experimental Design
For the induction of neurodegeneration, ROT (2.5 mg/kg body weight) was administered intraperitoneally (i.p.) once daily for 4 weeks. It was first dissolved in dimethyl sulfoxide (DMSO) to obtain 50X stock solution and further diluted in sunflower oil to attain a final concentration of 2.5 mg/ml for administration to rats. The above regimen used in the current study for the induction of Parkinsonism in rats following ROT administration was adopted with slight modification from a previous report (Fujikawa et al. 2005). To test the neuroprotective efficacy of GA, it was dissolved in sterile water and injected i.p. at a dose of 50 mg/kg body weight once daily for 4 weeks, 30 min prior to each dose of ROT. The control group received an equal amount of vehicle only. Animals were sacrificed 48 h after the last injection of ROT/GA/vehicle to wash out the effects of ROT/GA. The rats were divided into four experimental groups, each containing eight rats. The experimental groups were as follows:
- Group I:
-
Vehicle-injected control group (CONT)
- Group II:
-
ROT-injected and vehicle-treated group (ROT)
- Group III:
-
ROT-injected and GA-treated group (ROT + GA)
- Group IV:
-
GA-only injected group (GA)
Tissue Preparation for Biochemical Studies
At the end of the experiments, the animals were anesthetized with pentobarbital (40 mg/kg body weight) and cardiac perfusion was carried out using 0.01 M phosphate-buffered saline (PBS) at pH 7.4 to wash out blood. The brains were quickly removed and placed on an ice-plate where the two hemispheres were separated. The midbrain region was dissected from one hemisphere and immediately frozen in liquid nitrogen for further use. The other hemisphere was post-fixed in 4 % paraformaldehyde solution for 48 h and subsequently transferred to 10 % sucrose solution for three consecutive days at 4 °C before being prepared for cryostat sectioning.
Biochemical Studies
The midbrains from each group were homogenized in KCl buffer (Tris–HCl, 10 mM NaCl, 140 mM KCl, 300 mM EDTA, 1 mM Triton-X-100 0.5 %) at pH 8.0 supplemented with protease and phosphatase inhibitor. The tissue homogenates were centrifuged at 14,000×g for 20 min at 4 °C to obtain the post-mitochondrial supernatant for estimation of markers of oxidative stress, lipid peroxidation, and pro-inflammatory cytokines using spectrophotometric measurements and enzyme-linked immunosorbent assays (ELISA).
Estimation of Lipid Peroxidation
The concentration of the lipid peroxidation product, malondialdehyde (MDA), was estimated using the detection kit according to the manufacturer’s instructions (North West Life Science, Vancouver, WA, USA). Briefly, 250 µl samples or calibrator were incubated in the presence of 250 µl acid reagent and 250 µl thiobarbituric acid and vortexed vigorously. Samples were incubated for 60 min at 60 °C and then centrifuged at 10,000×g for 2–3 min. The reaction mixture was transferred to a cuvette and the spectra were recorded at 532 nm. The results were expressed as µM MDA/mg protein.
Estimation of Reduced Glutathione (GSH)
The level of GSH was estimated using a commercially available kit following the manufacturer’s instructions (Sigma-Aldrich). Briefly, the samples were first deproteinized with 5 % 5-sulfosalicylic acid solution and centrifuged to remove the precipitated protein and then the supernatant was used to measure glutathione concentration. Samples (10 µl) and standards of different concentrations were incubated for 5 min with 150 µl of working mixture (assay buffer + 5,5′-Dithiobis (2-nitrobenzoic acid) + glutathione reductase) in a 96-well plate. Diluted nicotinamide adenine dinucleotide phosphate solution (50 µl) was added to each well and mixed thoroughly. Absorbance of the samples was measured at 412 nm with kinetics for 5 min using a microplate reader. The results were expressed as µM GSH/mg protein.
Estimation of the Activity of Antioxidant Enzymes
The activity of the antioxidant enzymes, superoxide dismutase (SOD) and catalase (CAT), was estimated using commercially available assay kits according to the manufacturer’s instructions (Cayman Chemical Company, Ann Arbor, MI, USA). Briefly, CAT was estimated by adding 20 µl samples or standards of different concentrations to 100 µl assay buffer and 30 µl methanol in a 96-well plate. H2O2 (20 µl) was added to initiate the reaction and samples were incubated for 20 min at room temperature (RT). Potassium hydroxide (30 µl) was added to terminate the reaction and subsequently 30 µl CAT purpald and 10 µl CAT potassium periodate were added. The plate was incubated for 5 min at RT on a shaker and absorbance was read at 540 nm using a micro plate reader. For SOD measurement, 10 µl samples or standard were added in each well of a 96-well plate. Xanthine oxidase (20 µl) was added to each well to initiate the reaction. The plate was shaken for a few seconds and incubated for 30 min at RT. Absorbance was read at 450 nm using a microplate reader. CAT activity was expressed as nmol/min/mg protein and SOD activity was expressed as units/mg protein.
Estimation of Pro-inflammatory Cytokines Using ELISA Assays
The ELISA kits for interleukin-1β (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor-alpha (TNF-α) were purchased from R&D system, Minneapolis, MI, USA. The levels of IL-1β, IL-6, and TNF-α were estimated using commercial ELISA kits following the manufacturer’s instructions. Briefly, a 96-well plate was coated with 100 µl diluted capture antibody overnight at RT. Each well was aspirated and washed with wash buffer (0.05 % Tween 20 in PBS 0.01 M pH 7.4). The plate was blocked by adding 300 µl reagent diluent (1 % bovine serum albumin in PBS) for 1 h and washed with wash buffer. Samples or standards (100 µl) of different concentrations were added to the wells and incubated for 2 h. Each well was exchanged with 100 µl detection antibody and then incubated for 2 h at RT. Wells were then exchanged with 100 µl working solution (1:200) of streptavidin horseradish peroxidase and further incubated for 20 min. The wells were exchanged with 100 µl substrate solution and incubated for 20 min. Stop solution (2 N H2SO4; 50 µl) was added and the plate was gently tapped to ensure proper mixing. The optical density of each well was read immediately at 450 nm using a micro plate reader. The results were expressed as pg/mg protein.
Immunofluorescence Staining for Tyrosine Hydroxylase (TH), GFAP, and Iba-1
Immunofluorescence staining was performed on the coronal sections of the brain containing the SNc and striatum area to observe the expression sites of TH, GFAP, and Iba-1. Briefly, 14-μm serial, coronal brain sections were cut at the level of the striatum and SNc using a cryostat (Leica, Germany) and incubated with blocking reagent (10 % normal goat serum in PBS 0.3 % Triton-X 100) followed by incubation with the primary antibodies such as polyclonal rabbit anti-TH (1:500), anti-GFAP (1:1000) and anti-Iba-1 (1:1000) overnight at 4 °C. The sections were washed and incubated for 1 h with fluorescent secondary antibody alexa flour 488 or alexa flour 594 goat anti-rabbit at RT. Sections were then washed and mounted using Fluoroshield mounting media (Sigma-Aldrich). Images were taken with a fluorescent microscope (Olympus, Hamburg, Germany).
Assessment of TH-Immunoreactive (TH-ir) Dopaminergic Neurons and TH-ir Dopamine Nerve Fiber Loss
To determine the loss of TH-ir neurons in the SNc area, coronal sections at three different levels (−4.8, −5.04, and −5.28 mm from the bregma) of the medial terminal nucleus (MTN) region were counted and the average was presented as a percentage. Loss of striatal nerve fibers was evaluated by measuring the optical density of TH-ir nerve fibers in the striatum using NIH Image J software (NIH, Bethesda, USA). The optical density of TH-ir fibers at three different fields in each section (three sections/rat) with equal areas within the striatum (adjacent to 0.30 mm from the bregma) was measured for each rat and an average of the three areas was calculated and represented as a percentage. The optical density of the overlying cortex was taken as a background measure and subtracted from the value generated from the striatum. The counting of TH-ir neurons and measurement of optical density of the TH-ir fibers were carried out by an investigator blind to the experimental groups.
Assessment of Activated Astrocytes and Microglia in the Striatum
A minimum of three coronal sections at similar levels of the striatum from each group were used to analyze the number of activated astrocytes and microglia. From each section, activated astrocytes and microglia were counted from three different randomly chosen fields of equal area using Image J software.
Western Blot Analysis of COX-2 and iNOS
The striatum was collected from the rats of each experimental group. Cytosolic extracts were prepared by homogenizing the tissues in radioimmunoprecipitation assay buffer supplemented with protease and phosphatase inhibitor and centrifuged at 15,000 rpm for 20 min. Samples of cytoplasmic fractions containing equal amounts of protein (35 μg) were separated using 10 % sodium dodecyl sulfate–polyacrylamide gel electrophoresis. The proteins were transferred onto polyvinylidene difluoride membranes and incubated overnight at 4 °C with specific primary rabbit polyclonal antibodies against COX-2 (1:1000) and iNOS (1:500) followed by horseradish peroxidase-conjugated secondary antibody. The protein recognized by the antibody was visualized using an enhanced chemiluminescence pico kit (Thermo Scientific, Rockford, IL, USA). The blots were stripped and re-probed for β-actin (1:5000, monoclonal mouse; Millipore, MA, USA) as a loading control. The intensity of the bands was measured by densitometry and quantified using Image J software.
Protein Estimation
The protein content was estimated using the Pierce bicinchoninic acid protein assay kit (Thermo Scientific) following the manufacturer’s instructions.
Statistical Analyses
The data were expressed as the mean value ± SEM. The data for all studies were analyzed using one-way analysis of variance (ANOVA) followed by Tukey’s test to calculate the statistical significance between various groups. The software used was GraphPad InStat (La Jolla, CA, USA). In all tests, the criterion for any statistically significant difference was set at p < 0.05.GA.
Results
Effect of GA on Dopamine Neurons and Striatal Dopamine Nerve Terminal Loss
Following administration of ROT, a significant (p < 0.05) loss of dopamine neurons was observed compared with that in vehicle-injected control rats (Fig. 2a, c). Interestingly, GA pretreatment of ROT-injected rats was associated with significant protection of dopamine neurons when compared with that in ROT-injected rats. The dopamine neurons in the SNc project their processes to the striatum where the dopamine nerve terminal fibers are enriched and are immunoreactive to tyrosine hydroxylase (TH) antibody. Therefore, we examined whether the loss of dopamine neurons in the SNc area correlated with terminal loss of striatal dopamine fiber intensity as stained by the TH antibody. As anticipated, we observed a significant decrease in striatal nerve terminal density in ROT-treated animals when compared with that in vehicle-treated controls (Fig. 2b, d). However, we did not observe any marked loss of dopamine neurons and striatal fibers in the GA alone injected rats. Thus, our results suggest that treatment with GA protects dopamine neurons from ROT-neurotoxicity.

Immunofluorescence staining of tyrosine hydroxylase-immunoreactive (TH-ir) neurons to quantify the number of dopaminergic (DA) neurons in the substantia nigra (SNc) (a) and to detect the expression of TH-ir dopamine nerve terminal fibers in the striatum (b). The scale bar is 100 µm. a The number of TH-ir neurons were reduced in the SNc area of rotenone (ROT)-injected rats as compared with that in the control (CONT) group. Glycyrrhizic acid (GA) treatment shows profound protection of TH-ir neurons in ROT + GA-injected rats relative to ROT rats. GA alone injected rats did not show the any remarkable loss of dopamine neurons. b The expression of TH-ir striatal dopamine nerve terminals in the striatum of CONT, ROT, ROT + GA, and GA alone group rats. The CONT group shows higher immunoreactivity of TH in the striatum compared to the ROT group. GA treatment increased immunoreactivity of TH in the striatum of the ROT + GA group relative to the ROT group. c The number of DA neurons in the SNc was counted in three different sections of the SNc at the level of −4.8, −5.04, and −5.3 mm from the bregma for each animal. The number of DA neurons was significantly (*p < 0.05) higher in the SNc of the CONT group when compared to the ROT group. GA treatment significantly (# p < 0.05) protected the DA neurons from the ROT-induced neuronal death. d The expression of TH-ir dopamine nerve terminals was significantly (*p < 0.01) reduced in the striatum of rotenone (ROT)-injected rats as compared with that in the control (CONT) group. GA treatment shows significant (# p < 0.01) attenuation of TH-ir dopamine nerve terminals in ROT + GA-injected rats relative to ROT rats. Values are expressed as percentage of mean ± SEM relative to 100 % control (n = 3)
Effect of GA on Lipid Peroxidation and Glutathione Level
The rats administered ROT showed a significant (p < 0.01) increase in the lipid peroxidation product, MDA, as compared to the control group (Fig. 3a). Concomitantly, ROT administration caused a significant (p < 0.01) decrease in GSH levels when compared to the control group (Fig. 3b). However, the ROT-treated rats that received GA significantly (p < 0.05) attenuated the rise in MDA level (Fig. 3a) and exhibited improved GSH levels (Fig. 3b) when compared to the ROT group.

Estimation of MDA, GSH, SOD, and CAT in the midbrain tissue. Rotenone (ROT) injections caused significant (*p < 0.01) increases in malondialdehyde (MDA) (a) and decreased the level of reduced glutathione (GSH) (b) in the midbrain of ROT rats relative to control (CONT) group rats. Glycyrrhizic acid (GA) treatment in the ROT + GA group significantly (# p < 0.05) decreased the level of MDA and increased (# p < 0.05) the level of GSH. ROT injection also caused a significant (*p < 0.01) decrease in the activity of superoxide dismutase (SOD) (c) and catalase (CAT) (d) relative to the CONT group. GA treatment significantly (# p < 0.05) improved the ROT-induced decrease in SOD and CAT activity relative to the ROT-injected rats. Values are expressed as mean ± SEM (n = 6–8)
Effect of GA on Antioxidant Enzyme Activity
The activity of the antioxidant enzymes, SOD (Fig. 3c) and CAT (Fig. 3d), was decreased significantly (p < 0.01) in ROT-injected animals in comparison with control animals. However, treatment with GA significantly (p < 0.05) increased the activity of SOD (Fig. 3c) and CAT (Fig. 3d) when compared to the ROT-treated group. We did not observe any significant changes in SOD (Fig. 3c) or CAT (Fig. 3d) activity between controls and animals injected with GA.
Effect of GA on the Expression of Iba-1 and GFAP
It has been well documented that GFAP and Iba-1 are considered as markers of reactive oxygen species production and inflammatory processes. In the immunohistochemical analysis, remarkably high expression of GFAP, which indicates increased numbers of astrocytes with astrocytic hypertrophy, was observed in ROT-injected rats when compared to that in control rats (Fig. 4a, c). However, treatment with GA attenuated the expression of hypertrophied astrocytes in rats administered ROT when compared to the ROT challenged animals (Fig. 4a, c). A similarly significant increase in the expression of Iba-1, indicating activation of microglia, was observed as an index of the inflammatory response in ROT-injected rats (Fig. 4b, d). Treatment with GA significantly reduced the ROT-induced microglial activation represented by Iba-1 expression (Fig. 4b, d). These observations clearly suggest that GA inhibits the activation of microglia and astrocytes.

Immunofluorescence staining to detect the expression of glial fibrillary acidic protein (GFAP)-positive astrocytes (green) and ionized calcium-binding adaptor molecule-1 (Iba-1)-positive microglia (green) in the striatum of control (CONT), rotenone (ROT), and ROT + glycyrrhizic acid (GA) rats. Profound expression of GFAP-positive astrocytes (a) and Iba-1-positive microglia (b) was found in the ROT-treated rats relative to the CONT rats. GA administration to ROT-injected rats led to moderate staining of GFAP and Iba-1 in the ROT + GA rats relative to ROT-injected rats (scale bar 200 µm). Quantitative analysis of activated astrocytes (c) and microglia (d) revealed a significant (*p < 0.05) increase in the number of activated astrocytes and microglia in rats in the ROT-treated group relative to CONT rats. GA administration significantly (# p < 0.05) reduced the number of activated astrocytes and microglia in the ROT + GA group rats relative to ROT rats. Values are expressed as percentage of mean ± SEM (n = 3) (Color figure online)
Effect of GA on the Induction of Pro-inflammatory Cytokines
We also measured the concentration of the pro-inflammatory cytokines, IL-1β, IL-6, and TNF-α in response to ROT administration. Significant (p < 0.01) increases in the levels of IL-1β (Fig. 5a), IL-6 (Fig. 5b), and TNF-α (Fig. 5c) were observed in the ROT-injected animals when compared to the control group. However, GA treatment of ROT-injected animals significantly (p < 0.05) decreased the levels of all of these cytokines when compared to control group animals (Fig. 5a–c). Rats treated with GA alone did not show any significant changes in the levels of pro-inflammatory cytokines when compared to control group animals.

IL-1β, IL-6, and TNF-α were measured by enzyme-linked immunosorbent assay (ELISA) in the midbrain of control (CONT), rotenone (ROT), and ROT + glycyrrhizic acid (GA) group rats. The level of IL-1β (a), IL-6 (b), and TNF-α (c) was found to be significantly (*p < 0.05) increased in the ROT group relative to CONT rats. GA treatment significantly (# p < 0.05) decreased the ROT-induced increase of these cytokines in ROT + GA rats. Values are expressed as mean ± SEM (n = 6–8)
Effect of GA on Inflammatory Mediators: COX-2 and iNOS Expression
We further investigated the expression of COX-2 and iNOS by Western blots using the tissue lysates isolated from the striatum region. A significant (p < 0.05) increase in COX-2 expression (Fig. 6a, b) was observed in response to ROT injection (201.13 %) when compared with that in the control group (100 %). However, following treatment with GA in ROT-administered rats, a modest reduction in the level of COX-2 (21.17 %) was observed. Similarly, we also observed a significant (p < 0.05) increase in iNOS expression (Fig. 6a, c) in the ROT-injected animals when compared to the control group (168.35 vs. 100 %). Similar to the reduction in COX-2 following treatment with GA, a notable decrease (46.91 %) in iNOS induction (Fig. 6c) was observed.

Expression levels of striatal cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS) were measured by Western blot (a). A significant (*p < 0.05) increase in COX-2 (201.13 %) was observed in the rotenone (ROT) group relative to the control (CONT) group. Glycyrrhizic acid (GA) treatment followed by ROT injection remarkably decreased the expression of COX-2 by 21.71 % relative to the ROT group (b). Similarly, iNOS expression was increased (168.35 %) significantly (*p < 0.05) in ROT group relative to the CONT group. GA treatment notably decreased the iNOS expression by 46.91 % relative to the ROT group (c). Values are expressed as percentage of mean ± SEM relative to 100 % CONT (n = 3)
Discussion
The present study was undertaken to investigate the role of GA in an ROT-induced rat model of neurodegeneration. There are several neurotoxins such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), ROT, paraquat, and 6-hydroxydopamine (6-OHDA), which are well known for producing selective dopaminergic neurodegeneration in animal models of PD (Litteljohn et al. 2011; Johnson and Bobrovskaya 2014). However, among all existing models, the ideal animal model is one that recapitulates the majority of the relevant clinical and pathological features of the disease. The ROT model has some advantages over other available animal models as it produces most of the pathophysiological features of PD (Cannon et al. 2009; Betarbet et al. 2000). ROT is a potent naturally occurring environmental toxicant used as an insecticide and pesticide. It induces many of the key pathological features of human PD through multiple mechanisms including mitochondrial complex I inhibition, oxidative stress, inflammation, and glial activation. It also causes selective loss of dopaminergic neurons within the SNc area, depletion of striatal dopamine, and aggregation of α-synuclein with formation of Lewy body-like inclusions (Betarbet et al. 2000; Sherer et al. 2003; Cannon et al. 2009; Litteljohn et al. 2011; Johnson and Bobrovskaya 2014). Thus, oxidative injury and neuroinflammation contribute significantly to PD pathogenesis. In order to investigate alternative and early intervention approaches to prevent or halt the progressive nature of PD, we evaluated the potential of GA in ROT-induced neurodegeneration.
GA, which is composed of one molecule of glycyrrhetinic acid and two molecules of glucuronic acid, is abundantly present in the roots of the genus Glycyrrhiza glabra (Licorice or sweet root, family: Leguminosae). The herb licorice is credited with many pharmacological properties and is one of the most widely used herbal medicines and food additives (Asl and Hosseinzadeh 2008; Ojha et al. 2013). Recently, formulations of the licorice herb have been reported to be beneficial in neurodegenerative diseases, an effect that has been attributed to several of its active constituents, particularly GA (Shishkina et al. 2006; Kawakami et al. 2010; Mazzio et al. 2013; Gong et al. 2014). Although the beneficial effects of GA have been reported in in vitro models of amyloid neurotoxicity (Kawakami et al. 2010), glutamate excitotoxicity (Wang et al. 2014), and MPP+ neurotoxicity (Teng et al. 2014), it is not known whether GA is protective in an in vivo ROT-induced model of PD. Hence, the present study is the first to demonstrate the neuroprotective effect of GA in an in vivo chronic model of PD.
As treatment of PD focuses on symptomatic relief rather than amelioration of pathogenesis progression, the development of disease-modifying agents based on antioxidant and anti-inflammatory strategies along with other pharmacological properties would be useful in halting the development and progression of PD (Albarracin et al. 2012; Koppula et al. 2012; Song et al. 2012; Takeda et al. 2014; Al Dakheel et al. 2014; Fu et al. 2015). The severity of motor dysfunction has been shown to be caused by selective loss of dopamine neurons, which is at least in part associated with severe oxidative stress and inflammation (Dexter and Jenner 2013; Niranjan 2014). Several antioxidants have been shown to confer substantial protection against ROT-induced PD (Sherer et al. 2003; Verma and Nehru 2009; Xiong et al. 2012). Therefore, in the current study, we evaluated the neuroprotective potential of GA in an ROT model of PD. The dose of GA used in our study was selected based on our preliminary experiments and previous studies (Ojha et al. 2013; Song et al. 2013).
Immunohistochemical staining of coronal brain sections revealed significant degeneration of dopaminergic neurons indicated by a decrease in TH-ir neurons in the SNc following chronic administration of ROT (Fig. 2a, c). TH is a rate-limiting enzyme and a marker for DA neuron survival. In keeping with previous studies, the reduction in TH-ir in the SNc dopaminergic neurons following repeated administration of ROT indicates a significant effect of ROT on SNc neurons (Litteljohn et al. 2011; Xiong et al. 2012). In addition to dopamine neuron loss, we observed a significant decrease in dopamine nerve terminals that project to the striatum (Fig. 2b, d). The loss of dopamine neurons in the SNc and reduction in the density of nerve terminals in the striatum are considered to be pathological indices of PD (Sherer et al. 2003). Interestingly, GA treatment prior to ROT injection provided protection to dopamine neurons as well as their nerve terminals, clearly indicating the neuroprotective properties of GA (Fig. 2a–d).
Oxidative stress generated by free radicals and subsequent lipid peroxidation plays an important role in PD pathogenesis (Sherer et al. 2003; Litteljohn et al. 2011). During oxidative injury, accumulation of oxidants makes the cell membrane more susceptible to injury and results in formation of the lipid peroxidation product, MDA (Verma and Nehru 2009). The brain has less efficient antioxidant defense mechanisms and is rich in polyunsaturated fatty acids, so it is more sensitive to oxidative damage than other tissue. Our results showed that lipid peroxidation resulting from oxidative stress in the midbrain was clearly increased after the ROT challenge, in agreement with previous studies (Sherer et al. 2003; Verma and Nehru 2009; Litteljohn et al. 2011; Xiong et al. 2012; Johnson and Bobrovskaya 2014). The decrease in MDA level following treatment with GA can be ascribed to the enhanced activity of antioxidant defense mechanisms, as evidenced by increased activity of antioxidant enzymes and increased glutathione availability.
Endogenous antioxidant defense networks consist of enzymatic (SOD and CAT) and non-enzymatic (GSH) molecules that neutralize the oxygen free radicals that lead to oxidative stress if the antioxidant system is compromised (Anderson and Maes 2014; Celardo et al. 2014; Niranjan 2014). Perturbation of antioxidant defense system components such as GSH, SOD, and CAT has been well documented in the PD brain (Verma and Nehru 2009; Xiong et al. 2012; Anderson and Maes 2014; Celardo et al. 2014; Niranjan 2014). The decrease in GSH content following ROT administration in the current study likely occurred to minimize the deleterious consequence of oxidative stress. However, following GA pre-treatment to ROT-administered rats, significant recovery or restoration of GSH levels clearly demonstrates the antioxidant and free radical scavenging activity of GA. An increase in oxidative damage is often correlated with a simultaneous decline in the activity of the intracellular antioxidant enzymes, SOD and CAT. Following ROT administration, a significant reduction in the activity of SOD and CAT was observed in the midbrain tissues in our study (Fig. 3c, d). Low activity of SOD and CAT in ROT-treated rats may result from inactivation of the enzymes by H2O2 (Pigeolet et al. 1990). However, after simultaneous administration of GA and ROT, the significant improvement in the activity of SOD and CAT demonstrates the antioxidant activity of GA. Consistent with previous observations, our present findings suggest that the neuroprotective action of GA can be attributed to its direct free radical quenching properties or augmentation of antioxidant enzymes (Asl and Hosseinzadeh 2008). GA has been shown to protect dopaminergic neurons in vitro, an effect that was attributed to its free radical scavenging activity (Teng et al. 2014). However, this was the first report to demonstrate the antioxidant activity of GA in an in vivo rat model of PD.
To explore the possible effects of GA on the inflammatory pathways, the levels of various inflammatory molecules and pro-inflammatory cytokines were investigated. The central inflammatory process, which includes the activation of microglia and secretion of pro-inflammatory cytokines, is a key player in the neurodegenerative processes of PD (Niranjan 2014). Activated microglia exert their neurotoxic effects by releasing pro-inflammatory cytokines, such as TNFα, IL-1β, and IL-6. Following ROT administration, increased levels of IL-1β, IL-6, and TNF-α in the midbrain tissues were observed in the current study, in keeping with numerous previous reports (Sherer et al. 2003; Litteljohn et al. 2011; Xiong et al. 2012). Interestingly, GA treatment of ROT-administered rats significantly reduced levels of IL-1β, IL-6, and TNF-α concomitant with decreased microglial activation. This suggests that GA might counteract the activation process of microglia, thereby controlling the levels of IL-1β, IL-6, and TNF-α, which is consistent with previous studies (Zhu et al. 2012; Song et al. 2013; Gong et al. 2014). These pro-inflammatory cytokines have been shown to cause phosphorylation and degradation of the inhibitory factor, inhibitor of kappa B (IκB), which results in the activation of the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling cascade (Litteljohn et al. 2011). The activation of NF-κB also promotes the induction of iNOS from activated microglia. This in turn increases production of nitric oxide (NO), which plays a pivotal role in mediating the early stages of immune-inflammatory responses (Hartlage-Rübsamen et al. 1999; Lawrence et al. 2001; Yoke Yin et al. 2010). In our study, evidence for the anti-inflammatory activity of GA is further supported by attenuation of iNOS expression, which can be explained by its NF-κB inhibitory activity (Kawakami et al. 2010; Zhu et al. 2012; Gong et al. 2014; Wang et al. 2014).
Rats challenged with ROT show activation of microglia, which produce a myriad of neurotoxic and inflammatory mediators (COX-2 and iNOS) that subsequently contribute to DA neurodegeneration through immune activation and production of inflammatory mediators (Sherer et al. 2003; Xiong et al. 2012). In our study, we observed significant increases in COX-2 after ROT administration, consistent with earlier reports (Verma and Nehru 2009; Xiong et al. 2012). However, GA treatment to ROT-injected rats reduced the up-regulation of COX-2, which is also known to catalyze the oxidation of dopamine to generate toxic dopamine-quinone, a common feature of the PD brain (Sherer et al. 2003; Litteljohn et al. 2011; Johnson and Bobrovskaya 2014). In our study, down-regulation of COX-2 along with attenuation of pro-inflammatory cytokines can be ascribed to the multimodal anti-inflammatory properties of GA in affording protection against ROT-induced dopaminergic neuronal loss.
Recently, several studies have reported GA to be an activator of the transcription factors peroxisome proliferated activator receptors (PPAR-γ), which regulate inflammation by counteracting the oxidative pathway (Yoke Yin et al. 2010). The beneficial effect of PPAR-γ agonists on motor dysfunction and neurochemical deficits in PD is mediated by augmentation of antioxidants and attenuation of inflammatory mediators (Martin et al. 2012). Thus, the amelioration of inflammatory mediators such as COX-2, iNOS, and pro-inflammatory cytokines observed in the present study supports the PPAR-γ-mediated mechanism of GA in its neuroprotective effects.
In addition to oxidative stress and neuroinflammation, the activation of microglia, also known as the macrophages of the brain, is a common and early hallmark of neurodegenerative diseases and contributes to virtually all neuropathology (Anderson and Maes 2014; Celardo et al. 2014; Niranjan 2014). Several studies have reported that activation of glial cells enhances the release of several pro-inflammatory cytokines such as IL-1β, IL-6, and TNF-α and subsequently causes dopaminergic neurodegeneration following ROT administration (Sherer et al. 2003; Verma and Nehru 2009; Litteljohn et al. 2011; Xiong et al. 2012; Johnson and Bobrovskaya 2014). In our study, consistent with previous reports (Sherer et al. 2003; Verma and Nehru 2009; Litteljohn et al. 2011; Xiong et al. 2012; Johnson and Bobrovskaya 2014), we also observed microglial activation upon chronic ROT administration as evidenced by increased expression of Iba-1 and GFAP, which are considered to indicate microglial activation and subsequent neuroinflammation (Fig. 4a–d). GFAP is a marker for astrocytes known to be induced during central nervous system degeneration and to be more highly expressed in the aged brain. The density of GFAP-positive cells increases with the severity of the dopaminergic neuronal loss in the SNc (Damier et al. 1993). Similarly, in our study, we observed activation of GFAP in ROT-injected rats, which is suggestive of an inflammatory response (Damier et al. 1993; Sherer et al. 2003; Martin et al. 2012). However, treatment with GA significantly attenuated the activation of microglia and astrocytes as evidenced by reduced expression of GFAP and Iba-1, respectively. These results clearly suggest that GA rescues dopaminergic neurons and suppresses microglial and astrocyte activation, a starting point in dopaminergic neurodegeneration and neuronal loss.
In keeping with previous reports that have considered the benefits of GA in behavioral disorders (Shishkina et al. 2006; Song et al. 2013) the results of the present study are further suggestive of its therapeutic potential against ROT-induced dopaminergic neurodegeneration. In addition to its efficacy, the absence of adverse effects on brain tissue from GA alone further supports its relative safety. The preventive effects observed in this study could be further extrapolated to provide a means of protection against environmental toxicants that lead to neurodegeneration.
Conclusions
Based on the present study findings, we conclude that GA impedes ROT-induced dopaminergic neurodegeneration by restoration of the antioxidant system, inhibition of lipid peroxidation and inflammation, and preservation of dopaminergic neurons. Taken together, the present findings and those from previous reports indicate that GA appears to be a promising agent of natural origin for protection against PD and neurodegeneration. However, the exact molecular mechanism by which GA alters the antioxidant capacity or inflammatory response warrants further investigation.
Abbreviations
- ABC:
-
Avidin biotin complex
- COX-2:
-
Cyclooxygenase-2
- DAB:
-
Diamino benzidine
- GA:
-
Glycyrrhizic acid
- GFAP:
-
Glial fibrillary acidic protein
- GSH:
-
Reduced glutathione
- IBA-1:
-
Ionized calcium-binding adaptor molecule-1
- iNOS:
-
Inducible nitric oxide synthase
- MDA:
-
Malondialdehyde
- PD:
-
Parkinson’s disease
- ROT:
-
Rotenone
- SOD:
-
Superoxide dismutase
- TH:
-
Tyrosine hydroxylase
References
Al Dakheel A, Kalia LV, Lang AE (2014) Pathogenesis-targeted disease-modifying therapies in Parkinson disease. Neurotherapeutics 11:6–23
Albarracin SL, Stab B, Casas Z, Sutachan JJ, Samudio I, Gonzalez J, Gonzalo L, Capani F, Morales L, Barreto GE (2012) Effects of natural antioxidants in neurodegenerative disease. Nutr Neurosci 15:1–9
Anderson G, Maes M (2014) Neurodegeneration in Parkinson’s disease: interactions of oxidative stress, tryptophan catabolites and depression with mitochondria and sirtuins. Mol Neurobiol 49:771–783
Asl MN, Hosseinzadeh H (2008) Review of pharmacological effects of Glycyrrhiza sp. and its bioactive compounds. Phytother Res 22:709–724
Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT (2000) Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci 3:1301–1306
Cannon JR, Tapias V, Na HM, Honick AS, Drolet RE, Greenamyre JT (2009) A highly reproducible Rotenone model of Parkinson’s disease. Neurobiol Dis 34:279–290
Celardo I, Martins LM, Gandhi S (2014) Unravelling mitochondrial pathways to Parkinson's disease. Br J Pharmacol 171(8):1943–1957
Damier P, Hirsch EC, Zhang P, Agid Y, Javoy-Agid F (1993) Glutathione peroxidase, glial cells and Parkinson’s disease. Neuroscience 52:1–6
Dexter DT, Jenner P (2013) Parkinson disease: from pathology to molecular disease mechanisms. Free Radic Biol Med 62:132–144
Fu SP, Wang JF, Xue WJ, Liu HM, Liu BR, Zeng YL, Li SN, Huang BX, Lv QK, Wang W, Liu JX (2015) Anti-inflammatory effects of BHBA in both in vivo and in vitro Parkinson’s disease models are mediated by GPR109A-dependent mechanisms. J Neuroinflammation 12:9
Fujikawa T, Kanada N, Shimada A, Ogata M, Suzuki I, Hayashi I, Nakashima K (2005) Effect of sesamin in Acanthopanax senticosus HARMS on behavioral dysfunction in rotenone-induced parkinsonian rats. Biol Pharm Bull 28:169–172
Gong G, Xiang L, Yuan L, Hu L, Wu W, Cai L, Yin L, Dong H (2014) Protective effect of glycyrrhizin, a direct HMGB1 inhibitor, on focal cerebral ischemia/reperfusion-induced inflammation, oxidative stress, and apoptosis in rats. PLoS ONE 9:e89450
Hartlage-Rübsamen M, Lemke R, Schliebs R (1999) Interleukin-1β, inducible nitric oxide synthase, and nuclear factor-κB are induced in morphologically distinct microglia after rat hippocampal lipopolysaccharide/interferon-γ injection. J Neurosci Res 57:388–398
Johnson ME, Bobrovskaya L (2014) An update on the rotenone models of Parkinson’s disease: their ability to reproduce the features of clinical disease and model gene-environment interactions. Neurotoxicology 46C:101–116
Kawakami Z, Ikarashi Y, Kase Y (2010) Glycyrrhizin and its metabolite 18 beta-glycyrrhetinic acid in glycyrrhiza, a constituent herb of yokukansan ameliorate thiamine deficiency-induced dysfunction of glutamate transport in cultured rat cortical astrocytes. Eur J Pharmacol 626:154–158
Koppula S, Kumar H, More SV, Lim HW, Hong SM, Choi DK (2012) Recent updates in redox regulation and free radical scavenging effects by herbal products in experimental models of Parkinson’s disease. Molecules 17:11391–11420
Lawrence T, Gilroy DW, Colville-Nash PR, Willoughby DA (2001) Possible new role for NF-kappaB in the resolution of inflammation. Nat Med 7:1291–1297
Litteljohn D, Mangano E, Clarke M, Bobyn J, Moloney K, Hayley S (2011) Inflammatory mechanisms of neurodegeneration in toxin-based models of Parkinson’s disease. Parkinson’s Dis 2010:713517
Luo L, Jin Y, Kim ID, Lee JK (2013) Glycyrrhizin attenuates kainic Acid-induced neuronal cell death in the mouse hippocampus. Exp Neurobiol 22:107–115
Martin HL, Mounsey RB, Mustafa S, Sathe K, Teismann P (2012) Pharmacological manipulation of peroxisome proliferator-activated receptor γ (PPARγ) reveals a role for anti-oxidant protection in a model of Parkinson’s disease. Exp Neurol 235:528–538
Mazzio E, Deiab S, Park K, Soliman KF (2013) High throughput screening to identify natural human monoamine oxidase B inhibitors. Phytother Res 27:818–828
Niranjan R (2014) The role of inflammatory and oxidative stress mechanisms in the pathogenesis of Parkinson’s disease: focus on astrocytes. Mol Neurobiol 49:28–38
Ojha S, Golechha M, Kumari S, Bhatia J, Arya DS (2013) Glycyrrhiza glabra protects from myocardial ischemia-reperfusion injury by improving hemodynamic, biochemical, histopathological and ventricular function. Exp Toxicol Pathol 65:219–227
Pigeolet E, Corbisier P, Houbion A, Lambert D, Michiels C, Raes M, Zachary MD, Remacle J (1990) Glutathione peroxidase, superoxide dismutase, and catalase inactivation by peroxides and oxygen derived free radicals. Mech Ageing Dev 51:283–297
Schapira AH, Olanow CW, Greenamyre JT, Bezard E (2014) Slowing of neurodegeneration in Parkinson’s disease and Huntington’s disease: future therapeutic perspectives. Lancet 384:545–555
Sherer TB, Betarbet R, Kim JH, Greenamyre JT (2003) Selective microglial activation in the rat rotenone model of Parkinson’s disease. Neurosci Lett 341:87–90
Shishkina GT, Dygalo NN, Yudina AM, Kalinina TS, Tolstikova TG, Sorokina IV, Kovalenko IL, Anikina LV (2006) The effects of fluoxetine and its complexes with glycyrrhizic acid on behavior in rats and brain monoamine levels. Neurosci Behav Physiol 36:329–333
Song JX, Sze SC, Ng TB, Lee CK, Leung GP, Shaw PC, Tong Y, Zhang YB (2012) Anti-Parkinsonian drug discovery from herbal medicines: what have we got from neurotoxic models? J Ethnopharmacol 139:698–711
Song JH, Lee JW, Shim B, Lee CY, Choi S, Kang C, Sohn NW, Shin JW (2013) Glycyrrhizin alleviates neuroinflammation and memory deficit induced by systemic lipopolysaccharide treatment in mice. Molecules 18:15788–15803
Takeda A, Nyssen OP, Syed A, Jansen E, Bueno-de-Mesquita B, Gallo V (2014) Vitamin A and carotenoids and the risk of Parkinson’s disease: a systematic review and meta-analysis. Neuroepidemiology 42:25–38
Taylor JM, Main BS, Crack PJ (2013) Neuroinflammation and oxidative stress: co-conspirators in the pathology of Parkinson’s disease. Neurochem Int 62:803–819
Teng L, Kou C, Lu C, Xu J, Xie J, Lu J, Liu Y, Wang Z, Wang D (2014) Involvement of the ERK pathway in the protective effects of glycyrrhizic acid against the MPP + -induced apoptosis of dopaminergic neuronal cells. Int J Mol Med 34:742–748
Verma R, Nehru B (2009) Effect of centrophenoxine against rotenone-induced oxidative stress in an animal model of Parkinson’s disease. Neurochem Int 55:369–375
Wang D, Guo TQ, Wang ZY, Lu JH, Liu DP, Meng QF, Xie J, Zhang XL, Liu Y, Teng LS (2014) ERKs and mitochondria-related pathways are essential for glycyrrhizic acid-mediated neuroprotection against glutamate-induced toxicity in differentiated PC12 cells. Braz J Med Biol Res 47:773–779
Xiong N, Huang J, Chen C, Zhao Y, Zhang Z, Jia M, Zhang Z, Hou L, Yang H, Cao X, Liang Z, Zhang Y, Sun S, Lin Z, Wang T (2012) Dl-3-n-butylphthalide, a natural antioxidant, protects dopamine neurons in rotenone models for Parkinson’s disease. Neurobiol Aging 33:1777–1791
Yoke Yin C, So Ha T, Abdul Kadir K (2010) Effects of glycyrrhizic acid on peroxisome proliferator-activated receptor gamma (PPAR-gamma), lipoprotein lipase (LPL), serum lipid and HOMA-IR in rats. PPAR Res 2010:530265
Zhu X, Chen C, Ye D, Guan D, Ye L, Jin J, Zhao H, Chen Y, Wang Z, Wang X, Xu Y (2012) Diammonium glycyrrhizinate upregulates PGC-1α and protects against Aβ1-42-induced neurotoxicity. PLoS ONE 7:e35823
Acknowledgments
The research grant support from the United Arab Emirates University and the National Research foundation, United Arab Emirates to MEH and SO are duly acknowledged. The authors would also like to acknowledge Mahmoud Hag Ali, Animal Research Facility controller for his help with animal care and welfare.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
There are no patents, products in development, or marketed products to declare. This study was supported by grants from the College of Medicine & Health Sciences, UAE University, UAE. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Ethical approval
All procedures performed in studies involving animals were in accordance with the ethical standards of the Animal Research Ethics Committee, United Arab Emirates University, UAE.
Additional information
Shreesh Ojha and Hayate Javed have contributed equally to this work.
Rights and permissions
About this article

Cite this article
Ojha, S., Javed, H., Azimullah, S. et al. Glycyrrhizic acid Attenuates Neuroinflammation and Oxidative Stress in Rotenone Model of Parkinson’s Disease. Neurotox Res 29, 275–287 (2016). https://doi.org/10.1007/s12640-015-9579-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-015-9579-z