
Abstract
Neuroinflammation plays a key role in the pathogenesis of Parkinson’s disease (PD). Epidemiologic, animal, human, and therapeutic studies support the role of oxidative stress and inflammatory cascade in initiation and progression of PD. In Parkinson’s disease pathophysiology, activated glia affects neuronal injury and death through production of neurotoxic factors like glutamate, S100B, tumor necrosis factor alpha (TNF-α), prostaglandins, and reactive oxygen and nitrogen species. As disease progresses, inflammatory secretions engage neighboring cells, including astrocytes and endothelial cells, resulting in a vicious cycle of autocrine and paracrine amplification of inflammation leading to neurodegeneration. The exact mechanism of these inflammatory mediators in the disease progression is still poorly understood. In this review, we highlight and discuss the mechanisms of oxidative stress and inflammatory mediators by which they contribute to the disease progression. Particularly, we focus on the altered role of astroglial cells that presumably initiate and execute dopaminergic neurodegeneration in PD. In conclusion, we focus on the molecular mechanism of neurodegeneration, which contributes to the basic understanding of the role of neuroinflammation in PD pathophysiology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neuroinflammation results primarily from the presence of chronically activated glial cells (astrocytes and microglia) in the brain and is a common feature in the pathology of PD [1]. The role of reactive astrocytes and microglia is further emphasized by the fact that neuroscientists initially spoke of “Reactive gliosis” instead of neuroinflammation in describing glial responses of the central nervous system (CNS) tissue to injury [2]. Reactive gliosis, is the cellular manifestation of neuroinflammation and characterized by formation of glial cell soma, and accumulation and activation of enlarged glial cells, notably microglia and astrocytes, appearing immediately after injury [3]. Best considered as a local tissue response without any involvement of the peripheral immune system, neuroinflammation in PD is a complex process with multiple mediators, signaling pathways, and feedback loops. Further, various studies demonstrated beneficial effects of nonsteroidal anti-inflammatory drugs in treatment of CNS and other neurodegenerative diseases, which supported the role of neuroinflammation in the progressive degeneration of neurons [4, 5]. However, neuroinflammation and oxidative stress are connected to each other, but still they play very distinct and independent roles in disease progression. Among the neuroinflammatory mediators, cytokines and prostaglandins play important roles in causation and progression of neurodegenerative diseases [6, 7]. Oxidative stress-induced mediators include reactive oxygen species (ROS) and reactive nitrogen species (RNS). In this review, we discuss the role of inflammatory and oxidative stress as evidenced by various studies in the causation and progression of PD pathogenesis [8]. Last but not least, we also describe and highlight the potential role of astrocytes in the causation of neurodegeneration in Parkinson’s disease.
Some Important Aspects of the Parkinson’s Disease Pathophysiology
Parkinson’s disease is the second most common neurodegenerative disorder after Alzheimer’s dementia [9]. Parkinson’s disease is a common neurodegenerative disorder of unknown cause which cardinal clinical features include tremor, stiffness, slowness of movement, and postural instability [10]. Although the etiology of idiopathic Parkinson’s disease is unknown, this neurodegenerative disease is characterized by the degeneration of dopamine (DA)-producing neurons. The origin or central cause of this neuronal degeneration is unknown and may involve several molecular and cellular cascade of events, including oxidative stress, inflammation, accumulation of altered proteins, excitotoxicity, proapoptotic mechanisms, and mitochondrial dysfunction [11, 12]. It has recently been suggested that a glial reaction and inflammatory processes also participate in the cascade of events leading to neuronal degeneration [12–14]. First, an astroglial reaction was generally described at neuropathological examination in the substantia nigra (SN) of patients with PD and has been confirmed quantitatively using glial fibrillary acidic protein (GFAP) immunostaining [15]. An increased number of activated microglial cells has consistently been reported in PD [16]. The role played by these glial cells is not fully understood and still under debate [11, 13, 17]. Activated microglial cells may have a deleterious effect on DA neurons. Microglial cells represent resident brain macrophage cells and can be transformed into activated immunocompetent antigen-presenting cells during the pathological process [11]. Further, it has been reported that, in PD, there is an increased density of glial cells expressing proinflammatory cytokines including tumor necrosis factor alpha (TNF-α) and interleukin-1β [18]. Aside from increased levels of cytokines such as TNF-α, several investigators have also reported interleukins in the brain parenchyma or cerebrospinal fluid of PD patients [18, 19]. In addition to astrocytes and microglial, other cells may also participate in the neuroinflammatory processes in PD [20]. One may, thus, speculate that after a primary insult of environmental or genetic origin, the glial reaction may maintain the degeneration of DA neurons. However, this question, and more generally the role of inflammation in the pathogenesis of PD, cannot be addressed by postmortem studies alone [20]. Therefore, epidemiological and genetic studies also suggest a role of inflammatory processes in susceptibility to Parkinson’s disease [21]. Data from an epidemiological study regarding the use of nonsteroidal anti-inflammatory drugs (NSAIDs) and the risk of PD emphasize the role of inflammation [4, 22]. Interestingly, participants who reported regular use of nonaspirin, nonsteroidal anti-inflammatory drugs at the beginning of the study had a significantly lower risk of PD than nonregular users during the follow-up [22]. However, a majority of data from clinical studies and from experimental models show the potential use of NSAIDs in the treatment of Parkinson’s disease, but there are also few exceptional studies that contradict with the use of NSAIDs [23, 24].
The Evidences of Inflammation in Various Animal Models of PD
It is important to note that major cell types involved in the process of neuroinflammation-associated neurodegeneration are the microglia and astroglia [13]. In some parts, other types of cells can also participate in the initiation and progression of neuroinflammation-associated neurodegeneration [13, 25]. It is now further evident that various models of PD that ultimately result in the dopaminergic neurodegeneration involved the neuroinflammation and activation of resident glial cells. Almost a decade ago, intranigral delivery of lipopolysaccharide (LPS) was first shown to induce an inflammatory reaction that activated microglia and induced selective and irreversible damage to nigral dopaminergic (DA) neurons [26–28]. A study from a transgenic animal model of TNF-α expression has showed a significant loss in tyrosine hydroxylase activity and grooming behavior [29]. This study strongly supported the notion that DA neurons are intrinsically more susceptible to inflammatory stimuli. In recent years, two additional bacteriotoxin-induced inflammatory models of PD consisting of chronic low-dose LPS infusion into substantia nigra pars compacta (SNpc) of rats or intrauterine exposure to LPS were reported and shown to induce delayed, chronic, and progressive loss of DA neurons in the adult SNpc or in the offspring, respectively [30, 31]. Together, these inflammogen models of PD lend further support for a role of toxin-induced inflammation in the degeneration of the nigrostriatal pathway. Several exogenous compounds that inhibit specific protein complexes along the mitochondrial electron transport chain and cause DA neurotoxicity have been used to generate animal models of PD and all of them have a robust associated glial reaction [11, 32]. The best characterized models include 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), 6-hydroxydopamine (6-OHDA), N-methyl(R) salsolinol, rotenone, and paraquat [33, 34]. The enzyme monoamine oxidase B is present in astrocytes and microglia, where it converts MPTP into MPP+ [1-methyl-4-phenyl-pyridinium], a free radical oxidant that is taken up by DA transporters in neurons [35]. MPP+ enters the mitochondria, where it inhibits complex I function, thereby disrupting the first step in the electron transport chain required to sustain oxidative phosphorylation and triggering a circular cascade of oxidative stress that culminates in activation of the mitochondrial cell death machinery [36]. Although each neurotoxin may trigger different initial cascades of events, they all consistently involve oxidative stress as the critical mechanism that elicits the death of DA neurons [12]. Thus, general antioxidants are being intensely investigated for their ability to offer DA neuroprotection in experimental models of PD [37]. Evidence that the cycle of neuroinflammation triggered by exogenous neurotoxins persists long after the initial insult and may contribute to the progressive degeneration. Loss of DA neurons in MPTP models is associated with a glial response that peaks prior to the death of neurons [38]. MPTP is capable of inducing massive and prolonged microglia activation after single exposure in mice, monkeys, and humans [3]. Consistent with a critical role of the glial reaction in MPTP-mediated nigral neurotoxicity, anti-inflammatory drugs such as pioglitazone (a PPARγ agonist) and minocycline (a immunosuppressive and anti-inflammatory tetracycline derivative) have been shown to provide clinical benefit in MPTP-intoxicated mice. Numerous other anti-inflammatory compounds are under intense investigation as potential neuroprotective agents in experimental models of PD (Fig. 1). The neurotoxin of choice for inducing nigrostriatal degeneration in rats is the neurotoxic dopamine analog 6-hydroxydopamine (6-OHDA) and several studies support the importance of glial activity and inflammatory mediators in the neurodegenerative activity of this DA neurotoxin. 6-OHDA can be injected directly into the SN, forebrain bundle, or the striatal terminals, where it induces a retrograde degeneration and leads to the apoptotic death of DA neuron cell bodies in the SNpc [39]. The microglial activation and delayed loss of DA neurons induced by direct administration of 6-OHDA into the SN of mice can be blocked by the potent anti-inflammatory tetracycline derivative minocycline. Similarly, the cyclooxygenase-2 (COX-2) inhibitor celecoxib attenuated microglia activation induced by intrastriatal 6-OHDA administration and the delayed and progressive phase of DA neuron loss [40]. Although 6-OHDA-lesioned rats have been demonstrated to have increased levels of cytokines, in particular TNF, in both SN and striatum, the identity of the inflammatory mechanisms that mediate 6-OHDA-induced DA neurodegeneration has not been clearly established [41]. However, pharmacologic evidence implicates TNF-dependent events in death of DA neurons. Specifically, chronic pharmacological inhibition of soluble TNF signaling for 2 weeks with a dominant negative TNF inhibitor attenuated 6-OHDA-induced DA neuron loss detected at 3 weeks postlesion by approximately 50 % [41].

All chemical-induced models of PD produce inflammation. A diagrammatic representation of inflammatory mechanisms involved in PD models. All chemically induced models of PD produce inflammation in the brain and astroglial activation. Activated astroglia produces toxic inflammatory mediators that contribute to PD neurodegeneration [110]
Role of Mediators of Inflammation in the PD Pathogenesis
Evidence implicating cytokines in nigrostriatal pathway degeneration and from postmortem analyses indicated that the levels of several cytokines including TNF-α and interleukin 1-beta (IL-1β) are significantly elevated in the area of substantia nigra [42]. In the same area, maximal destruction of vulnerable melanin-containing dopamine-producing neurons occurs in PD patients [43–45]. In a research report, it has been evident that the serum concentrations of some cytokines are upregulated in the PD patients compared to the healthy individuals [19]. Among the cytokines, more importantly, are TNF and IL-1β and the downstream signaling pathways activated by them may represent common pathways on which divergent stimuli converge to elicit death of vulnerable neuronal populations through mechanisms that include mitochondrial toxicity (as described for TNF below) and caspase-dependent apoptosis or other forms of programmed cell death [46]. In addition to these, IL-1α and IL-6 are also involved in the pathophysiology of PD and play a very important role in dopaminergic neurodegeneration by the modulation of either glial or neuronal cell functions [47, 48].
Strong evidence implicates TNF-α as an early and late player in the pathophysiology of PD [49]. TNF-α signals through one of two receptors. Tnfrsf1a (formerly TNFR1) is expressed in many cell types including nigrostriatal DA neurons [42, 50]. Tnfrsf1b (formerly TNFR2) is expressed primarily by cells of the immune system including microglia, but its expression has also been reported in heart and endothelial cells as well as dopaminergic, cortical, and hippocampus neurons [51, 52]. Moreover, mice lacking TNF-α receptors are generally resistant towards dopaminergic neurodegeneration [53, 54]. Levels of TNF in the healthy adult brain are generally very low and produced primarily by neurons [52]. In contrast, high levels of TNF and soluble Tnfrsf1a have been detected in CSF and in the SN of postmortem brains of PD patients [44]. SN dopaminergic neurons are extremely sensitive to TNF [49, 51]. In addition, TNF can activate the abundant numbers of microglia in the midbrain, potentiating inflammatory responses that lead to autoamplification of ROS, NO, and superoxide radicals to form highly oxidizing peroxynitrite species [55]. TNF-dependent microglia activation in the SN creates an environment of oxidative stress through activation of NADPH oxidase that further hastens oxidative damage of DA neurons [52, 56, 57]. Lastly, single-nucleotide polymorphisms (SNPs) in the promoter of the TNF gene have been studied in 1patients with sporadic PD [58]. Collectively, findings obtained from histopathologic, genetic, and pharmacologic studies strongly implicate a role for TNF-dependent mechanisms and downstream targets in neurotoxin- and bacteriotoxin-induced loss of nigral DA neurons and suggest that high TNF levels in the midbrain may increase susceptibility for Parkinson’s disease in humans [59]. After an initial toxic insult, a self-perpetuating cycle of microglia activation would contribute to persistently elevated levels of toxic mediators in the nigral environment. Under either of these scenarios and independent of trigger that elicits its production, we hypothesize TNF action in the high oxidant environment of the SN is likely to result in (1) depletion of endogenous antioxidant capacity in vulnerable DA neurons, (2) potentiation of microglial production of ROS via NADPH oxidase-dependent pathways, and (3) activation of cell death pathways in oxidative stressed neurons (Fig. 2).

Mechanisms of oxidative stress and inflammation in Parkinson’s disease. Left side of the figure focuses on the oxidative stress-mediated pathway linked to neurodegeneration. Right side of the figure shows the inflammation-mediated pathway. Here we focus that inflammation-induced pathway to the neurodegeneration are linked to the oxidative stress leads to proteasome dysfunctions which in turn may lead to lewy body formation and alpha synuclein dysfunction resulting in the excessive oxidative damage causing neurodegeneration
It is already known that IL-1β significantly affects dopaminergic neuronal functions in many ways [45]. There is also conflicting data regarding the role of IL-1β in dopaminergic neuron survival, but due to its high concentrations in cerebrospinal fluid (CSF) and postmortem brains of PD patients, it has been implicated in the pathophysiology of the disease [9]. IL-1β has been shown to increase the intracellular calcium through IL-1β receptor in C6 rat astrocytes [60]. Moreover, IL-1β regulates C/EBP, homologous protein 10 (CHOP) transcriptional activity that, in turn, regulates a number of genes involved in the pathogenesis of PD [61]. In addition to TNF-α and IL-1β, other cytokines like IL1-α and IL6 are also predominantly involved in the process of neuroinflammation-mediated dopaminergic neurodegeneration. However, much less is known about the role of IL-1α and IL-6 in the pathogenesis of PD. One important role of IL-1α and IL-6 is that they promote the growth of neuronal terminal in the mouse [62]. Further, IL-6 and IL-6 receptor have also been involved in the regulation of VCAM-1 gene expression that is a major gene responsible for the astrocyte function [63]. Therefore, targeted inhibition of the glial reaction and inflammatory processes triggered by various stimuli may represent an attractive therapeutic approach to slow down or delay progression of PD.
If the initial trigger that triggers microglial activation is not resolved (as in the case of a genetic mutation or a prolonged or repeated environmental exposure), a loop of neuroinflammation is likely to ensue and contribute to neuronal dysfunction and eventual death of vulnerable neuronal populations [64]. In support of this idea, experimental, clinical, and epidemiological studies indicate that activation of resident microglial populations may be occurring in parallel with the neuronal dysfunction underlying the disease process in certain neurodegenerative diseases. The two common ways by which neuroinflammation induces oxidative stress are via production of high levels of ROS by activated glia such as microglia and astrocytes and via arachidonic acid signaling through the activation of cyclooxygenase and lipoxigenase (LOX) pathways [65–67]. The brain expresses both COX-1 and COX-2; COX-1 is the key cyclooxygenase enzyme in microglia that is upregulated during inflammatory responses, whereas COX-2 levels are dynamically regulated by proinflammatory mediators under physiological conditions [40, 65, 68]. In addition, COX-2 has been shown to catalyze oxidation of cytosolic DA and its expression is upregulated in neurons and astrocytes in response to CNS injury [69, 70]. Consistent with these evidences, examination of brain samples from humans or mice exposed to MPTP confirms the presence of elevated COX-2 levels and extensive DA quinone formation as well as formation of protein-bound 5-cysteinyl-DA adducts [71, 72]. COX-2 ablation or pharmacological inhibition protects against MPTP- and 6-OHDA-induced nigral DA neuron loss [73]. ProstaglandinE-2 (PGE2), produced by COX-2, can induce an intraneuronal toxic effect directly on DA neurons [70]. Prostaglandins of the J2 series also induce oxidative stress by causing a decrease in glutathione and glutathione peroxidase activity, a decrease in mitochondrial membrane potential, and overproduction of protein-bound lipid peroxidation products including acrolein and 4-hydroxy-2-nonenal (HNE) [74]. These effects suggest that prostaglandins of the J2 series are either a source of markedly increased ROS generation or modulators of ROS sensitivity. The high density of microglia in midbrain relative to other parts of the brain coupled with expression of major histocompatability complex-1 (MHC1) and beta2-microglobulin in SN, DA neurons may promote microglial-derived oxidative stress in this region of the brain by facilitating presentation of misfolded, aggregated, or oxidized proteins or protein fragments to microglia, thereby enhancing their phagocytic activity against DA neurons [75–77]. Collectively, all of the above conditions of the midbrain microenvironment may act in concert to confer increased susceptibility to the dopamine-producing neurons to inflammatory stimuli [78]. The existence of inflammatory processes in PD is supported by evidences of activated microglia, accumulation of cytokines, nuclear factor kappa B (NF-κB) pathway activation, and oxidative damage to proteins in the CSF of live patients as well as in postmortem brain samples of Parkinson’s diseases and most experimental models of PD [19, 44, 79].
Even if neuroinflammation does not occur in the early stages of DA neuron dysfunction, the release of chemoattractants by the dying DA neurons would be expected to lead to greater infiltration of the region by activated microglia coming to remove neuronal debris and may be a contributing factor in progression of disease as the respiratory bursts associated with phagocytic activities would further enhance oxidative stress for the remaining population of DA neurons. Perhaps the most convincing and compelling evidence to support the claim that inflammatory mechanisms are likely to contribute to PD risk comes from epidemiological studies. Specifically, a large prospective study of hospital workers indicated that the incidence of idiopathic Parkinson’s disease in chronic users of over-the-counter nonsteroidal anti-inflammatory drugs (NSAIDs) that scavenge free oxygen radicals and inhibit cyclooxygenase activity was 46 % lower than that of age-matched nonusers. Inhibitions of COX-mediated DA oxidation as well as inhibition of microglial-derived toxic mediator production are likely to be among the mechanisms that contribute to decreased incidence of PD in chronic NSAID users [4, 73]. This and other evidence relating to the protective effects of aspirin or other NSAIDs on DA neurons in animal models of PD as well as epidemiological data exploring the effectiveness of NSAIDs in the prevention of PD has been reviewed recently [73]. Results of these studies are less surprising in light of the fact that it is well-documented that certain neuron–glia interactions can lead to neuronal death [80]. Mechanistically, these PD risk-lowering effects of NSAIDs strongly suggest that neuroinflammatory processes contribute to DA neuron loss and development of PD in humans [13]. Although the protective effect of NSAIDs are likely to be primarily mediated by COX inhibition, multiple mechanisms, including the Rho kinase pathway, cannot be ruled out at this time in mediating the beneficial effects of NSAIDs [4].
From a neuropathological standpoint, microglial activation is indicative of an active, ongoing process of cell death [81]. While the presence of activated microglia in PD is consistent with the fact that PD is a progressive condition, their demonstration in postmortem samples from MPTP-intoxicated individuals who came to autopsy several decades after being exposed to the toxin challenges the notion that MPTP produces a ‘hit-and-run’ kind of damage [5]. Conversely, this important observation suggests that a single acute insult to the SNpc by MPTP could set in motion a self-sustained cascade of events with long-lasting deleterious effects [82]. Looking at mice injected with MPTP and killed at different time points thereafter, it appears that the time course of reactive astrocyte formation parallels that of dopaminergic structure destruction in both the striatum and the SNpc, and that GFAP expression remains upregulated even after the main wave of neuronal death has passed [78, 83]. This is supported by the demonstration that blockade of MPP+ uptake into dopaminergic neurons completely prevents not only SNpc dopaminergic neuronal death but also GFAP upregulation [78]. Remarkably, activation of microglial cells, which is also quite strong in the MPTP mouse model, occurs much earlier than that of astrocytes and, more importantly, reaches a maximum before the peak of dopaminergic neurodegeneration [13].
The Role of Oxidative Stress in the PD Pathophysiology
At the cellular level, a significant body of evidences supported the “oxidative stress hypothesis” for initiation of nigral dopamine-producing neuronal loss [12, 84, 85]. Oxidative stress generate when there is an intracellular accumulation of reactive oxygen and nitrogen species (ROS/RNS) due to reduced endogenous antioxidant capacity and/or overproduction of ROS within the cell [12, 86]. Clearly, all aerobic organisms are susceptible towards oxidative stress because ROS (primarily superoxide and hydrogen peroxide) are produced by mitochondria during respiration [87, 88]. In fact, the brain is considered to be abnormally sensitive to oxidative damage, in part, because oxygen consumption by the brain constitutes 20 % of the total oxygen consumption in the body, and the brain is enriched in the more easily peroxidizable fatty acids while its antioxidant defenses (such as catalase, superoxide dismutase, glutathione, and glutathione peroxidase) are relatively sparse [86, 89]. It is very important that the SN appears to be among the most vulnerable regions primarily because it operates under a prooxidative state relative to other parts of the brain even in healthy individuals [90]. Specifically, the substantia nigra has a high metabolic rate combined with a high content of oxidizable species that include DA, DA-derived ROS, neuromelanin, polyunsaturated fatty acids, iron, and a low content of antioxidants (glutathione in particular), all of which render this brain region highly vulnerable to the effects of peroxynitrite and sulfite [86]. In summary, evidence of enhanced oxidative stress in the brains of PD patients includes increased oxidation of lipids, DNA, and proteins and has been documented in a large number of studies [91]. Further, in support of oxidative stress, extensive evidence also supports the involvement of impaired mitochondrial function in PD [92].
Mitochondria generate ROS as by-products of molecular oxygen consumption in the electron transport chain. Mitochondrial dysfunction of complex I of the respiratory transport chain has been reported from PD brain, particularly in the substantia nigra. A defect in complex I function would likely increase production of superoxide anion and free radical production by impairing electron flow from NADH to ubiquinone, building up oxidative stress through subsequent superoxide dismutase. Mutations in somatic mitochondrial DNA have been reported in PD brain and also in the aging brain. These findings are important because the mitochondrial DNA encodes components of the respiratory transport chain complexes, and such mutations may impair efficient electron flow from NADH to molecular oxygen. Oxidative phosphorylation in the substantia nigra has been shown to become increasingly impaired as a function of age. Clinically, untreated PD patients display decreased mitochondrial activity of complexes I and I/III [93]. It has been proposed that DA neurons may have an intrinsic sensitivity to complex I defects or agents that compromise its function on the basis of studies that demonstrate selective toxicity of the pesticide rotenone for DA neurons despite the fact that rotenone inhibits mitochondrial complex I throughout the brain [93]. Homozygous mutations linked to familial PD affect the activity of its kinase domain that has high homology to that of Ca2+/calmodulin-dependent serine/threonine kinases and prevent its ability to preserve mitochondrial membrane potential in conditions known to compromise mitochondrial integrity [93–95]. Among the mediators of neuroinflammation, NO contributes to DA neuron death through mechanisms that, while not completely understood, are likely to involve mediation of excitotoxicity, activation of PARP-1 (poly-ADP ribose polymerase), DNA damage, activation of caspase-dependent and independent cell death, and/or nitrosylation of proteins including α-synuclein and Parkin [86]. Mitochondria are not only sources of ROS but have also been shown to be subcellular targets of cytokines such as TNF that promote overproduction of ROS in mitochondria [96]. Glutathione is the only antioxidant in the cell available to metabolize hydrogen peroxide and a small fraction of the total cellular GSH pool is sequestered in mitochondria (mGSH) by the action of a carrier that transports it from the cytosol to the mitochondrial matrix [97]. Other evidence consistent with an important role of mitochondrial function in the etiology of PD includes the observation that inhibition of the mitochondrial respiratory complex I after exposure to the neurotoxin MPTP gives rise to PD-like pathologies in humans, non-human primates, mice, and rats [93, 98, 99].
Particularly, here, it is important to know that astroglial-mediated inflammatory and oxidative stress mechanism may be more important than the microglia or neurons, since they are the more abundant cell type in the brain and extensively involved in the nourishment of neurons. A small change in the surrounding astroglial cells may effectively cause neuronal cell death compared to any other type of the cells in the brain. In addition to the multiple factors such as cytokines and fatty acid metabolites produced from activated astroglia cells, ROS and nitrite may be the key mediator of glia-facilitated MPTP neurotoxicity. Dopaminergic neurons in the nigra are known to be particularly vulnerable to oxidative stress, presumably due to their lower antioxidant capacity, increased accumulation of ion and oxidation-prone dopamine (DA), and possible defects in mitochondria. In addition to oxidative and nitrative stresses, Ca2++ ion release in astrocytes is an important event and involved in the regulation of a number of mechanisms including release of S100B and other neurotoxins. However, earlier studies have reported alteration in the calcium ion homeostasis in PD, but the state of intracellular Ca2++ ion level in astrocytes in response to MPTP and melatonin was not demonstrated (Fig. 2).
Astrocytes Play an Important and Distinct Role in the Initiation and Progression of Parkinson’s Disease
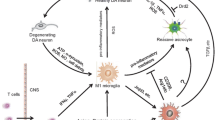
Astrocytes play direct, important, active, and critical roles in mediating neuronal survival and function in Parkinson’s disease (Fig. 3). [100]. This role of astrocytes is well accepted in the progression of amyotrophic lateral sclerosis (ALS), in which the removal of glutamate from the extracellular space by astrocytes confers neuroprotection, whereas astrocytic release of soluble, potentially toxic molecules promotes neurodegeneration [101]. In recent years, this context-dependent dual role of astrocytes has also been documented in experimental models of PD [100]. Astrocytes, in particular, are more susceptible and recognize various signals from activated microglia in forms of soluble chemokines and cytokines and function differentially [13]. Particularly, here, it is important to know that astroglial-mediated inflammatory and oxidative stress mechanism may be more important than the microglia or neurons, since they are the more abundant cell type in the brain and extensively involved in the nourishment of the neurons [102]. A small change in the surrounding astroglial cells may effectively cause neuronal cell death compared to any other type of cells in the brain. More recently, we have demonstrated a distinct role of astrocyte-mediated oxidative stress and inflammation in the models of PD [102].

Astrocytes play an important role in the neuronal death in PD. Here, we want to stress that altered astrocyte functions may be an important mechanism in the initiation and progression of dopaminergic neurons. Microglia and other microenvironment in the brain can assist in trigging astrocyte function that generates a massive quality of neurotoxic factors and lack of growth factors to the neurons causing, ultimately, dopaminergic neuronal demise. Figure shows that astrocytes functions are altered in the parkinson's disease pathophysiology. As described in the top portion of the figure, chemically induced models of PD shows increase in the production of inflammatory meditators from microglia. These inflammatory mediaters alone or in conjugation of other agents may trigger altered astrocytes functions leading to neurodegeneration
The neuroprotective and neurodegenerative functions of astrocytes depend largely on the molecules that they release into and uptake from the extracellular space, also sometimes described as the microenvironment that astrocytes and neurons commonly share [100]. For example, it is well documented that astrocytes can release and supply neurons with neurotrophic factors such as nerve growth factor (NGF), glial cell line-derived neurotrophic factor (GDNF), and mesencephalic astrocyte-derived neurotrophic factor (MANF), neurotrophin-3, and basic fibroblast growth factor (bFGF) as well as metabolic substrates such as lactate and the antioxidant glutathione for the survival and proper functioning of neurons [100, 103]. Astrocytes also confer neuroprotection by siphoning away excess extracellular excitotoxic agents such as glutamate, potassium, and calcium. On the other hand, when astrocytes undergo a state of gliosis in response to neuronal injury or toxic insults, together with microglia, they release cytokines and chemokines that are deleterious to neurons. In addition to the multiple factors such as fatty acid metabolites, cytokines produced from activated astroglial cells, reactive oxygen species (ROS), and nitrite may be the key mediator of glia-facilitated MPTP neurotoxicity [104]. In addition to oxidative and nitrative stresses, Ca2++ ion release in astrocytes is an important event and is involved in the regulation of a number of mechanisms including release of S100B and other neurotoxins [105] (Fig. 3).
In addition to these functions, there have been some more advances in the recent understanding of astroglial role in the pathogenesis of Parkinson’s disease. In a recent study, it has been confirmed that astroglial-derived GDNF is a potent inhibitor of microglial activation, suggesting an important and regulatory role of astroglial cells [103]. Another recent study based on the genomic analysis of the astroglia shows that there are two different kinds of reactive astrogliosis that means there may be more that one kind of phenotypes existing in pathogenesis [106]. A study that provides a direct role of astrocytes in neuroinflammation-associated pathophysiology in Parkinson’s disease has also been recently confirmed [107]. Interestingly, it has been shown that interferon regulatory factor 3 inhibits astrocyte inflammatory gene expression through suppression of the proinflammatory miR-155 and miR-155, exploring newer targets to reduce astrocyte-specific neuroinflammation [108]. More recently, it has been shown that cobalt(II) beta-ketoaminato complexes can be novel inhibitors of astrocyte-specific neuroinflammation [109]. Importantly, it has been demonstrated that alteration in Parkinson-associated gene DJ-1 mutation is directly linked to astrocyte neuroinflammation [110].
Conclusion
In conclusion, in this review, we have tried to give a perspective on the wide variety of interactions between inflammatory mediators with etiology of Parkinson’s disease. Inflammatory and oxidative stress mechanisms are likely to play an important role in the neurodegenerative pathogenesis of PD. Defects in mitochondrial functions, suggested by the presence of mutations in mitochondrial DNA and impaired respiratory transport chain function in PD brain further provides connection to redox metabolism to PD. Understanding the role of, particularly, astrocytes in PD pathology may lead to development of mechanism-based therapeutics and improved pharmacotherapy for PD.
Abbreviations
- NGF:
-
Nerve growth factor
- GDNF:
-
Glial cell line-derived neurotrophic factor
- MANF:
-
Mesencephalic astrocyte-derived neurotrophic factor
- bFGF:
-
Basic fibroblast growth factor
- PD:
-
Parkinson’s disease
- ROS:
-
Reactive oxygen species
- RNS:
-
Reactive nitrogen species
- TNF-α:
-
Tumor necrosis factor-α
- NF-κB:
-
Nuclear factor kappa-B
- COX-2:
-
Cyclooxygenase-2
- GFAP:
-
Glial fibrillary acidic protein
- CHOP:
-
C/EBP homologous protein 10
- iNOS:
-
Inducible nitric oxide synthase
- IL-1α:
-
Interleukin-1α
- IL-1β:
-
Interleukin-1β
- IL-6:
-
Interleukin-6
- P-p38 MAPK:
-
Phosphorylated p38 mitogen-activated protein kinase
- NO:
-
Nitrite
References
Minagar A, Shapshak P, Fujimura R, Ownby R, Heyes M, Eisdorfer C (2002) The role of macrophage/microglia and astrocytes in the pathogenesis of three neurologic disorders: HIV-associated dementia, Alzheimer disease, and multiple sclerosis. J Neurol Sci 202:13–23
Balasingam V, Tejada-Berges T, Wright E, Bouckova R, Yong VW (1994) Reactive astrogliosis in the neonatal mouse brain and its modulation by cytokines. J Neurosci 14:846–856
Hu X, Zhang D, Pang H, Caudle WM, Li Y, Gao H, Liu Y, Qian L, Wilson B, Di Monte DA, Ali SF, Zhang J, Block ML, Hong JS (2008) Macrophage antigen complex-1 mediates reactive microgliosis and progressive dopaminergic neurodegeneration in the MPTP model of Parkinson’s disease. J Immunol 181:7194–7204
Asanuma M, Miyazaki I (2008) Nonsteroidal anti-inflammatory drugs in experimental parkinsonian models and Parkinson’s disease. Curr Pharm Des 14:1428–1434
Esposito G, Scuderi C, Savani C, Steardo L Jr, De Filippis D, Cottone P, Iuvone T, Cuomo V, Steardo L (2007) Cannabidiol in vivo blunts beta-amyloid induced neuroinflammation by suppressing IL-1beta and iNOS expression. Br J Pharmacol 151:1272–1279
Wight RD, Tull CA, Deel MW, Stroope BL, Eubanks AG, Chavis JA, Drew PD, Hensley LL Resveratrol effects on astrocyte function: relevance to neurodegenerative diseases. Biochemical and biophysical research communications 426:112-115
Maccioni RB, Rojo LE, Fernandez JA, Kuljis RO (2009) The role of neuroimmunomodulation in Alzheimer’s disease. Ann N Y Acad Sci 1153:240–246
Sekiyama K, Sugama S, Fujita M, Sekigawa A, Takamatsu Y, Waragai M, Takenouchi T, Hashimoto M (2012) Neuroinflammation in Parkinson's disease and related disorders: a lesson from genetically manipulated mouse models of alpha-synucleinopathies. Parkinson's Dis 2012:271732
Tansey MG, Goldberg MS (2009) Neuroinflammation in Parkinson’s disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol Dis
Albrecht S, Buerger E (2009) Potential neuroprotection mechanisms in PD: focus on dopamine agonist pramipexole. Curr Med Res Opin 25:2977–2987
Klegeris A, McGeer PL (2000) R-(−)-Deprenyl inhibits monocytic THP-1 cell neurotoxicity independently of monoamine oxidase inhibition. Exp Neurol 166:458–464
Guillot TS, Richardson JR, Wang MZ, Li YJ, Taylor TN, Ciliax BJ, Zachrisson O, Mercer A, Miller GW (2008) PACAP38 increases vesicular monoamine transporter 2 (VMAT2) expression and attenuates methamphetamine toxicity. Neuropeptides 42:423–434
Mena MA, Garcia de Yebenes J (2008) Glial cells as players in parkinsonism: the “good”, the “bad”, and the “mysterious” glia. Neuroscientist Rev J Bringing Neurobiol Neurol Psychiatry 14:544–560
Niranjan R, Kamat PK, Nath C, Shukla R (2010) Evaluation of guggulipid and nimesulide on production of inflammatory mediators and GFAP expression in LPS stimulated rat astrocytoma, cell line (C6). J Ethnopharmacol 127:625–630
Solano RM, Casarejos MJ, Menendez-Cuervo J, Rodriguez-Navarro JA, Garcia de Yebenes J, Mena MA (2008) Glial dysfunction in parkin null mice: effects of aging. J Neurosci 28:598–611
Tambuyzer BR, Ponsaerts P, Nouwen EJ (2009) Microglia: gatekeepers of central nervous system immunology. J Leukoc Biol 85:352–370
Niranjan R, Nath C, Shukla R (2011) Guggulipid and nimesulide differentially regulated inflammatory genes mRNA expressions via inhibition of NF-κB and CHOP activation in LPS-stimulated rat astrocytoma cells, C6. Cell Mol Neurobiol 31:755–764
Rogers J, Mastroeni D, Leonard B, Joyce J, Grover A (2007) Neuroinflammation in Alzheimer’s disease and Parkinson’s disease: are microglia pathogenic in either disorder? Int Rev Neurobiol 82:235–246
Brodacki B, Staszewski J, Toczylowska B, Kozlowska E, Drela N, Chalimoniuk M, Stepien A (2008) Serum interleukin (IL-2, IL-10, IL-6, IL-4), TNFalpha, and INFgamma concentrations are elevated in patients with atypical and idiopathic parkinsonism. Neurosci Lett 441:158–162
Dheen ST, Kaur C, Ling EA (2007) Microglial activation and its implications in the brain diseases. Curr Med Chem 14:1189–1197
Khandhar SM, Marks WJ (2007) Epidemiology of Parkinson’s disease. Dis Mon 53:200–205
Ton TG, Heckbert SR, Longstreth WT Jr, Rossing MA, Kukull WA, Franklin GM, Swanson PD, Smith-Weller T, Checkoway H (2006) Nonsteroidal anti-inflammatory drugs and risk of Parkinson’s disease. Mov Disord Off J Mov Disord Soc 21:964–969
Etminan M, Carleton BC, Samii A (2008) Non-steroidal anti-inflammatory drug use and the risk of Parkinson disease: a retrospective cohort study. J Clin Neurosci 15:576–577
van Staa TP, Smeeth L, Persson I, Parkinson J, Leufkens HG (2008) What is the harm-benefit ratio of Cox-2 inhibitors? Int J Epidemiol 37:405–413
Hirsch EC, Hunot S, Damier P, Faucheux B (1998) Glial cells and inflammation in Parkinson’s disease: a role in neurodegeneration? Ann Neurol 44:S115–S120
Castano A, Herrera AJ, Cano J, Machado A (2002) The degenerative effect of a single intranigral injection of LPS on the dopaminergic system is prevented by dexamethasone, and not mimicked by rh-TNF-alpha, IL-1beta and IFN-gamma. J Neurochem 81:150–157
Tian YY, An LJ, Jiang L, Duan YL, Chen J, Jiang B (2006) Catalpol protects dopaminergic neurons from LPS-induced neurotoxicity in mesencephalic neuron-glia cultures. Life Sci 80:193–199
Santiago M, Hernandez-Romero MC, Machado A, Cano J (2009) Zocor Forte (simvastatin) has a neuroprotective effect against LPS striatal dopaminergic terminals injury, whereas against MPP+ does not. Eur J Pharmacol 609:58–64
Aloe L, Fiore M (1997) TNF-alpha expressed in the brain of transgenic mice lowers central tyroxine hydroxylase immunoreactivity and alters grooming behavior. Neurosci Lett 238:65–68
Wenk GL, McGann-Gramling K, Hauss-Wegrzyniak B, Ronchetti D, Maucci R, Rosi S, Gasparini L, Ongini E (2004) Attenuation of chronic neuroinflammation by a nitric oxide-releasing derivative of the antioxidant ferulic acid. J Neurochem 89:484–493
Carvey PM, Chang Q, Lipton JW, Ling Z (2003) Prenatal exposure to the bacteriotoxin lipopolysaccharide leads to long-term losses of dopamine neurons in offspring: a potential, new model of Parkinson’s disease. Front Biosci 8:s826–s837
Lane EL, Soulet D, Vercammen L, Cenci MA, Brundin P (2008) Neuroinflammation in the generation of post-transplantation dyskinesia in Parkinson’s disease. Neurobiol Dis 32:220–228
Grunblatt E, Mandel S, Youdim MB (2000) MPTP and 6-hydroxydopamine-induced neurodegeneration as models for Parkinson’s disease: neuroprotective strategies. J Neurol 247(Suppl 2):II95–II102
Meredith GE, Totterdell S, Potashkin JA, Surmeier DJ (2008) Modeling PD pathogenesis in mice: advantages of a chronic MPTP protocol. Parkinsonism Relat Disord 14(Suppl 2):S112–S115
Siddiqui A, Mallajosyula JK, Rane A, Andersen JK Ability to delay neuropathological events associated with astrocytic MAO-B increase in a parkinsonian mouse model: implications for early intervention on disease progression. Neurobiology of disease 40:444-448
Speciale SG (2002) MPTP: insights into parkinsonian neurodegeneration. Neurotoxicol Teratol 24:607–620
Prasad KN, Cole WC, Kumar B (1999) Multiple antioxidants in the prevention and treatment of Parkinson’s disease. J Am Coll Nutr 18:413–423
Bjarkam CR, Nielsen MS, Glud AN, Rosendal F, Mogensen P, Bender D, Doudet D, Moller A, Sorensen JC (2008) Neuromodulation in a minipig MPTP model of Parkinson disease. Br J Neurosurg 22(Suppl 1):S9–S12
Wilms H, Zecca L, Rosenstiel P, Sievers J, Deuschl G, Lucius R (2007) Inflammation in Parkinson’s diseases and other neurodegenerative diseases: cause and therapeutic implications. Curr Pharm Des 13:1925–1928
Reksidler AB, Lima MM, Zanata SM, Machado HB, da Cunha C, Andreatini R, Tufik S, Vital MA (2007) The COX-2 inhibitor parecoxib produces neuroprotective effects in MPTP-lesioned rats. Eur J Pharmacol 560:163–175
Ros-Bernal F, Hunot S, Herrero MT, Parnadeau S, Corvol JC, Lu L, Alvarez-Fischer D, Carrillo-de Sauvage MA, Saurini F, Coussieu C, Kinugawa K, Prigent A, Hoglinger G, Hamon M, Tronche F, Hirsch EC, Vyas S Microglial glucocorticoid receptors play a pivotal role in regulating dopaminergic neurodegeneration in parkinsonism. Proc Natl Acad Sci USA 108:6632-6637
Pieper HC, Evert BO, Kaut O, Riederer PF, Waha A, Wullner U (2008) Different methylation of the TNF-alpha promoter in cortex and substantia nigra: Implications for selective neuronal vulnerability. Neurobiol Dis 32:521–527
Bessler H, Djaldetti R, Salman H, Bergman M, Djaldetti M (1999) IL-1 beta, IL-2, IL-6 and TNF-alpha production by peripheral blood mononuclear cells from patients with Parkinson’s disease. Biomed Pharmacother 53:141–145
Nagatsu T, Mogi M, Ichinose H, Togari A (2000) Changes in cytokines and neurotrophins in Parkinson’s disease. J Neural Transm Suppl:277-290
Bian MJ, Li LM, Yu M, Fei J, Huang F (2009) Elevated interleukin-1beta induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine aggravating dopaminergic neurodegeneration in old male mice. Brain Res 1302:256–264
Kraft C, Reggiori F, Peter M (2009) Selective types of autophagy in yeast. Biochim Biophys Acta 1793:1404–1412
Hofmann KW, Schuh AF, Saute J, Townsend R, Fricke D, Leke R, Souza DO, Portela LV, Chaves ML, Rieder CR (2009) Interleukin-6 serum levels in patients with Parkinson’s disease. Neurochem Res 34:1401–1404
Allan SM, Pinteaux E (2003) The interleukin-1 system: an attractive and viable therapeutic target in neurodegenerative disease. Curr Drug Targets CNS Neurol Disord 2:293–302
McCoy MK, Ruhn KA, Blesch A, Tansey MG TNF: a key neuroinflammatory mediator of neurotoxicity and neurodegeneration in models of Parkinson’s disease. Advances in experimental medicine and biology 691:539-540
Gomez-Santos C, Ferrer I, Santidrian AF, Barrachina M, Gil J, Ambrosio S (2003) Dopamine induces autophagic cell death and alpha-synuclein increase in human neuroblastoma SH-SY5Y cells. J Neurosci Res 73:341–350
Ramirez SH, Hasko J, Skuba A, Fan S, Dykstra H, McCormick R, Reichenbach N, Krizbai I, Mahadevan A, Zhang M, Tuma R, Son YJ, Persidsky Y (2012) Activation of cannabinoid receptor 2 attenuates leukocyte-endothelial cell interactions and blood–brain barrier dysfunction under inflammatory conditions. J Neurosci Off J Soc Neurosci 32:4004–4016
Tansey MG, Frank-Cannon TC, McCoy MK, Lee JK, Martinez TN, McAlpine FE, Ruhn KA, Tran TA (2008) Neuroinflammation in Parkinson’s disease: is there sufficient evidence for mechanism-based interventional therapy? Front Biosci 13:709–717
Hirsch EC, Breidert T, Rousselet E, Hunot S, Hartmann A, Michel PP (2003) The role of glial reaction and inflammation in Parkinson’s disease. Ann N Y Acad Sci 991:214–228
Sriram K, Lin GX, Jefferson AM, Roberts JR, Chapman RS, Chen BT, Soukup JM, Ghio AJ, Antonini JM Dopaminergic neurotoxicity following pulmonary exposure to manganese-containing welding fumes. Arch Toxicol 84:521-540
Breedveld FC (2005) Tumour necrosis factor antagonists: infliximab, adalimumab and etanercept. Ned Tijdschr Geneeskd 149:2273–2277
Cheret C, Gervais A, Lelli A, Colin C, Amar L, Ravassard P, Mallet J, Cumano A, Krause KH, Mallat M (2008) Neurotoxic activation of microglia is promoted by a nox1-dependent NADPH oxidase. J Neurosci 28:12039–12051
Li B, Guo YS, Sun MM, Dong H, Wu SY, Wu DX, Li CY (2008) The NADPH oxidase is involved in lipopolysaccharide-mediated motor neuron injury. Brain Res 1226:199–208
Liang X, Wang Q, Hand T, Wu L, Breyer RM, Montine TJ, Andreasson K (2005) Deletion of the prostaglandin E2 EP2 receptor reduces oxidative damage and amyloid burden in a model of Alzheimer’s disease. J Neurosci 25:10180–10187
Sethi V, Yousry TA, Muhlert N, Ron M, Golay X, Wheeler-Kingshott C, Miller DH, Chard DT (2012) Improved detection of cortical MS lesions with phase-sensitive inversion recovery MRI. J Neurol Neurosurg Psychiatry 83:877–882
Ward RJ, Lallemand F, de Witte P, Crichton RR, Piette J, Tipton K, Hemmings K, Pitard A, Page M, Della Corte L, Taylor D, Dexter D Anti-inflammatory actions of a taurine analogue, ethane beta-sultam, in phagocytic cells, in vivo and in vitro. Biochem Pharmacol 81:743-751
Svotelis A, Doyon G, Bernatchez G, Desilets A, Rivard N, Asselin C (2005) IL-1 beta-dependent regulation of C/EBP delta transcriptional activity. Biochem Biophys Res Commun 328:461–470
Parish CL, Finkelstein DI, Tripanichkul W, Satoskar AR, Drago J, Horne MK (2002) The role of interleukin-1, interleukin-6, and glia in inducing growth of neuronal terminal arbors in mice. J Neurosci 22:8034–8041
Purohit DP, Perl DP, Haroutunian V, Powchik P, Davidson M, Davis KL (1998) Alzheimer disease and related neurodegenerative diseases in elderly patients with schizophrenia: a postmortem neuropathologic study of 100 cases. Arch Gen Psychiatry 55:205–211
Chen LC, Smith A, Ben Y, Zukic B, Ignacio S, Moore D, Lee N (2004) Temporal gene expression patterns in G93A/SOD1 mouse. Amyotroph Lateral Scler Other Mot Neuron Disord 5:164–171
Stefanova N, Kaufmann WA, Humpel C, Poewe W, Wenning GK (2012) Systemic proteasome inhibition triggers neurodegeneration in a transgenic mouse model expressing human alpha-synuclein under oligodendrocyte promoter: implications for multiple system atrophy. Acta Neuropathol 124:51–65
Hensley K, Mhatre M, Mou S, Pye QN, Stewart C, West M, Williamson KS (2006) On the relation of oxidative stress to neuroinflammation: lessons learned from the G93A-SOD1 mouse model of amyotrophic lateral sclerosis. Antioxid Redox Signal 8:2075–2087
Klegeris A, McGeer PL (2002) Cyclooxygenase and 5-lipoxygenase inhibitors protect against mononuclear phagocyte neurotoxicity. Neurobiol Aging 23:787–794
Choi SH, Bosetti F (2009) Cyclooxygenase-1 null mice show reduced neuroinflammation in response to beta-amyloid. Aging (Albany NY) 1:234–244
Nogawa S, Zhang F, Ross ME, Iadecola C (1997) Cyclo-oxygenase-2 gene expression in neurons contributes to ischemic brain damage. J Neurosci 17:2746–2755
Consilvio C, Vincent AM, Feldman EL (2004) Neuroinflammation, COX-2, and ALS—a dual role? Exp Neurol 187:1–10
Teismann P, Tieu K, Choi DK, Wu DC, Naini A, Hunot S, Vila M, Jackson-Lewis V, Przedborski S (2003) Cyclooxygenase-2 is instrumental in Parkinson’s disease neurodegeneration. Proc Natl Acad Sci USA 100:5473–5478
Minghetti L (2004) Cyclooxygenase-2 (COX-2) in inflammatory and degenerative brain diseases. J Neuropathol Exp Neurol 63:901–910
Iuvone T, Esposito G, De Filippis D, Bisogno T, Petrosino S, Scuderi C, Di Marzo V, Steardo L (2007) Cannabinoid CB1 receptor stimulation affords neuroprotection in MPTP-induced neurotoxicity by attenuating S100B up-regulation in vitro. J Mol Med 85:1379–1392
Tzeng SF, Hsiao HY, Mak OT (2005) Prostaglandins and cyclooxygenases in glial cells during brain inflammation. Curr Drug Targets Inflamm Allergy 4:335–340
Yasuda Y, Shinagawa R, Yamada M, Mori T, Tateishi N, Fujita S (2007) Long-lasting reactive changes observed in microglia in the striatal and substantia nigral of mice after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Brain Res 1138:196–202
Kim HJ, Fan X, Gabbi C, Yakimchuk K, Parini P, Warner M, Gustafsson JA (2008) Liver X receptor beta (LXRbeta): a link between beta-sitosterol and amyotrophic lateral sclerosis–Parkinson’s dementia. Proc Natl Acad Sci USA 105:2094–2099
Sugama S, Takenouchi T, Kitani H, Fujita M, Hashimoto M (2009) Microglial activation is inhibited by corticosterone in dopaminergic neurodegeneration. J Neuroimmunol 208:104–114
Aoki E, Yano R, Yokoyama H, Kato H, Araki T (2009) Role of nuclear transcription factor kappa B (NF-kappaB) for MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahyropyridine)-induced apoptosis in nigral neurons of mice. Exp Mol Pathol 86:57–64
Gatev P, Wichmann T (2009) Interactions between cortical rhythms and spiking activity of single basal ganglia neurons in the normal and parkinsonian state. Cereb Cortex 19:1330–1344
Graeber MB, Streit WJ Microglia: biology and pathology. Acta Neuropathol 119:89-105
McGeer PL, McGeer EG (2008) Glial reactions in Parkinson’s disease. Mov Disord 23:474–483
Vroon A, Drukarch B, Bol JG, Cras P, Breve JJ, Allan SM, Relton JK, Hoogland PV, Van Dam AM (2007) Neuroinflammation in Parkinson’s patients and MPTP-treated mice is not restricted to the nigrostriatal system: microgliosis and differential expression of interleukin-1 receptors in the olfactory bulb. Exp Gerontol 42:762–771
Schneider JS, Denaro FJ (1988) Astrocytic responses to the dopaminergic neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) in cat and mouse brain. J Neuropathol Exp Neurol 47:452–458
Yuan H, Zheng JC, Liu P, Zhang SF, Xu JY, Bai LM (2007) Pathogenesis of Parkinson’s disease: oxidative stress, environmental impact factors and inflammatory processes. Neurosci Bull 23:125–130
Katunina EA, Malykhina EA, Kuznetsov NV, Avakian GN, Gusev E, Nerobkova LN, Voronina TA, Barskov IV (2006) Antioxidants in complex treatment of Parkinson’s disease. Zh Nevrol Psikhiatr Im S S Korsakova 106:22–28
Zhou C, Huang Y, Przedborski S (2008) Oxidative stress in Parkinson’s disease: a mechanism of pathogenic and therapeutic significance. Ann N Y Acad Sci 1147:93–104
Jenner P (2003) Oxidative stress in Parkinson's disease. Ann Neurol 53(Suppl 3):S26–S36, discussion S36-28
Ahmed M, Luggen M, Herman JH, Weiss KL, Decourten-Myers G, Quinlan JG, Khanna D (2006) Hypertrophic pachymeningitis in rheumatoid arthritis after adalimumab administration. J Rheumatol 33:2344–2346
Favier A (2006) Oxidative stress in human diseases. Ann Pharm Fr 64:390–396
Lee WS, Tsai WJ, Yeh PH, Wei BL, Chiou WF (2006) Divergent role of calcium on Abeta- and MPTP-induced cell death in SK-N-SH neuroblastoma. Life Sci 78:1268–1275
Ortiz-Ortiz MA, Moran JM, Bravosanpedro JM, Gonzalez-Polo RA, Niso-Santano M, Anantharam V, Kanthasamy AG, Soler G, Fuentes JM (2009) Curcumin enhances paraquat-induced apoptosis of N27 mesencephalic cells via the generation of reactive oxygen species. Neurotoxicology 30:1008–1018
Fukae J, Mizuno Y, Hattori N (2007) Mitochondrial dysfunction in Parkinson’s disease. Mitochondrion 7:58–62
Beal MF (2003) Mitochondria, oxidative damage, and inflammation in Parkinson’s disease. Ann N Y Acad Sci 991:120–131
Jung BD, Shin EJ, Nguyen XK, Jin CH, Bach JH, Park SJ, Nah SY, Wie MB, Bing G, Kim HC Potentiation of methamphetamine neurotoxicity by intrastriatal lipopolysaccharide administration. Neurochem Int 56:229-244
Akundi RS, Huang Z, Eason J, Pandya JD, Zhi L, Cass WA, Sullivan PG, Bueler H Increased mitochondrial calcium sensitivity and abnormal expression of innate immunity genes precede dopaminergic defects in Pink1-deficient mice. PloS one 6:e16038
Hunter RL, Dragicevic N, Seifert K, Choi DY, Liu M, Kim HC, Cass WA, Sullivan PG, Bing G (2007) Inflammation induces mitochondrial dysfunction and dopaminergic neurodegeneration in the nigrostriatal system. J Neurochem 100:1375–1386
Lee M, Kwon BM, Suk K, McGeer E, McGeer PL Effects of obovatol on GSH depleted glia-mediated neurotoxicity and oxidative damage. Journal of neuroimmune pharmacology : the official journal of the Society on NeuroImmune Pharmacology 7:173-186
Drouin-Ouellet J, Gibrat C, Bousquet M, Calon F, Kriz J, Cicchetti F The role of the MYD88-dependent pathway in MPTP-induced brain dopaminergic degeneration. Journal of neuroinflammation 8:137
Niranjan R, Nath C, Shukla R The mechanism of action of MPTP-induced neuroinflammation and its modulation by melatonin in rat astrocytoma cells, C6. Free radical research 44:1304-1316
Rappold PM, Tieu K (2010) Astrocytes and therapeutics for Parkinson’s disease. Neurother J Am Soc Exp Neuro Ther 7:413–423
Hauser DN, Cookson MR (2011) Astrocytes in Parkinson’s disease and DJ-1. J Neurochem 117:357–358
Niranjan R, Rajasekar N, Nath C, Shukla R (2012) The effect of guggulipid and nimesulide on MPTP-induced mediators of neuroinflammation in rat astrocytoma cells, C6. Chemico-biological interactions
Rocha SM, Cristovao AC, Campos FL, Fonseca CP, Baltazar G (2012) Astrocyte-derived GDNF is a potent inhibitor of microglial activation. Neurobiol Dis 47:407–415
Niranjan R, Nath C, Shukla R (2012) Melatonin attenuated mediators of neuroinflammation and alpha-7 nicotinic acetylcholine receptor mRNA expression in lipopolysaccharide (LPS) stimulated rat astrocytoma cells, C6. Free Radic Res 46:1167–1177
Niranjan R, Nath C, Shukla R (2010) The mechanism of action of MPTP-induced neuroinflammation and its modulation by melatonin in rat astrocytoma cells, C6. Free Radic Res 44:1304–1316
Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, Barres BA (2012) Genomic analysis of reactive astrogliosis. J Neurosci Off J Soc Neurosci 32:6391–6410
Karpuk N, Burkovetskaya M, Kielian T (2012) Neuroinflammation alters voltage-dependent conductance in striatal astrocytes. J Neurophysiol 108:112–123
Tarassishin L, Loudig O, Bauman A, Shafit-Zagardo B, Suh HS, Lee SC (2011) Interferon regulatory factor 3 inhibits astrocyte inflammatory gene expression through suppression of the proinflammatory miR-155 and miR-155*. Glia 59:1911–1922
Madeira JM, Beloukhina N, Boudreau K, Boettcher TA, Gurley L, Walker DG, McNeil WS, Klegeris A (2012) Cobalt(II) beta-ketoaminato complexes as novel inhibitors of neuroinflammation. Eur J Pharmacol 676:81–88
Waak J, Weber SS, Waldenmaier A, Gorner K, Alunni-Fabbroni M, Schell H, Vogt-Weisenhorn D, Pham TT, Reumers V, Baekelandt V, Wurst W, Kahle PJ (2009) Regulation of astrocyte inflammatory responses by the Parkinson’s disease-associated gene DJ-1. FASEB J Off Publ Fed Am Soc Exp Biol 23:2478–2489
Acknowledgments
Lab space and facilities provided by Dr. Anil Mishra at the School of Medicine, Division of Gastroenterology and Liver Disease, Case Western Reserve University, Cleveland, OH, USA, is gratefully acknowledged.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Niranjan, R. The Role of Inflammatory and Oxidative Stress Mechanisms in the Pathogenesis of Parkinson’s Disease: Focus on Astrocytes. Mol Neurobiol 49, 28–38 (2014). https://doi.org/10.1007/s12035-013-8483-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-013-8483-x