
Abstract
Parkinson disease is an inexorably progressive neurodegenerative disorder. Multiple attempts have been made to establish therapies for Parkinson disease which provide neuroprotection or disease modification—two related, but not identical, concepts. However, to date, none of these attempts have succeeded. Many challenges exist in this field of research, including a complex multisystem disorder that includes dopaminergic and non-dopaminergic features; poorly understood and clearly multifaceted disease pathogenic mechanisms; a lack of reliable animal models; an absence of effective biomarkers of disease state, progression, and target engagement; and the confounding effects of potent symptomatic therapy. In this article, we will review previous, ongoing, and potential future trials designed to alter the progressive course of the disease from the perspective of the targeted underlying pathogenic mechanisms.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Parkinson disease (PD) is a progressive neurodegenerative disease leading to motor deficits mainly in the form of tremor, rigidity, bradykinesia, and gait impairment. These motor features are largely attributed to the progressive loss of dopaminergic neurons in the substantia nigra pars compacta (SNc); hence, current treatment strategies for PD have targeted the dopamine system. This approach is remarkably effective in ameliorating the dopamine-dependent signs, especially early in the disease. However, with time, it is associated with a variety of motor complications. Also, non-dopaminergic motor and nonmotor features emerge or worsen and dominate the late stages of the illness, resulting in increasing treatment-resistant disability. Having treatments that are neuroprotective or disease-modifying would prevent the severely debilitating complications of advanced PD [1].
There are several obstacles to the successful development of neuroprotective or disease-modifying therapies. The underlying pathogenesis of PD has not yet been fully elucidated, but likely involves various different cellular processes (Fig. 1). Hence, a single agent may not be effective against the range of abnormal pathways that lead to cell dysfunction and death. Preclinical studies in PD are limited by the lack of animal models that accurately reflect the clinical course and recapitulate the neuropathological findings of the human disease. For clinical trials, major hurdles include establishing the proper drug doses to be tested and selecting the appropriate outcome measures, as none has the capacity to accurately assess all aspects of the complex neurodegenerative process. Furthermore, the variable progression of clinical signs and symptoms, as well as the availability of very effective symptomatic therapies for PD complicate the assessment of efficacy of any neuroprotective or disease-modifying treatment.

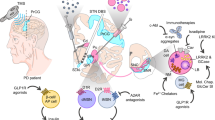
Proposed pathogenic mechanisms that may be targeted for neuroprotective therapies in Parkinson disease (PD). The underlying pathogenesis of PD has not yet been fully elucidated but several key mechanisms have been implicated, including 1) neuroinflammation; 2) N-methyl-D-aspartate (NMDA) receptor-mediated excitotoxicity; 3) altered calcium homeostasis due to increased CaV1.3 channel expression; 4) mitochondrial dysfunction, oxidative stress, and apoptotic cell death; 5) loss of neurotrophic support; 6) α-synuclein (α-syn) misfolding, accumulation, and aggregation into toxic oligomers with propagation via a prion-like mechanism; and 7) increased leucine-rich repeat kinase 2 (LRRK2) kinase activity. ATP = adenosine 5′-triphosphate; Ca2+ = calcium; GDNF = glial-derived neurotrophic factor; GFRα = GDNF family receptor α; JNK = c-Jun N-terminal kinase; Ret = rearranged during transfection receptor tyrosine kinase; ROS = reactive oxygen species. Pointed arrows indicate positive regulation and blunted arrows indicate negative regulation
Although sometimes used interchangeably, the concepts of neuroprotection and disease-modification are not the same. Neuroprotection implies an intervention that affects disease pathogenesis and hence preserves neurons from further degeneration, resulting in a cessation or reduction in evolution of the disease. Disease modification is also associated with a positive effect on clinical progression, but does not require this link with the underlying neurodegenerative process. In contrast, symptomatic treatments only ease the symptoms of the disease for as long as they may provide a pharmacological effect without influencing its course. Success with the development of neuroprotective or disease-modifying treatments would have an important beneficial impact on the lives of patients with PD above and beyond the short-term benefits the symptomatic treatment provides.
Previous and Ongoing Clinical Studies of Neuroprotection and Disease Modification
Understanding the mechanisms involved in the underlying pathogenesis of PD is pivotal for development of neuroprotective therapies. Several key mechanisms have been implicated, including oxidative stress and mitochondrial dysfunction, protein misfolding and aggregation, inflammation, excitotoxicity, apoptotic cell death, and loss of trophic support [2] (Fig. 1). Drugs targeting some of these mechanisms have demonstrated benefit in preclinical studies. Based on these findings, several clinical trials for patients with PD have been conducted (Table 1). We discuss below the previous clinical trials evaluating neuroprotective or disease-modifying therapies for PD based on their putative mechanisms of action.
Antioxidants
Oxidative stress results from an increased presence of reactive free radicals that occurs either because of an overproduction of these free radicals or a failure of mechanisms that limit their accumulation. Oxidative damage has been found in the SNc of PD patients [3, 4], and oxidative stress can be caused by reactive oxygen species (ROS) that are produced as a result of dopamine metabolism [5].
Based on the evident role of oxidative stress in PD pathogenesis, several clinical trials have been conducted using several antioxidants, including vitamin E and the monoamine oxidase B (MAO-B) inhibitors selegiline and rasagiline. The first major clinical neuroprotective trial in PD was the DATATOP study [6]. It assessed the effect of vitamin E and selegiline in patients with early PD. Vitamin E was selected because it is a powerful lipid soluble antioxidant. Selegiline was studied because, in addition to enhancing striatal dopamine, MAO-B inhibition also lessens oxidative stress due to dopamine metabolism. Furthermore, the MAO-B inhibitory effect of selegiline prevents 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced neurotoxicity in numerous animal models by blocking its conversion to the toxic 1‐methyl‐4‐phenylpyridinium ion (MPP+). In addition, selegiline has a propargylamine moiety that appears to have anti-apoptotic effects through blockade of glyceraldehyde-3-phosphate dehydrogenase. The primary endpoint of the DATATOP study was the time until patients’ required L-dopa treatment, a milestone of disease progression. Patients treated with vitamin E experienced no benefit, while those treated with selegiline experienced a significant delay in the need for L-dopa in comparison with placebo patients, consistent with a disease-modifying effect. However, the unanticipated confounder was that selegiline itself likely had mild symptomatic effects that improved motor symptoms in PD [6].
Selegiline was also examined in the SINDEPAR study in which untreated PD patients were randomly assigned to receive selegiline or placebo in combination with dopaminergic therapy for 12 months followed by a 2-month washout period [7]. Patients receiving selegiline had significantly less deterioration from baseline than did those on placebo, consistent with a neuroprotective effect. However, it is possible that the washout period was not sufficiently long to completely eliminate a long duration symptomatic effect. Therefore, again it was not possible to establish that the drug had an effect on the rate of disease progression. Another study examined the long term effects of selegiline as monotherapy and in combination with L-dopa in the early phase of PD over a period of 7 years [8]. Similar to the earlier trials, the findings from this suggested that selegiline may slow the progression of PD. However, the results were hard to interpret as Unified Parkinson’s Disease Rating Scale (UPDRS) scores were significantly lower in the selegiline group after 48 months, but not after 60 months. Furthermore, these scores were only available for 19 patients in the selegiline arm and 28 on placebo.
Rasagiline is a newer propargylamine derivative and more potent MAO-B inhibitor than selegiline. Some reports also suggest that the aminoindan metabolite of rasagiline confer additional neuroprotective activity [9]. Rasagiline has been studied using a delayed-start clinical trial design intended to reduce the confounding effect of symptomatic efficacy. The TEMPO study evaluated PD patients treated with placebo or rasagiline [10]. This study’s results suggested that earlier treatment confers benefit, but the overall duration of this study was relatively short and the group sizes were modest. A larger and longer study (ADAGIO study) was conducted to verify these initial results [11]. There was significant improvement of the UPDRS motor and activities of daily living (ADL) subscales in the early-start groups for both 1 mg and 2 mg of rasagiline versus placebo. However, at week 72, the only significant difference between early-start and delayed-start groups was for ADL subscores with the 1 mg dose [12]. The rate of UPDRS deterioration was less than was anticipated from previous studies and correlated with baseline severity. The failure of the 2 mg dose and the success of the 1 mg dose in all three predefined variables have made the interpretation of these results regarding disease modification difficult. Further biological plausibility for the modifying effects of rasagiline have been proposed based on the evidence that mitochondrial impairment contributes to dopaminergic neuronal loss in PD and rasagiline has been found to affect numerous mitochondrial mechanisms that prevent apoptotic cell death [13].
Mitochondrial Enhancers
Production of ROS and resulting oxidative stress can result from mitochondrial dysfunction. The autosomal recessive forms of parkinsonism related to mutations in Parkin and PINK1 have been shown to be linked to mitochondrial abnormalities; in particular, impairment in mitochondrial autophagy (i.e., mitophagy) [14]. The mechanism responsible for mitochondrial dysfunction in sporadic PD patients is not yet well understood. However, several studies have shown that sporadic PD patients have a reduction in mitochondrial complex I levels and activity in their SNc [15–17].
Clinical trials have pursued the investigation of mitochondrial enhancers, including co-enzyme Q10 (Co-Q10), mitoquinone, and creatine, as potential neuroprotective agents in PD. Co-Q10 is an electron carrier for complexes I and II, and a free radical scavenger [18]. A pilot study showed that patients who received the highest dose of Co-Q10 (1200 mg/day) had significantly less deterioration in UPDRS scores than patients taking placebo [19]. However, patients in the high-dose Co-Q10 group also showed short -term improvement in ADL scores after introduction of the drug, consistent with a symptomatic effect. A study of high dose Co-Q10 (2400 mg/day) was terminated prematurely owing to lack of efficacy [20]. A powerful mitochondrial antioxidant mitoquinone failed to slow the progression of PD over a 12-month period [21].
Creatine is a naturally-occurring compound which, when converted to phosphocreatine, acts as a short term energy source in tissues with high energy requirements, such as brain. Creatine supplementation has demonstrated neuroprotective features in cellular and animal models of neurodegenerative diseases, including PD [22]. The beneficial effects are believed to be due to improvement in overall bioenergetics and/or mitochondrial deficits. A pilot study of early PD patients compared creatine to placebo for 2 years [23]. No difference was evident in UPDRS scores or in striatal 123I‐2β‐carbomethoxy‐3β‐(4‐iodophenyl)tropane (β-CIT) single-photon emission computed tomography (SPECT) uptake between the 2 groups. Neuroprotection Exploratory Trials in Parkinson’s Disease (NET‐PD), sponsored by the National Institute of Neurological Disorders and Stroke (NINDS), initially found no evidence for futility of creatine therapy [24]. Subsequently, this group has undertaken a simple, long-term study strategy in which patients are randomized to creatine or placebo and followed for a long duration (i.e., 5–7 years) while concurrently receiving other necessary PD medications. This blinded placebo-controlled trial of creatine in PD will use a composite endpoint involving a global statistical test encompassing 5 clinical rating scales to provide a multidimensional assessment of disease progression and potentially provide higher power to test the hypothesis [25]. Since the review and acceptance of this manuscript, the NINDS has stopped the NET‐PD long‐term study of creatine for treatment of early stage PD. According to the NET‐PD website, an interim analysis revealed evidence for futility. Continued follow‐up of subjects was not expected to demonstrate a statistically significant difference between creatine and placebo. Hence, the study’s Data Safety Monitoring Board recommended termination of the study.
Dopamine Agonists and L-dopa
Laboratory studies have shown protective effects of dopamine agonists on dopamine neurons and their ability to inhibit apoptosis [26]. Two earlier prospective parallel design double-blind trials have been performed to evaluate this effect. Both trials used the rate of decline in surrogate neuroimaging biomarkers of nigrostriatal function as the primary endpoint to avoid the confounding factor of potential symptomatic benefit of the medications. The REAL-PET study compared ropinirole with L-dopa on PD progression [27], while the CALM-PD study compared the effect of pramipexole with L-dopa in early PD patients [28]. Both studies demonstrated that L-dopa was associated with an approximate 30 % greater rate of decline in measures of the biomarker in comparison with the dopamine agonist. This suggested that possible disease modification could be attributed to dopamine agonists. However, there were several important limitations, including the lack of a placebo control, lack of an extended washout of the medications and the fact that the clinical status of L-dopa treated patients improved to a greater extent than that of the dopamine agonist treated group. Another significant limitation was the inability to prove that these neuroimaging measures reflect neuroprotection of dopamine systems [29]. The same results could have been due to L-dopa-induced toxicity [30] although there has been ongoing debate regarding the effect of L-dopa on neurons in PD patients. Some believe that treatment of PD patients with L-dopa potentially promotes neurodegeneration owing to the production of free radicals secondary to dopamine catabolism [31, 32], while others have found a potential neuroprotective effect in animal models [33, 34].
The ELLDOPA study was designed to try to determine whether L-dopa is toxic and accelerates the progression of PD [35]. Patients receiving L-dopa had less clinical deterioration from baseline than did placebo patients, suggesting a protective, rather than a toxic, effect. However, SPECT studies performed as part of this clinical trial demonstrated that patients who received L-dopa had an increased rate of decline in uptake of striatal β-CIT (a dopamine transporter ligand) compared with placebo, suggestive of a toxic, rather than a protective, effect. Therefore, the ELLDOPA study did not resolve the issue of whether or not L-dopa is toxic in PD [36]. Given the very robust beneficial effects of L-dopa for motor symptoms, the discrepant decline in imaging markers may be better explained by the artifactual effects on the ligands assessed (see below). The clinical effects of L-dopa are being studied currently in a delayed-start design (LEAP study) being conducted in the Netherlands (R. de Bie, personal communication). This same study design combined with 123I-FP-CIT SPECT, in the recently reported PROUD study, failed to support the earlier suggestions of disease-modifying effects of pramipexole as the clinical and neuroimaging measures showed no significant difference between early and delayed pramipexole [37].
Trophic Factors
A contributor to cell death in PD has been the loss of neurotrophic factors. Nerve growth factor, brain-derived neurotrophic factor, and glial-derived neurotrophic factor (GDNF) have been found to be reduced in the SNc of PD patients [38–40]. GDNF is a potent neurotrophic factor that supports the survival of dopaminergic nigral neurons and has been shown to be neuroprotective in animal models of PD [41, 42]. However, a recent study raises concerns about the potential for treatments such as GDNF to provide benefit in PD. It has been shown that α-synuclein-induced downregulation of the transcription factor Nurr1 reduces the expression of RET (“rearranged during transfection”), the receptor tyrosine kinase that mediates GDNF signaling, resulting in a blockade of the effects of GDNF [43]. The applicability of these findings in humans with PD remains uncertain.
An open-label trial of direct delivery of recombinant GDNF into the putamen suggested effectiveness [44], while a randomized controlled study in later stage patients was halted owing to lack of efficacy [45]. Further studies with GDNF are ongoing, including investigation into viral-mediated gene delivery using adeno-associated virus encoding GDNF (AAV2-GDNF) for advanced PD [46]. A gene therapy approach using neurturin, a member of the GDNF family, has been studied [47]. AAV2-neurturin injected initially into the putamen and later both into the putamen and SNc failed to provide significant benefit in later stage PD patients. Recently, PYM50028 (Cogane), a synthetic agent designed to promote the release of both GDNF and brain-derived neurotrophic factor, failed to show benefit in a large, placebo-controlled phase II study [48].
Neuroimmunophilins are intracellular receptor proteins that bind immunosuppressive drugs like cyclosporine and FK506. They have been shown to promote neuronal growth and have demonstrated neuroprotection in animal models [49–51]. Their exact mechanism of action is not known; however, it may be associated with glutathione, which has antioxidant properties and can stimulate neurotrophic factors [52, 53]. A phase II trial of the neuroimmunophilin ligand GPI-1485 showed no significant difference of motor symptoms in patients with mild-to-moderate PD compared with placebo. However, there was a non-significant trend of increased β-CIT uptake in patients who were on the highest dose of GPI-1485 [54].
Other Pharmacologic Agents
Glutamate-mediated excitotoxicity has been implicated as a pathogenic mechanism in PD and other neurodegenerative diseases. Excessive activation of N-methyl-D-aspartate (NMDA) receptors by glutamate results in increased intracellular calcium levels that can activate cell death pathways and lead to apoptosis [55]. Dopaminergic neurons in the SNc have high levels of glutamate receptors and receive glutamatergic innervation from the subthalamic nucleus and cortex. The potential for excitotoxic damage in PD has led to trials of agents that interfere with this mechanism. NMDA receptor antagonists have been reported to induce protective effects in some, but not all, studies in models of neurodegenerative diseases such as PD. Riluzole is a drug with multiple mechanisms of action, including inhibition of glutamate release. It has been found to be well tolerated in a trial for PD patients [56]. However, a large multicenter study of riluzole in PD patients was discontinued because of lack of benefit in the interim data analysis [57].
Anti-apoptotic drugs such as CEP-1347 and TCH346 [58, 59] could have neuroprotective effects based on their capacity to block the c-Jun N-terminal kinase signaling pathway and glyceraldehyde-3-phosphate dehydrogenase translocation, respectively. Unfortunately, trials with these agents were negative. The failure of TCH346, a propargylamine without MAO-B inhibitory effects, suggests that the possible benefit obtained with selegiline and rasagiline could be largely dopaminergic via MAO-B inhibition. Minocycline, a second-generation tetracycline long used as an antimicrobial agent, is another anti-apoptotic drug that has been considered a potential candidate for neuroprotection. Minocycline is a caspase inhibitor that has neuroprotective effects against excitotoxicity by inhibiting activation and proliferation of microglia [60, 61]. It has demonstrated protection against dopaminergic cell loss in both the MPTP and 6‐hydroxydopamine (6-OHDA) animal models [62, 63]. An initial phase II trial showed that minocycline was well tolerated and could not be rejected as futile, setting the stage for possible phase III trials [24].
GM1 ganglioside is an endogenous sphingolipid that has been shown to stimulate the repair of the nigrostriatal system in a variety of animal models. The mechanisms by which GM1 ganglioside may induce neurorestorative or neuroprotective effects are not yet clearly defined. A recent randomized controlled delayed start study confirmed earlier evidence for symptomatic effects, but provided little support for convincing disease-modifying effects [64].
Lessons Learned from Previous Studies
None of the clinical trials for PD have shown clear disease-modifying effects, even though several different neuroprotective and disease-modifying strategies are promising. These studies have provided important insights into many of the potential limitations of developing and testing for disease-modifying therapies in PD.
Animal Models
Toxin-based models using 6-OHDA in rodents and MPTP in mice and primates have been widely employed in preclinical studies for PD. Both of these models demonstrate degeneration of dopaminergic nigral neurons and thus have proven useful in evaluating dopamine-based therapies [65, 66]. However, these models require the acute administration of toxins and likely do not reflect the chronic and progressive neurodegenerative course of PD. With the identification of roles for α-synuclein, parkin, leucine-rich repeat kinase 2 (LRRK2), and other proteins in the pathogenesis of PD through human genetic studies, transgenic models based on these genetic causes of parkinsonism have been developed with some advantages over the acute toxin models [67, 68]. Another recent approach is the induction of cell-to-cell transmission of α-synuclein aggregates using brain inoculation with α-synuclein fibrils [69]. Regardless, there is no one animal model for PD that mimics the full pathology and clinical symptomology of the illness. More accurate animal models would provide greater insights into the pathogenesis of PD. It is also hoped that more clinically relevant models used in the development of new neuroprotective therapies would allow for improved translation from preclinical studies to future human clinical trials.
Clinical Trial Design
One of the major challenges to clinical trials for PD in determining whether the benefit of a study drug was a result of a neuroprotective effect has been the potentially confounding symptomatic or regulatory effects of the study agent. Refinements in clinical trial design have been made to try to address the issue of separating early symptomatic benefit from disease-modifying effect of a study intervention. Some trials have incorporated a washout period before assessing the primary outcome measure. However, symptomatic drug effects may be longstanding and outlast the washout period [70]. An alternative approach has been to use a delayed start design where one group of patients is started on the therapy several months before the comparative group [71]. This approach is based on the assumption that the symptomatic effects would be similar across the groups at the end of the study. There are still some potential problems with this approach as longer treatment may result in increased sensitivity to the drug and may also result in different rates of drop out between groups, as patients randomized to placebo in the initial phase of the trial are more likely to drop out [10].
Another major challenge in these clinical trials has been determining how to test neuroprotection in a living patient. Traditionally, clinical measures based on neurological examination have been used to assess progression (or lack of progression) of PD. The UPDRS includes clinical examination of motor function and scales rating patients’ subjective views of function in daily activities. There are several limitations to this scale [72]. For instance, available symptomatic treatments have a large effect on the UPDRS score, which may obscure evidence of neuroprotection. The scale is also heavily weighted towards motor dysfunction, particularly tremor-related symptoms. Much of the disability associated with PD is not considered in the scale, such as that related to autonomic dysfunction and fatigue. The Movement Disorder Society has recently revised the UPDRS to make it more sensitive to subtle motor changes and to capture non-motor features that are frequently present in the early stages of the disease [73]. This may make the scale more useful in assessing symptomatic therapies, as well as effective in detecting change associated with putative neuroprotective or disease-modifying agents. Given the challenges in defining neuroprotection, the complexity of the disease, the limitations of earlier trial designs, and the confounding effects of symptomatic therapy, the NINDS NET-PD investigators have chosen to utilize a “long-term simple study” design with composite endpoints assessing quality of life, as well as dopaminergic and non-dopaminergic clinical features [71]. All of the problems outlined above could be resolved or improved by the identification of a validated biomarker that could be used to confirm the diagnosis or to objectively measure disease progression and drug efficacy.
Biomarkers
Biomarkers are objectively measured characteristics that can act as indicators of the underlying pathogenic process. Currently, no biomarkers have been validated for PD, but there is ongoing research to identify biomarkers that could assist in more accurate and early diagnosis of PD, as well as allow for monitoring disease progression and response to therapeutic interventions. SPECT and positron-emission tomography imaging have been used as default measures of dopamine cell numbers, but these imaging techniques have their limitations. A concern with both approaches is that the underlying chemistry of the radioactive tracers used may be altered by the pharmacological effects of the treatments under investigation such that imaging changes may not necessarily reflect changes in dopaminergic neuron counts. Also, some of the features of advanced PD do not have a major dopaminergic basis and, accordingly, will not be captured by dopaminergic tracers [74]. These imaging techniques have not yet been validated as appropriate surrogate measures of neuroprotection through correlation with neuropathology and/or clinical symptoms [29].
There are several potential candidate markers apart from neuroimaging. These include protein-based markers, markers of oxidative stress or inflammation, and measurement of patterns of variations in genes and proteins (i.e., genomics and proteomics, respectively). A study evaluating a panel of seven cerebrospinal fluid (CSF) markers (α-synuclein, DJ-1, total tau, phosphorylated tau, amyloid β peptide 1–42, Flt3 ligand, and fractalkine levels) demonstrated utility in diagnosing PD, separating PD from controls and other neurodegenerative diseases, as well as detecting changes that correlate with disease progression [75]. Studies in PD have found elevated levels of circulating interleukins (IL)-6, IL-10, and IL-12 [76], but more data are needed before markers of oxidative stress can be considered potential biomarkers. Genomic and proteomic measurements are promising, but technically demanding. It is likely that in the near future a biomarker panel might be developed, possibly combing clinical batteries, imaging, and “omics”-based measures, as well as other disease-state-related markers [77].
A biomarker could also be valuable in guiding proper dosing in drug trials. Typically, doses of study drugs selected for clinical trials are based on an attempt to reproduce plasma concentrations in animal models. However, plasma concentrations of a drug do not necessarily mirror the concentration of the drug in the brain. Furthermore, a given agent may be effective only within a narrow dose range. Thus, it is possible that negative studies with promising drugs have occurred owing to the selection of the wrong dose rather than the futility of the intervention. Choosing an adaptive clinical trial design can help in this regard, as it allows for many different doses of the same medication to be tested in a single trial without negatively affecting the integrity of the trial [78].
Clinicians need to understand the potential pitfalls associated with the use of biomarkers, either as diagnostic measures or as surrogate markers. While combining various biomarkers would hopefully improve diagnostic accuracy, it might risk diagnosing unaffected people as having PD. Biomarkers expected to represent disease state could be utilized as the end point of a clinical trial. However, there could be dissociation between this surrogate marker and the clinical end-point it is trying to represent; the treatment might alter the biomarker, but not the disease, or it might not alter the biomarker even though it does alter an important aspect of the disease pathophysiology that is not assayed by the biomarker [79].
Potential Neuroprotective and Disease-Modifying Therapies
Our increasing understanding of PD pathogenesis, which is built upon prior research findings and further augmented by genetic and epidemiologic discoveries, is leading to novel approaches to neuroprotection for PD (Fig. 1). Further studies into antioxidants and trophic factors are continuing. Novel targets for neuroprotection in PD that are actively being investigated include the adenosine system, inflammatory pathways, calcium channels, α-synuclein aggregation, and LRRK2 kinase activity.
Antioxidants
Free radical damage can be associated with increased iron levels, which are seen in the SNc of PD patients [80]. Cliquinol is an iron chelator that is associated with a reduction in neuronal death in the MPTP mouse model. Currently, a phase II/III study is evaluating deferiprone, another oral iron chelator, on iron overload in the SNc (as assessed by the MRI T2* sequence) with respect to the clinical progression of PD [81]. If the results are positive, a larger neuroprotection study could be pursued.
Epidemiological studies have shown that uric acid might be a potential neuroprotective agent in PD. Uric acid acts as an antioxidant by scavenging ROS and reactive nitrogen species [82]. Studies have shown a decreased incidence of PD among patients with high serum urate levels [83, 84] and among patients with gout [85]. In early PD patients, those with higher plasma urate levels show evidence for slower disease progression [86], and a recent study showed that patients on diets that promote high urate levels have a reduced risk of developing PD [87]. However, the potential benefits of a urate-rich diet have to be weighed against the risk of developing gout and cardiovascular disease. Inosine is an antioxidant that raises urate levels. The SURE-PD trial is currently evaluating the disease-modifying potential of this agent in early PD patients [88].
Zonisamide is an anti-epileptic drug that has been shown to provide symptomatic benefit in PD patients. In vitro and animal models have demonstrated that zonisamide has modulator effects on oxidative stress, intracellular Ca2+ signaling, and caspase-3 activity [89], as well as neuroprotective effects in MPTP-induced dopaminergic neuronal degeneration [90]. Currently, the ZONIST trial is investigating the neuroprotection by zonisamide in early PD [91].
Excessive free radical formation and depletion of glutathione (GSH), the brain’s primary antioxidant, have been demonstrated in PD patients. Some clinicians have been using GSH intravenously for PD patients. A randomized, placebo-controlled trial of intravenous GSH in PD proved safe and well tolerated; however, there was no improvement evidenced in any outcome variable [92]. Currently, there is lack of evidence for efficacy and, indeed, lack of data that GSH actually crosses the blood–brain barrier [93]. Intranasal GSH is a novel method of delivery that bypasses the obstacles associated with other delivery methods. Currently, a phase I study is ongoing in PD patients [94]. Another study is also underway using proton magnetic resonance spectroscopy to determine whether levels of GSH are decreased in PD patients and whether GSH levels increase following daily supplementation with N-acetylcysteine, a GSH precursor [95]. If successful, this study will provide a justification for larger controlled clinical trials.
Polyphenols in green tea are natural antioxidants that may protect dopamine neurons through inhibition of nitric oxide and ROS production. Green tea polyphenols were found to be protective against 6-OHDA in rodents [96] and hence a randomized controlled trial is currently ongoing to assess the neuroprotective effect of green tea in de novo PD patients [97].
Trophic Factors
Trophic factors continue to hold significant promise for the future. The strength of this approach is that the biology of the factors themselves is well known, and it does not rely on a detailed understanding of the mechanisms of cell death in PD. Thus, neurotrophic factors may enhance dopaminergic survival, regardless of the mechanism of cell death. As indicated above, further studies with GDNF are ongoing. Further analysis of the results of the AAV2-neurturin studies may provide insights that will allow this, and related, therapies to become more effective. One critical issue that may have compromised these treatments in earlier studies is their use in the later stages of disease (largely justified by the necessary surgical intervention). However, to be effective, trophic therapies may have to be applied at a relatively early stage when there is a sufficient number of surviving nigral neurons continuing to innervate the striatum. By the time these treatments were applied in previous studies, nigrostriatal degeneration may have been too advanced to have benefited from the provision of trophic support.
Adenosine Receptor Antagonists
Epidemiological studies have shown that caffeine consumption has been associated with a reduced risk of PD. The association is established especially in men, while in women it is uncertain, possibly because of interaction with hormone replacement therapy [98, 99]. Caffeine is a non-selective adenosine A1/A2A receptor antagonist that acts in the brain primarily at A2A receptors. Hence, there has been growing interest in evaluating adenosine receptor antagonists as potential neuroprotective agents [100]. A2A receptors are highly expressed within the striatum where their blockade leads to locomotor activation by reducing inhibitory output of the basal ganglia indirect pathway. Currently, there are several selective A2A receptor antagonists in development. Istradefylline has been studied in several phase II and phase III trials [101]. Other A2A receptor antagonists in development are preladenant, tozadenant, vipadenant, and V81444 [101].These agents are largely being studied for their symptomatic effects, although phase III trials of preladenant have been recently terminated owing to lack of efficacy compared with placebo [102]. Whether A2A receptor antagonists have disease-modifying effects remains to be seen.
Anti-Inflammatory Agents
Neuroinflammation has been recognized as an important mechanism involved in PD pathogenesis [103, 104]. Microglial activation has been found in PD animal models, as well as in the SNc and striatum of PD patient brains [105–107]. Pro-inflammatory cytokines, such as IL-1β, IL-6, and tumor necrosis factor-α, are elevated in the CSF and basal ganglia of PD patients [108]. Elevated serum levels of complement proteins have also been detected in PD [109]. It is not clear whether neuroinflammation plays a primary role in disease pathogenesis or is entirely secondary. It is also not certain whether activation of these pathways accentuates or might even partially retard the degenerative process.
Anti-inflammatory agents are being pursued as potential disease-modifying treatments for PD. Several animal models had demonstrated that certain nonsteroidal anti-inflammatory drugs have neuroprotective qualities [110]. However, epidemiological studies have provided conflicting results. An initial study showed that nonsteroidal anti-inflammatory drug use lowers the risk of PD by 45 % [111] and a follow-up study by the same group showed that only ibuprofen had this neuroprotective effect [112]. Other epidemiological studies examining this association have shown nonsignificant trends [113, 114].
An alternative approach to targeting neuroinflammation may be the use of statins. In addition to lowering cholesterol, statins have anti-inflammatory effects, including reduction of tumor necrosis factor-α, nitric oxide, and superoxide production by microglia [115], and may also scavenge free radicals [116]. Simvastatin also reduces dopamine loss in MPTP animal models [115]. Epidemiological studies have shown that statin use, particularly simvastatin, is associated with reduced PD incidence [117, 118]. However, other studies have suggested low low-density lipoprotein cholesterol levels are associated with increased PD risk [119, 120].
Targeting the peroxisome proliferator activated receptor-γ has the potential for reducing the production of pro-inflammatory cytokines by harmful activated microglia, while sparing or inducing beneficial activated microglia [121]. The FS-Zone study conducted by the NINDS-NET investigation is currently evaluating pioglitazone, a proliferator activated receptor-γ agonist, for potential disease-modifying effects [122].
Calcium Channel Blockers
It has been shown that ventral tier SNc dopaminergic neurons, as well as other selectively vulnerable neurons in PD, have calcium-dependent pacemaking properties that put them at risk of damage by oxidative stress. Antagonizing the CaV1.3 channels using the L-type calcium channel blocker isradipine reverts dopaminergic neurons to a latent juvenile pacemaking mechanism and protects these cells from both 6-OHDA and MPTP toxicity [123]. Isradipine has been used as an antihypertensive agent and there are variable epidemiological data supporting a positive effect of dihydropyridine calcium channel blockers on the progression of PD [124, 125]. Preliminary studies have assessed the safety and tolerability of isradipine in PD [126] and a larger trial evaluating its disease-modifying effects is being planned.
α-Synuclein-Directed Therapies
There is abundant evidence implicating the protein α-synuclein in the pathogenesis of PD [127]. Missense mutations in the α-synuclein gene (SNCA), as well as duplications and triplications of the locus containing SNCA, are associated with rare familial forms of PD. Polymorphisms in SNCA have also been identified as risk factors for sporadic PD. The identification of α-synuclein as a major component of Lewy bodies and Lewy neurites—the protein aggregates that are neuropathological hallmarks of PD—led to the discovery of α-synuclein aggregation as a key event in the disease process. Soluble oligomers of α-synuclein aggregates are most likely the toxic forms of α-synuclein that cause neuronal dysfunction and death in PD. The finding of Lewy bodies within dopaminergic neurons from healthy fetal mesencephalic grafts transplanted into the striatum of PD patients has suggested that α-synuclein pathology may be transmissible. Additional results from cell culture and animal studies have supported prion-like spread of α-synuclein [128]. Thus, targeting the formation, accumulation, and/or spread of toxic forms of α-synuclein may prove to be neuroprotective in PD [129].
Vaccine-based therapies are being pursued as potential α-synuclein-directed therapies. Studies on passive or active immunization using a transgenic mouse model of PD that overexpresses human α-synuclein have demonstrated reductions in α-synuclein oligomer levels [129]. Currently, there is an ongoing phase I study of active immunization of early PD patients with PD01A, which elicits α-synuclein antibodies [130].
Other strategies to reduce toxic α-synuclein and prevent neuronal death, which are not yet being tested in clinical trials, include directly blocking α-synuclein aggregation with monoclonal antibodies, short peptides, or small molecules [131, 132]. Promotion of chaperone function could also encourage α-synuclein clearance [133], as could upregulation of the proteasomal and/or autophagy–lysosomal systems [134]. Reduction in the levels of harmful proteins using RNA interference technology is a promising strategy for treatment of neurodegenerative diseases, such as Huntington’s disease [135] and possibly PD. All of these strategies are at a relatively early stage of development and await further study before proceeding to human intervention trials.
Kinase Inhibitors
Mutations in LRKK2 are the most common genetic cause of autosomal dominant PD to date, resulting in about 2 % of all cases of PD [136, 137] and up to 40 % in some isolated populations, such as those in North African regions [138, 139]. LRRK2 is a large multidomain protein that contains serine/threonine kinase activity. G2019S, the most common pathogenic mutation of LRRK2, occurs within the kinase domain and is associated with increased kinase activity [140, 141]. Kinases are generally good targets for small molecule therapies, and have recently shown promise in clinical studies. A LRRK2 inhibitor, CZC-25146, prevents mutant LRRK2-induced injury of cultured rodent and human neurons with mid-nanomolar potency [142]. Two other inhibitors, GW5074 and sorafenib, showed protection against LRRK2-induced neurodegeneration in Caenorhabditis elegans and Drosophila. These findings have suggested that increased kinase activity of LRRK2 is neurotoxic and hence inhibition of LRRK2 activity could have a disease-modifying effect [143].
Other Pharmacologic Agents
Exenatide, a glucagon like peptide-1 receptor agonist currently used in the treatment of type II diabetes, has been shown to have neuroprotective/neurorestorational effects in 6-OHDA and MPTP animal models. A proof-of-concept single blind trial design was conducted in PD [144] and this treatment will almost certainly be pursued for potential disease-modifying effects.
Nicotine use had been proposed as neuroprotective based on the observation of low prevalence of smoking in PD patients. However, controlled studies have provided conflicting results. Currently, two phase II, randomized, controlled studies of transdermal nicotine are ongoing and will allow the evaluation of potential neuroprotective effects [145, 146].
Erythropoietin (EPO), a hematopoietic cytokine, has been demonstrated to protect nigral dopaminergic neurons from cell death induced by 6-OHDA in rodents and it has been hypothesized that anti-inflammation may be one of mechanisms responsible for EPO neuroprotection [147]. A phase I study showed EPO to be safe and well tolerated in PD patients [148].
Granulocyte colony-stimulating factor, a hematopoietic growth factor, has demonstrated neuroprotection in a PD rodent model [149]. Currently, a phase II study is evaluating the potential disease-modifying effect of recombinant granulocyte colony-stimulating factor (filgrastim) in early stage PD [150].
Nonpharmacologic Strategies
Animal studies have shown that “intensive” exercise improves motor function and may have neuroprotective properties. Currently, the ParkCycle study is evaluating the effects of aerobic exercise on cognitive and adaptive plasticity in PD [151].
Surgical Interventions
Deep brain stimulation (DBS) of the subthalamic nucleus (STN) induces a marked long-term improvement in motor fluctuations, dyskinesia, and overall quality of life in advanced PD patients [152, 153]. DBS of bilateral globus pallidus interna is as efficacious as STN DBS for the management of both motor fluctuations and dyskinesia [154, 155]. The mechanism by which DBS imparts its beneficial effects in PD remains unclear, but it has been postulated that disinhibition and overactivity of the STN due to dopamine deficiency in PD may be reduced by bilateral STN DBS [156]. In animal models, it had been suggested that this abnormal STN activity results in excessive amounts of glutamate release in the SNc leading to NMDA receptor-mediated excitotoxic cell damage of dopaminergic neurons that further contributes to neuronal loss in PD [157, 158]. Therefore, it has been hypothesized that DBS of the STN may be neuroprotective in PD [159]. In support of this hypothesis, long-term bilateral STN DBS in the 6-OHDA rat model of PD showed rescue of dopaminergic SNc neurons [160]. Currently, there is an ongoing pilot study investigating whether bilateral STN DBS is a safe and effective treatment in slowing the progression in patients with early PD. The results will provide data for the design of a full-scale, multicenter trial to investigate this hypothesis [161].
Gene transfer of glutamic acid decarboxylase into the STN, which is believed to modulate the production of GABA and thus to reduce overactivity of this nucleus, has been demonstrated to provide symptomatic benefit in late-stage PD [162]. Theoretically, as proposed for STN DBS, this treatment might also reduce excitotoxic damage in the SNc. To date, this therapy has not been explored for this purpose.
Stem Cell Therapy
There has been a long standing hope that cell transplantation therapy could provide a long-term therapeutic option for PD owing to the perceived relatively specific and localized loss of neurons in PD. This was supported by animal studies that showed beneficial effects of such an intervention [163, 164]. The first clinical trial of transplantation therapy was performed in the mid-1980s [165]. More than 400 patients worldwide have now been treated with fetal cell transplantation [166]. Open-label studies showed beneficial effects in PD symptoms following such transplantations [167]; however, controlled clinical trials [168–170] showed little benefit compared with the placebo group. There has been a dramatic progress in stem cell therapy technology in the past decade. This includes the ability to generate induced pluripotent stem cells from patient fibroblasts and to differentiate these induced pluripotent stem cells into dopaminergic neurons that may be engrafted in vivo [171]. However, the long-term safety and proven efficacy is missing [172].
One important and critical limitation of cell-based therapies designed to address the loss of the nigrostriatal dopamine system is the widespread multisystems nature of PD and the fact that many of the most disabling and treatment-resistant features of late stage disease probably have little to do with this dopaminergic cell loss [173, 174].
Conclusions
Neuroprotection and disease modification in PD remain important, but elusive, goals. A successful neuroprotective or disease-modifying treatment could transform PD from a relentlessly progressive and disabling disease to a problem that might be managed with only a modest effect on quality of life. Current barriers include limited knowledge of the basic mechanisms of PD, and challenges in the methodology used to assess disease progression and study outcomes. Overall, however, the activity aimed at understanding and treating PD has grown and should, ultimately, result in therapies that will successfully modify the progressive disease course.
References
Lang AE. Clinical trials of disease-modifying therapies for neurodegenerative diseases: the challenges and the future. Nat Med 2010;16:1223–1226.
Yacoubian TA, Standaert DG. Biochim Biophys Acta 2009;1792:676–687.
Alam ZI, Jenner A, Daniel SE, et al. Oxidative DNA damage in the parkinsonian brain: an apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J Neurochem 1997;69:1196–1203.
Dexter DT, Sian J, Rose S, et al. Indices of oxidative stress and mitochondrial function in individuals with incidental Lewy body disease. Ann Neurol 1994;35:38–44.
Hastings TG, Lewis DA, Zigmond MJ. Reactive dopamine metabolites and neurotoxicity: implications for Parkinson’s disease. Adv Exp Med Biol 1996;387:97–106.
The Parkinson Study Group. Effect of deprenyl on the progression of disability in early Parkinson’s disease. N Engl J Med 1989;321:1364–1371.
Olanow CW, Hauser RA, Gauger L, et al. The effect of deprenyl and levodopa on the progression of signs and symptoms in Parkinson’s disease. Ann Neurol 1995;38:771–777.
Pålhagen S, Heinonen E, Hägglund J, et al. Selegiline slows the progression of the symptoms of Parkinson disease. Neurology 2006;66:1200–1206.
Bar-Am O, Weinreb O, Amit T, Youdim MB. The neuroprotective mechanism of 1-(R)-aminoindan, the major metabolite of the anti-parkinsonian drug rasagiline. J Neurochem 2010;112:1131–1137.
Parkinson Study Group. A controlled, randomized, delayed-start study of rasagiline in early Parkinson disease. Arch Neurol 2004;61:561–566.
Olanow CW, Rascol O, Hauser R, et al. A double-blind, delayed start trial of rasagiline in Parkinson’s disease. N Engl J Med 2009;361:1268–1278.
Rascol O, Fitzer-Attas CJ, Hauser R et al. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease (the ADAGIO study): prespecified and post-hoc analyses of the need for additional therapies, changes in UPDRS scores, and non-motor outcomes. Lancet Neurol 2011;10:415–423.
Jenner P, Langston JW. Explaining ADAGIO: a critical review of the biological basis for the clinical effects of rasagiline. Mov Disord 2011;26:2316–2323.
Deas E, Wood NW, Plun-Favreau H. Mitophagy and Parkinson’s disease: the PINK1-parkin link. Biochim Biophys Acta 2011;1813:623–633.
Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1989;1:1269.
Schapira AHV, Cooper JM, Dexter D, et al. Mitochondrial complex I deficiency in Parkinson’s disease. J Neurochem 1990;54:823–827.
Mizuno Y, Ohta S, Tanaka M, et al. Deficiencies in complex I subunits of the respiratory chain in Parkinson’s disease. Biochem Biophys Res Commun 1989;163:1450–1455.
Beal MF. Bioenergetic approaches for neuroprotection in Parkinson’s disease. Ann Neurol 2003;53:S39-S47.
Shults CW, Oakes D, Kieburtz K, et al. Effects of coenzyme Q10 in early Parkinson disease: evidence of slowing of the functional decline. Arch Neurol 2002;59:1541–1550.
Effects of Coenzyme Q10 (CoQ) in Parkinson Disease (QE3). Clinicaltrials.gov. Available at: www.http://clinicaltrials.gov/ct2/show/NCT00740714. Accessed August 16, 2013.
Snow BJ, Rolfe FL, Lockhart MM, et al. A double-blind, placebo controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease- modifying therapy in Parkinson’s disease. Mov Disord 2010;25:1670–1674.
Matthews RT, Ferrante RJ, Klivenyi P, et al. Creatine and cyclocreatine attenuate MPTP neurotoxicity. Exp Neurol 1999;157:142–149.
Bender A, Koch W, Elstner M, et al. Creatine supplementation in Parkinson disease: a placebo controlled randomized pilot trial. Neurology 2006;67:1262–1264.
NINDS NET-PD Investigators. A randomized, double-blind, futility clinical trial of creatine and minocycline in early Parkinson disease. Neurology 2006;66:664–671.
Elm JJ; NINDS NET-PD Investigators. Design innovations and baseline findings in a long-term Parkinson’s trial: the National Institute of Neurological Disorders and Stroke Exploratory Trials in Parkinson’s Disease Long-Term Study-1. Mov Disord 2012;27:1513–1521.
Olanow CW, Jenner P, Brooks D. Dopamine agonists and neuroprotection in Parkinson’s disease. Ann Neurol 1998;44:167–174.
Whone A, Watts R, Stoessl J, et al. Slower progression of Parkinson’s disease with ropinirole versus levodopa: the REAL-PET study: Ann Neurol 2003;54:93–101.
Parkinson Study Group. Dopamine transporter brain imaging to assess the effects of Pramipexole vs levodopa Parkinson disease progression. JAMA 2002;287:1653–1661.
Ravina B, Eidelberg D, Ahlskog JE, et al. The role of radiotracer imaging in Parkinson disease. Neurology 2005;64:208–215.
Olanow CW, Agid Y, Mizuno Y, et al. Levodopa in the treatment of Parkinson’s disease: current controversies. Mov Disord 2004;19:997–1005.
Fahn S. Is levodopa toxic? Neurology 1996;47:S184-195.
Barzilai A, Melamed E, Shirvan A. Is there a rationale for neuroprotection against dopamine toxicity in Parkinson’s disease? Cell Mol Neurobiol 2001;21:215–235.
Murer MG, Dziewczapolski G, Menalled LB et al. Chronic levodopa is not toxic for remaining dopamine neurons, but instead promotes their recovery, in rats with moderate nigrostriatal lesions. Ann Neurol 1998;43:561–575.
Datla KP, Blunt SB, Dexter DT. Chronic L-DOPA administration is not toxic to the remaining dopaminergic nigrostriatal neurons, but instead may promote their functional recovery, in rats with partial 6-OHDA or FeCl(3) nigrostriatal lesions. Mov Disord 2001;16:424–434.
Fahn S, Oakes D, Shoulson I, et al. Levodopa and the progression of Parkinson’s disease. N Engl J Med 2004;351:2498–2508.
Olanow CW, Jankovic J. Neuroprotective therapy in Parkinson’s disease and motor complications: a search for a pathogenesis-targeted, disease-modifying strategy. Mov Disord 2005;20:S3-10.
Schapira AH, McDermott MP, Barone P, et al. Pramipexole in patients with early Parkinson’s disease (PROUD): a randomised delayed-start trial. Lancet Neurol 2013;12:747–755.
Howells DW, Porritt MJ, Wong JY, et al. Reduced BDNF mRNA expression in the Parkinson’s disease substantia nigra. Exp Neurol 2000;166:127–135.
Chauhan NB, Siegel GJ, Lee JM. Depletion of glial cell line-derived neurotrophic factor in substantia nigra neurons of Parkinson’s disease brain. J Chem Neuroanat 2001;21:277–288.
Mogi M, Togari A, Kondo T, et al. Brain-derived growth factor and nerve growth factor concentrations are decreased in the substantia nigra in Parkinson’s disease. Neurosci Lett 1999;270:45–48.
Cheng FC, Ni DR, Wu MC, Kuo JS, Chia LG. Glial cell line-derived neurotrophic factor protects against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced neurotoxicity in C57BL/6 mice. Neurosci Lett 1998;252:87–90.
Fox CM, Gash DM, Smoot MK, Cass WA. Neuroprotective effects of GDNF against 6-OHDA in young and aged rats. Brain Res 2001;896:56–63.
Decressac M, Kadkhodaei B, Mattsson B, Laguna A, Perlmann T, Björklund A. α-Synuclein-induced down-regulation of Nurr1 disrupts GDNF signaling in nigral dopamine neurons. Sci Transl Med 2012;4:156–163.
Gill SS, Patel NK, Hotton GR, et al. Direct brain infusion of glial cell line-derived neurotrophic factor in Parkinson disease. Nat Med 2003;9:589–595.
Lang AE, Gill S, Patel NK, et al. Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson disease. Ann Neurol 2006;59:459–466.
AAV2-GDNF for Advanced Parkinson’s Disease. Clinicaltrials.gov. Available at: www.http://clinicaltrials.gov/ct2/show/NCT01621581. Accessed August 16, 2013.
Marks WJ Jr, Bartus RT, Siffert J, et al. Gene delivery of AAV2-neurturin for Parkinson’s disease: a double-blind, randomised, controlled trial. Lancet Neurol 2010;9:1164–1172.
Investigation of Cogane (PYM50028) in Early-stage Parkinson’s Disease (CONFIDENT-PD). Clinicaltrials.gov. Available at: www.http://clinicaltrials.gov/ct2/show/NCT01060878. Accessed August 16, 2013.
Steiner JP, Connolly MA, Valentine HL, et al. Neurotrophic actions of nonimmunosuppressive analogues of immunosuppressive drugs FK506, rapamycin and cyclosporin A. Nat Med 1997;3:421–428.
Khan Z, Ferrari G, Kasper M, et al. The nonimmunosuppressive immunophilin ligand GPI-1046 potently stimulates regenerating axon growth from adult mouse dorsal root ganglia cultured in Matrigel. Neuroscience 2002;114:601–609.
Zhang C, Steiner JP, Hamilton GS, Hicks TP, Poulter MO. Regeneration of dopaminergic function in 6-hydroxydopamine-lesioned rats by neuroimmunophilin ligand treatment. J Neurosci 2001;21:RC156.
Tanaka K, Yoshioka M, Miyazaki I, Fujita N, Ogawa N. GPI1046 prevents dopaminergic dysfunction by activating glutathione system in the mouse striatum. Neurosci Lett 2002;321:45–48.
Tanaka K, Fujita N, Ogawa N. Immunosuppressive (FK506) and non-immunosuppressive (GPI1046) immunophilin ligands activate neurotrophic factors in the mouse brain. Brain Res 2003;970:250–253.
Poulter MO, Payne KB, Steiner JP. Neuroimmunophilins: a novel drug therapy for the reversal of neurodegenerative disease? Neuroscience 2004;128:1–6.
Mody I, MacDonald JF. NMDA receptor-dependent excitotoxicity: the role of intracellular Ca2+ release. Trends Pharmacol Sci 1995;16:356–359.
Jankovic J, Hunter C. A double-blind, placebo-controlled and longitudinal study of riluzole in early Parkinson’s disease. Parkinsonism Relat Disord 2002;8:271–276.
Rascol O, Olanow W, Brooks D, Koch G, Truffinet P, Bejuit R. A 2-year multicenter, placebo-controlled, double-blind, parallel group study of the effect of riluzole on Parkinson’s disease progression. Mov Disord 2002;17 (Suppl. 5):A80.
Parkinson Study Group PRECEPT Investigators. Mixed lineage kinase inhibitor CEP-1347 fails to delay disability in early Parkinson disease. Neurology 2007;69:1480–1490.
Olanow CW, Schapira AH, LeWitt PA, et al. TCH346 as a neuroprotective drug in Parkinson’s disease: a double-blind, randomised, controlled trial. Lancet Neurol 2006;5:1013–1020.
Tikka T, Fiebich BL, Goldsteins G, Keinanen R, Koistinaho J. Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J Neurosci 2001;21:2580–2588.
Tikka TM, Koistinaho JE. Minocycline provides neuroprotection against N-methyl-D-aspartate neurotoxicity by inhibiting microglia. J Immunol 2001;166:7527–7533.
Du Y, Ma Z, Lin S, et al. Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson’s disease. Proc Natl Acad Sci U S A 2001;98:14669–14674.
He Y, Appel S, Le W. Minocycline inhibits microglial activation and protects nigral cells after 6-hydroxydopamine injection into mouse striatum. Brain Res 2001;909:187–193.
Schneider JS, Gollomp SM, Sendek S, Colcher A, Cambi F, Du W. A randomized, controlled, delayed start trial of GM1 ganglioside in treated Parkinson’s disease patients. J Neurol Sci 2013;324:140–148.
Bezard E, Yue Z, Kirik D, Spillantini MG. Animal models of Parkinson’s disease: limits and relevance to neuroprotection studies. Mov Disord 2013;57:61–70.
Olanow CW, Kordower J. Modeling Parkinson’s disease. Ann Neurol 2009;66:432–436.
Dawson TM, Ko HS, Dawson VL. Genetic animal models of Parkinson’s disease. Neuron 2010;66:646–661.
Stocchi F, Olanow CW. Obstacles to the development of a neuroprotective therapy for Parkinson’s disease. Mov Disord 2013;28:3–7.
Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, Lee VM. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012;338:949–953.
Kieburtz K. Issues in neuroprotection clinical trials in Parkinson’s disease. Neurology 2006;66:S50-57.
Lang AE, Melamed E, Poewe W, Rascol O. Trial designs used to study neuroprotective therapy in Parkinson’s disease. Mov Disord 2013;28:86–95.
The Unified Parkinson’s Disease Rating Scale (UPDRS): status and recommendations. Mov Disord 2003;18:738–750.
Goetz CG, Tilley BC, Shaftman SR, et al., for the Movement Disorder Society UPDRS Revision Task Force. Movement Disorder Society-sponsored revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS): scale presentation and clinimetric testing results. Mov Disord. 2008;23:2129–2170.
Lang AE, Obeso JA. Time to move beyond nigrostriatal dopamine deficiency in Parkinson’s disease. Ann Neurol 2004;55:761–765.
Shi M, Bradner J, Hancock AM, et al. Cerebrospinal fluid biomarkers for Parkinson disease diagnosis and progression. Ann Neurol 2011;69:570–580.
Rentzos M, Nikolaou C, Andreadou E, et al. Circulating interleukin-10 and interleukin-12 in Parkinson’s disease. Acta Neurol Scand 2009;119:332–337.
Agarwal PA, Stoessl AJ. Biomarkers for Trials of Neuroprotection in Parkinson’s Disease. Mov Disord 2013;28:71–85.
Dragalin V. An introduction to adaptive designs and adaptation in CNS trials. Eur Neuropsychopharmacol. 2011;21:153–158.
Michell AW, Lewis SJ, Foltynie T, Barker RA. Biomarkers and Parkinson’s disease. Brain 2004;127:1693–1705.
Dexter DT, Wells FR, Lees AJ, et al. Increased nigral iron content and alterations in other metal ions occurring in brain in Parkinson’s disease. J Neurochem 1989;52:1830–1836.
Efficacy and Safety of the Iron Chelator Deferiprone in Parkinson’s Disease (FAIR-PARK-I). Clinicaltrials.gov. Available at: www.clinicaltrials.gov/ct2/show/NCT00943748. Accessed August 16, 2013.
Ames BN, Cathcart R, Schwiers E, Hochstein P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: a hypothesis. Proc Natl Acad Sci U S A 1981;78:6858–6862.
de Lau LM, Koudstaal PJ, Hofman A, Breteler MM. Serum uric acid levels and the risk of Parkinson disease. Ann Neurol 2005;58:797–800.
Weisskopf MG, O’Reilly E, Chen H, Schwarzschild MA, Ascherio A. Plasma urate and risk of Parkinson’s disease. Am J Epidemiol 2007;166:561–567.
Alonso A, Rodriguez LA, Logroscino G, Hernan MA. Gout and risk of Parkinson disease: a prospective study. Neurology 2007;69:1696–1700.
Schwarzschild MA, Schwid SR, Marek K, et al. Serum urate as a predictor of clinical and radiographic progression in Parkinson disease. Arch Neurol 2008;65:716–723.
Gao X, Chen H, Choi HK, Curhan G, Schwarzschild MA, Ascherio A. Diet, urate, and Parkinson’s disease risk in men. Am J Epidemiol 2008;167:831–838.
Safety of Urate Elevation in Parkinson’s Disease (SURE-PD). ClinicalTrials.gov. Available at: www.clinicaltrials.gov/ct2/show/NCT00833690. Accessed August 16, 2013.
Yürekli VA, Gürler S, Nazıroğlu M, Uğuz AC, Koyuncuoğlu HR. Zonisamide attenuates MPP+−induced oxidative toxicity through modulation of Ca2+ signaling and caspase-3 activity in neuronal PC12 cells. Cell Mol Neurobiol 2013;33:205–212.
Choudhury ME, Moritoyo T, Kubo M, et al. Zonisamide-induced long-lasting recovery of dopaminergic neurons from MPTP-toxicity. Brain Res 2011;1384:170–178.
Study of Zonisamide in Early Parkinson Disease (ZONIST). ClinicalTrials.gov. Available at: www.clinicaltrials.gov/ct2/show/NCT01766128. Accessed August 16, 2013.
Hauser RA, Lyons KE, McClain T, et al. Randomized, doubleblind, pilot evaluation of intravenous glutathione in Parkinson’s disease. Mov Disord 2009;24:979–983.
Okun MS, Lang A, Jankovic J. Reply: Based on the available randomized trial patients should say no to glutathione for Parkinson’s disease. Mov Disord. 2010;25:961–962.
Intranasal Glutathione in Parkinson’s Disease (inGSH in PD). ClinicalTrials.gov. Available at: www.clinicaltrials.gov/ct2/show/NCT01398748. Accessed August 16, 2013.
N-Acetylcysteine for Neuroprotection in Parkinson’s Disease (NAC for PD). ClinicalTrials.gov. Available at: www.clinicaltrials.gov/ct2/show/NCT01470027. Accessed August 16, 2013.
Guo S, Yan J, Yang T, Yang X, Bezard E, Zhao B. Protective effects of green tea polyphenols in the 6-OHDA rat model of Parkinson’s disease through inhibition of ROS-NO pathway. Biol Psychiatry 2007;62:1353–1362.
Efficacy and Safety of Green Tea Polyphenol in De Novo Parkinson’s Disease Patients. ClinicalTrials.gov. Available at: http://www.clinicaltrials.gov/ct2/show/NCT00461942. Accessed August 16, 2013.
Ascherio A, Zhang SM, Hernan MA, et al. Prospective study of caffeine consumption and risk of Parkinson’s disease in men and women. Ann Neurol 2001;50:56–63.
Palacios N, Gao X, McCullough ML, et al. Caffeine and risk of Parkinson’s disease in a large cohort of men and women. Mov Disord 2012;27:1276–82.
Schwarzschild MA, Agnati L, Fuxe K, Chen JF, Morelli M. Targeting adenosine A2A receptors in Parkinson’s disease. Trends Neurosci 2006;29:647–654.
Kalia LV, Brotchie JM, Fox SH. Novel nondopaminergic targets for motor features of Parkinson’s disease: review of recent trials. Mov Disord 2013;28:131–144.
A Placebo- and Active-Controlled Study of Preladenant in Early Parkinson’s Disease (P05664 AM5). ClinicalTrials.gov. Available at: www.clinicaltrials.gov/ct2/show/NCT01155479. Accessed August 16, 2013.
Tansey MG, McCoy MK, Frank-Cannon TC. Neuroinflammatory mechanisms in Parkinson’s disease: potential environmental triggers, pathways, and targets for early therapeutic intervention. Exp Neurol 2007;208:1–25.
McGeer EG, McGeer PL. The role of anti-inflammatory agents in Parkinson’s disease. CNS Drugs 2007;21:789–797.
McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988;38:1285–1291.
McGeer PL, Schwab C, Parent A, Doudet D. Presence of reactive microglia in monkey substantia nigra years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine administration. Ann Neurol 2003;54:599–604.
Sherer TB, Betarbet R, Kim JH, Greenamyre JT. Selective microglial activation in the rat rotenone model of Parkinson’s disease. Neurosci Lett 2003;341:87–90.
Mogi M, Harada M, Kondo T, et al. Interleukin-1 beta, interleukin-6, epidermal growth factor and transforming growth factor-alpha are elevated in the brain from parkinsonian patients. Neurosci Lett 1994;180:147–150.
Goldknopf IL, Sheta EA, Bryson J, et al. Complement C3c and related protein biomarkers in amyotrophic lateral sclerosis and Parkinson’s disease. Biochem Biophys Res Commun 2006;342:1034–1039.
Esposito E, Di Matteo V, Benigno A, Pierucci M, Crescimanno G, Di Giovanni G. Non-steroidal anti-inflammatory drugs in Parkinson’s disease. Exp Neurol 2007;205:295–312.
Chen H, Zhang SM, Hernan MA, et al. Nonsteroidal anti-inflammatory drugs and the risk of Parkinson disease. Arch Neurol 2003;60:1059–1064.
Chen H, Jacobs E, Schwarzschild MA, et al. Nonsteroidal antiinflammatory drug use and the risk for Parkinson’s disease. Ann Neurol 2005;58:963–967.
Bower JH, Maraganore DM, Peterson BJ, Ahlskog JE, Rocca WA. Immunologic diseases, anti-inflammatory drugs, and Parkinson disease: a case–control study. Neurology 2006;67:494–496.
Hernan MA, Logroscino G, Garcia Rodriguez LA. Nonsteroidal anti-inflammatory drugs and the incidence of Parkinson disease. Neurology 2006;66:1097–1099.
Selley ML. Simvastatin prevents 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced striatal dopamine depletion and protein tyrosine nitration in mice. Brain Res 2005;1037:1–6.
Di Napoli P, Taccardi AA, Oliver M, De Caterina R. Statins and stroke: evidence for cholesterol independent effects. Eur Heart J 2002;23:1908–1921.
Wolozin B, Wang SW, Li NC, Lee A, Lee TA, Kazis LE. Simvastatin is associated with a reduced incidence of dementia and Parkinson’s disease. BMC Med 2007;5:20.
Wahner AD, Bronstein JM, Bordelon YM, Ritz B. Statin use and the risk of Parkinson disease. Neurology 2008;70:1418–1422.
de Lau LM, Koudstaal PJ, Hofman A, Breteler MM. Serum cholesterol levels and the risk of Parkinson’s disease. Am J Epidemiol 2006;164:998–1002.
Huang X, Chen H, Miller WC, et al. Lower low-density lipoprotein cholesterol levels are associated with Parkinson’s disease. Mov Disord 2007;22:377–381.
Carta AR, Pisanu A. Modulating microglia activity with PPAR-γ agonists: a promising therapy for Parkinson’s disease? Neurotox Res 2013;23:112–123.
Pioglitazone in Early Parkinson’s Disease. ClinicalTrials.gov. Available at: http://www.clinicaltrials.gov/ct2/show/NCT01280123. Accessed August 16, 2013.
Chan CS, Gertler TS, Surmeier DJ. A molecular basis for the increased vulnerability of substantia nigra dopamine neurons in aging and Parkinson’s disease. Mov Disord 2010;25:S63-70.
Pasternak B, Svanström H, Nielsen NM, Fugger L, Melbye M, Hviid A. Use of calcium channel blockers and Parkinson’s disease. Am J Epidemiol 2012;175:627–635.
Marras C, Gruneir A, Rochon P, et al. Dihydropyridine calcium channel blockers and the progression of parkinsonism. Ann Neurol 2012;71:362–369.
Simuni T, Borushko E, Avram MJ, et al. Tolerability of isradipine in early Parkinson’s disease: a pilot dose escalation study. Mov Disord 2010;25:2863–2866.
Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of α-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci 2013;14:38–48.
Visanji NP, Brooks PL, Hazrati LN, Lang AE. The prion hypothesis in Parkinson’s disease: Braak to the future. Acta Neuropathol Comm 2013;1:2.
Kalia LV, Kalia SK, McLean PJ, Lozano AM, Lang AE. α-Synuclein oligomers and clinical implications for Parkinson disease. Ann Neurol 2013;73:155–169.
Tolerability and Safety of Subcutaneous Administration of Two Doses of AFFITOPE® PD01A in Early Parkinson’s Disease. Clinicaltrials.gov. Available at: http://clinicaltrials.gov/ct2/show/NCT01568099. Accessed August 16, 2013.
Bodles AM, El-Agnaf OM, Greer B, Guthrie DJ, Irvine GB. Inhibition of fibril formation and toxicity of a fragment of alpha-synuclein by an N-methylated peptide analogue. Neurosci Lett 2004;359:89–93.
Amer DA, Irvine GB, El-Agnaf OM. Inhibitors of alpha-synuclein oligomerization and toxicity: a future therapeutic strategy for Parkinson’s disease and related disorders. Exp Brain Res 2006;173:223–233.
Kalia SK, Kalia LV, McLean PJ. Molecular chaperones as rational drug targets for Parkinson’s disease therapeutics. CNS Neurol Disord Drug Targets 2010;9:741–753.
Qiao L, Hamamichi S, Caldwell KA, et al. Lysosomal enzyme cathepsin D protects against alpha-synuclein aggregation and toxicity. Mol Brain 2008;1:17.
Yu D, Pendergraff H, Liu J, et al. Single-stranded RNAs use RNAi to potently and allele-selectively inhibit mutant huntingtin expression. Cell 2012;150:895–908.
Gilks WP, Abou-Sleiman PM, Gandhi S, et al. A common LRRK2 mutation in idiopathic Parkinson’s disease. Lancet 2005;365:415–416.
Healy DG, Falchi M, O’Sullivan SS, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case–control study. Lancet Neurol 2008;7:583–590.
Lesage S, Ibanez P, Lohmann E, et al. G2019S LRRK2 mutation in French and North African families with Parkinson’s disease. Ann Neurol 2005;58:784–787.
Ozelius LJ, Senthil G, Saunders-Pullman R, et al. LRRK2 G2019S as a cause of Parkinson’s disease in Ashkenazi Jews. N Engl J Med 2006;354:424–425.
West AB, Moore DJ, Biskup S, et al. Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A 2005;102:16842–16847.
West AB, Moore DJ, Choi C, et al. Parkinson’s disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum Mol Genet 2007;16:223–232.
Ramsden N, Perrin J, Ren Z, et al. Chemoproteomics-based design of potent LRRK2-selective lead compounds that attenuate Parkinson’s disease-related toxicity in human neurons. ACS Chem Biol 2011;6:1021–1028.
Liu Z, Hamamichi S, Lee BD, et al. Inhibitors of LRRK2 kinase attenuate neurodegeneration and Parkinson-like phenotypes in Caenorhabditis elegans and Drosophila Parkinson’s disease models. Hum Mol Genet 2011;20:3933–3942.
Aviles-Olmos I, Dickson J, Kefalopoulou Z, et al. Exenatide and the treatment of patients with Parkinson’s disease. J Clin Invest 2013 May 20 [Epub ahead of print].
Efficacy of Transdermal Nicotine, on Motor Symptoms in Advanced Parkinson’s Disease (NICOPARK2). Clinicaltrials.gov. Available at: www.clinicaltrials.gov/ct2/show/NCT00873392. Accessed August 16, 2013.
Disease-modifying Potential of Transdermal NICotine in Early Parkinson’s Disease (NIC-PD). Clinicaltrials.gov. Available at: www.clinicaltrials.gov/ct2/show/ NCT01560754. Accessed August 16, 2013.
Xue YQ, Zhao LR, Guo WP, Duan WM. Intrastriatal administration of erythropoietin protects dopaminergic neurons and improves neurobehavioral outcome in a rat model of Parkinson’s disease. Neuroscience 2007;146:1245–1258.
Pedroso I, Bringas ML, Aguiar A, et al. Use of Cuban recombinant human erythropoietin in Parkinson’s disease treatment. MEDICC Rev 2012;14:11–17.
McCollum M, Ma Z, Cohen E, et al. Post-MPTP treatment with granulocyte colony-stimulating factor improves nigrostriatal function in the mouse model of Parkinson’s disease. Mol Neurobiol 2010;41:410–419.
Study of the Neuro-protective Effect of Granulocyte-colony Stimulating Factor on Early Stage Parkinson’s Disease. Clinicaltrials.gov. Available at: www.clinicaltrials.gov/ct2/show/NCT01227681. Accessed August 16, 2013.
The ParkCycle Study. Clinicaltrials.gov. Available at: www.clinicaltrials.gov/ct2/show/ NCT01562496. Accessed August 16, 2013.
Pahwa R, Factor SA, Lyons KE, et al. Practice parameter: treatment of Parkinson disease with motor fluctuations and dyskinesia (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2006;66:983–995.
Fox SHKR, Lim SY, Ravina B, et al. The Movement Disorder Society evidence-based medicine review update: treatments for the motor symptoms of Parkinson’s disease. Mov Disord 2011;26:S2-S41.
Anderson VC, Burchiel KJ, Hogarth P, Favre J, Hammerstad JP. Pallidal vs subthalamic nucleus deep brain stimulation in Parkinson disease. Arch Neurol 2005;62:554–560.
Follett KA, Weaver FM, Stern M, et al. Pallidal versus subthalamic deep-brain stimulation for Parkinson’s disease. N Engl J Med 2010;362:2077–2091.
Lozano AM, Dostrovsky J, Chen R, Ashby P. Deep brain stimulation for Parkinson’s disease: disrupting the disruption. Lancet Neurol 2002;1:225–231.
Kita H., Kitai S.T. Efferent projections of the subthalamic nucleus in the rat: light and electron microscopic analysis with the PHA-L method. J. Comp. Neurol 1987;260:435–452.
Temel Y, Blokland A, Steinbusch HW, Visser-Vandewalle V. The functional role of the subthalamic nucleus in cognitive and limbic circuits. Prog Neurobiol 2005;76:393–413.
Rodriguez MC, Obeso JA, Olanow CW. Subthalamic nucleus-mediated excitotoxicity in Parkinson’s disease: a target for neuroprotection. Ann Neurol 1998;44:S175-S188.
Temela Y, Visser-Vandewalle V, Kapland S, et al. Protection of nigral cell death by bilateral subthalamic nucleus stimulation. Brain Res 2006;1120:100–105.
Deep Brain Stimulation (DBS) for Early Stage Parkinson’s Disease (PD). Clinicaltrials.gov. Available at: www.clinicaltrials.gov/ct2/show/NCT00282152. Accessed August 16, 2013.
LeWitt PA, Rezai AR, Leehey MA, et al. AAV2-GAD gene therapy for advanced Parkinson’s disease: a double-blind, sham-surgery controlled, randomised trial. Lancet Neurol 2011;10:309–319.
Park HJ, Lee PH, Bang OY, Lee G, Ahn YH. Mesenchymal stem cells therapy exerts neuroprotection in a progressive animal model of Parkinson’s disease. J Neurochem 2008;107:141–151.
Sadan O, Bahat-Stromza M, Barhum Y, et al. Protective effects of neurotrophic factor-secreting cells in a 6-OHDA rat model of Parkinson disease. Stem Cells Dev 2009;18:1179–1190.
Brundin P, Strecker RE, Lindvall O, et al. Intracerebral grafting of dopamine neurons. Experimental basis for clinical trials in patients with Parkinson’s disease. Ann N Y Acad Sci 1987; 495,473-496.
Nishimura K, Takahashi J. Therapeutic application of stem cell technology toward the treatment of Parkinson’s disease. Biol Pharm Bull 2013;36:171–175.
Mendez I, Dagher A, Hong M, et al. Simultaneous intrastriatal and intranigral fetal dopaminergic grafts in patients with Parkinson disease: a pilot study. Report of three cases. J Neurosurg 2002;96:589–596.
Gross RE, Watts RL, Hauser RA, et al. Intrastriatal transplantation of microcarrier-bound human retinal pigment epithelial cells versus sham surgery in patients with advanced Parkinson’s disease: a double-blind, randomised, controlled trial. Lancet Neurol 2011;10:509–519.
Freed CR, Greene PE, Breeze RE, et al. Transplantation of embryonic dopamine neurons for severe Parkinson’s disease. N Engl J Med 2001;344:710–719.
Olanow CW, Goetz CG, Kordower JH, et al. A double-blind controlled trial of bilateral fetal nigral transplantation in Parkinson’s disease. Ann Neurol 2003;54:403–414.
Kriks S, Shim JW, Piao J, et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 2011;480:547–551.
Karussis D, Petrou P, Kassis I. Clinical experience with stem cells and other cell therapies in neurological diseases. J Neurol Sci 2013;324:1–9.
Lang AE, Obeso JA. Challenges in Parkinson’s disease: restoration of the nigrostriatal dopamine system is not enough. Lancet Neurol 2004;3:309–316.
Olanow CW, Kordower JH, Lang AE, Obeso JA. Dopaminergic transplantation for Parkinson’s disease: current status and future prospects. Ann Neurol 2009;66:591–596.
Tetrud JW, Langston JW. The effect of deprenyl (selegiline) on the natural history of Parkinson’s disease. Science 1989;245:519–522.
The Parkinson Study Group. Effects of tocopherol and deprenyl on the progression of disability in early Parkinson’s disease. N Engl J Med 1993;328:176–183.
Myllylä VV, Sotaniemi KA, Vuorinen JA, Heinonen EH. Selegiline as initial treatment in de novo parkinsonian patients. Neurology 1992;42:339–343.
Parkinson Study Group. Impact of deprenyl and tocopherol treatment on Parkinson’s disease in DATATOP subjects not requiring levodopa. Ann Neurol 1996;39:29–36.
Pålhagen S, Heinonen EH, Hägglund J, et al. Selegiline delays the onset of disability in de novo parkinsonian patients. Swedish Parkinson Study Group. Neurology 1998;51:520–525.
NINDS NET-PD Investigators. A randomized clinical trial of coenzyme Q(10) and GPI-1485 in early Parkinson disease. Neurology 2007;68:20–28.
Parkinson Study Group. Pramipexole vs levodopa as initial treatment for Parkinson disease: A randomized controlled trial. Parkinson Study Group. JAMA 2000;284:1931–1938.
The GPI 1485 Study Group. GPI 1485, a neuroimmunophilin ligand, fails to alter disease progression in mild to moderate Parkinson’s disease. Mov Disord 2006;21:A1009.
Ribeiro MJ, Stoessl AJ, Brooks D, et al. Evaluation of a potential neurotrophic drug on the progression of Parkinson disease with 18FDOPA. J Nucl Med 2009;50(Suppl. 2):A125.
Schneider JS, Sendek S, Daskalakis C, Cambi F. GM1 ganglioside in Parkinson’s disease: results of a five year open study. J Neurol Sci 2010;292:45–51.
Acknowledgments
LVK is supported by a Canadian Institutes of Health Research Clinician-Scientist Award. AEL holds the Jack Clark Chair in Parkinson’s Disease Research at the University of Toronto. The authors have no real or perceived conflicts of interest. Full conflict of interest disclosures are available in the electronic supplementary material for this article.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Since the review and acceptance of this manuscript, the NINDS has stopped the NET‐PD long‐term study of creatine for treatment of early stage PD. According to the NET‐PD website, an interim analysis revealed evidence for futility. Continued follow‐up of subjects was not expected to demonstrate a statistically significant difference between creatine and placebo. Hence, the study’s Data Safety Monitoring Board recommended termination of the study.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 511 kb)
Rights and permissions
About this article
Cite this article
AlDakheel, A., Kalia, L.V. & Lang, A.E. Pathogenesis-Targeted, Disease-Modifying Therapies in Parkinson Disease. Neurotherapeutics 11, 6–23 (2014). https://doi.org/10.1007/s13311-013-0218-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-013-0218-1