
Abstract
Streptomyces griseorubens E44G is a chitinolytic bacterium isolated from cultivated soil in Saudi Arabia (a hot, arid climatic region). In vitro, antifungal potential of S. griseorubens E44G was assessed against the phytopathogenic fungus, Fusarium oxysporum f. sp. lycopersici (the causative agent of the Fusarium wilt disease of tomato). An inhibition zone of 24 mm was recorded. The chitinolytic activity of S. griseorubens E44G was proved when the colloidal chitin agar plate method was used. A thermostable chitinase enzyme of 45 kDa molecular weight was purified using gel filtration chromatography. The optimum activity was obtained at 60 °C and pH 5.5. The purified enzyme has shown a very pronounced activity against the phytopathogenic fungus, F. oxysporum. The molecular characterization of the chitinase gene indicated that it consists of 1218 bp encoding 407 amino acids. The phylogentic analysis based on the nucleotide DNA sequence and the deduced amino acids sequence showed high similarity percentages with other chitinases isolated from different Streptomyces species. In the field evaluation, application of both S. griseorubens E44G treatments significantly increased all tested growth and yield parameters and decreased the disease severity compared with the infected-untreated tomato plants suggesting potential as a biocontrol agent.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Fusarium wilt, caused by Fusarium oxysporum f. sp. lycopersici, is one of the most serious diseases of tomato over the world. It causes significant damages to the plant, leading to a high economic loss in tomato yield (Suárez-Estrella et al. 2007). Chemical fungicides, such as azoxystrobin, benomyl, prochloraz, and carbendazim, are available, but the use of the chemical fungicides is undesirable due to their negative effects on non-target organisms, human, and animals (Arcury and Quandt 2003). Biological control represents an alternative safe and potential option for plant diseases management (Gnanamanickam 2002).
Chitinases are glycosyl hydrolase enzymes which catalyze the hydrolytic cleavage of the β-1,4-linkages. Chitinases gained much significance due to the fact that they have a vast bio-processing and biotechnological aspects. In agriculture, they have a potential application in the biological control of the phytopathogenic fungi. Their antifungal activity is attributed to the hydrolytic effects on chitin, which constitutes the main structural component of fungi. Chitinase enzyme is known to be produced by several organisms, including bacteria and actinomycetes (Brzezinska et al. 2013; Saber et al. 2015), fungi (Karthik et al. 2014), plants, invertebrates and vertebrates (Siroski et al. 2014). Streptomyces strains are considered as the major producers of chitinases in soil. Due to their antifungal activity, Streptomyces strains have been widely studied as biological control agents against different plant pathogenic fungi (Al-Askar et al. 2011; Alekhya and Gopalakrishnan 2014; Al-Askar et al. 2015a).
In our previous researches, several actinomycetal strains isolated from soil in Saudi Arabia were screened for their antifungal potential. One of the promising chitinase producing isolates was identified as S. griseorubens E44G (Al-Askar et al. 2014; Al-Askar et al. 2015b). This study deals with 1) in vitro screening of S. griseorubens E44G for antifungal potentiality against the phytopathogenic fungus F. oxysporum, 2) purification and characterization of the chitinase enzyme from S. griseorubens E44G, and 3) evaluation of its application for the possible use as a biocontrol agent against Fusarium wilt disease of tomato under field conditions.
Materials and methods
Microorganisms
The fungal pathogen was isolated from tomato plants showing the Fusarium wilt disease symptoms. The pathogenic fungus was cultured on plates containing potato dextrose agar medium (PDA) (Difco, Detroit, MI, USA, Cat. No. 213400) and incubated for 72 h at 25 ± 2 °C, then purified using single spore technique. Identification of the fungal pathogen was done according to its cultural and morphological characteristics (Domsch et al. 1980). Stock cultures of F. oxysporum were kept on slants of PDA at 5 °C.
In a previous study, the used bacterial strain was isolated from a cultivated soil sample collected from Saudi Arabia. Identification of the isolated bacterium (S. griseorubens E44G) was done according to the morphological, biochemical and physiological characteristics (Buchanan and Gibbons 1974) and molecularly confirmed using 16S rRNA gene (Al-Askar et al. 2014). Stock culture of the bacterium was maintained on starch nitrate/NaCl slants at 4 °C.
Screening for antifungal potentiality
Starch nitrate agar plates (3 replicates) inoculated with F. oxysporum as a spore suspension before solidification were prepared. In the centre of the plate, a disk (6 mm diameter) from a newly grown culture of S. griseorubens E44G was placed and incubated at 31 °C. The antifungal potentiality was recorded in the form of a clear zone, if any, after 3 days of incubation.
Assay for chitinolytic potentiality
Chitinolytic potentiality was assessed using colloidal chitin agar plate (Mitsutomi et al. 1995) containing 0.1% colloidal chitin and 1.5% agar and incubated at 31 °C. After 72 h, the development of a clear zone around the disc was observed.
Culturing conditions
A culture of S. griseorubens E44G was cultured on ISP-2 broth media containing chitin (5 g.L−1, HiMedia, India, Cat. No. 1398–61-4) as a substrate and the pH was adjusted to 7. The bacterial culture was incubated at 31 °C for three days with agitation (150 rpm), and then centrifuged at 8000 rpm for 15 min at 5 °C. Using a sterile 0.45 Millipore filter membrane, the supernatant was filtered and then kept at 5 °C for further purification of the chitinase.
Chitinase activity assay
The chitinase activity in the culture supernatant and at all purification steps was estimated as described by Khan et al. (2010). The reaction solution for the chitinase activity contained 50 mM acetate buffer; 1 mL, 5% colloidal chitin; 1 mL at pH 5 and crude enzyme; 1 mL, and incubated at 50 °C for 1 h. To stop the reaction, boiling for 15 min was done then the solution was centrifuged for 15 min at 5000 rpm. The chitinase activity was measured spectrophotometrically at 540 nm using a spectrophotometer (UV-260, Shimadzu, Japan) based on the concentration of released GlcNAc (Monreal and Reese 1969). The quantity of enzyme that produces 1 mM.min−1 of GlcNAc under the described assay conditions was considered as the unit of chitinase activity.
Preparation of colloidal chitin
The colloidal chitin was prepared according to Khan et al. (2010). In 250 mL of conc. Hydrochloric acid, 20 g of chitin powder was dissolved with continuous stirring at 5 °C for 1 h. To remove the excess of the acid, the chitin was washed by distilled water 5 times, then filtered using filter paper. The collected colloidal chitin was kept as a paste at 5 °C.
Estimation of protein content
Estimation of the protein content was done as described by Bradford (1976). The reaction mixture was composed of the enzyme sample; 10 μ.mL−1, Bradford solution; 990 μ.mL−1 (Coomassie Brilliant Blue G-250, Serva, Heidelberg, Germany, Cat. No. 17524.01) and adjusted to volume of 200 mL using distilled water. After 3 min, the absorbance was recorded using a spectrophotometer at 595 nm. For blank sample, 0.9% NaCl (10 μ.mL−1) was used. To make the standard curve, Bovine serum albumin (Sigma, USA, Cat. No. A9056) was used.
Purification of chitinase
Enzyme precipitation
Using ammonium sulphate (70%) the protein content was precipitated in the supernatant. The crude enzyme was continuously stirred using magnetic stirrer and kept at 5 °C for 12 h, then centrifuged at 8000 rpm for 20 min, then the pellet was collected and resuspended in sodium acetate buffer (pH 5) and dialyzed overnight.
Gel filtration chromatography
The re-suspended protein pellet was filtrated by filter (0.45 μm pore size) then loaded into a Mono Q column (GE Healthcare Life science, Piscataway, NJ, USA) using 50 mM sodium acetate buffer at pH 5. Fractions (3 mL) were collected with flow rate of 1 mL.min−1. After the assessment of the collected fractions for chitinase enzyme, the positive fractions were pooled and loaded into a column of Sephadex G-100 (Sigma, USA, Cat. No. G100120) using 50 mM sodium acetate buffer at pH 5 with NaCl (100 mM) at 0.5 mL.min−1 flow rate.
Sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE)
The purified chitinase enzyme was subjected to polyacrylamide gel electrophoresis as described by Laemmli (1970). Gels were then stained over night (Coomassie Brilliant Blue G-250; 0.25 g.mL−1, methanol; 25% and acetic acid; 10%) and then destained in 25% methyl alcohol and acetic acid for 12 h. Standard proteins were used as markers (Fermentas Life Science, CA, USA).
Characterisation of chitinase enzyme
Effect of pH on chitinase activity and stability
The optimum pH for chitinase enzyme was estimated by varying the pH value of the reaction mixture between 3 and 9.5 at 0.5 pH unit intervals. To adjust the pH value the following buffers were used (Tris-HCl buffer (50 mM) for pH 8–9.5, phosphate buffer (50 mM) for pH 7, and acetate buffer (50 mM) for pH 3–6). For estimation of the enzyme activity, 2 mL of the buffer was added to the enzyme solution (100 mL) and incubated at 36 °C for 31 min. pH stability of the chitinase was estimated by incubation of the enzyme solution at different pHs at the same temperature without the substrate for 1 h.
Effect of temperature on chitinase activity and stability
To determine the optimum temperature for the chitinase activity, the reaction mixture was incubated at the best pH (5.5) at different temperatures (20–80 °C) for 35 min. Thermal stability of the purified enzyme was determined by estimating the residual enzyme activity after the incubation at the previous temperatures at pH 5.5 for 1 h without the substrate.
Antifungal potential of the purified enzyme
The antifungal potential was estimated for the purified enzyme using the agar well diffusion method (Taechowisan et al. 2003). A spore suspension of the tested fungi was prepared and adjusted at the concentration of 105 spore.mL−1 and inoculated to PDA plates before solidification. Wells (7 mm diameter) were made into the agar plate using sterile cork borer. Enzyme solution (50 μL) was added to the wells and heat boiled enzyme solution was used as a control with triplicate replicates. Enzyme activity was estimated after 3 days of incubation at 25 ± 2 °C as the diameter of the inhibition zone.
Molecular characterization of chitinase gene
Chitinase gene amplification using specific PCR, cloning and sequencing
Total DNA from S. griseorubens E44G was extracted using DNA extraction kit (Qiagen, USA) according to the manufacturer’s instructions. To amplify the chitinase gene, the extracted DNA was subjected to PCR amplification according to Taechowisan et al. (2004) using two oligonucleotide primers designed based on the family 19 chitinase genes of Streptomyces sp. (5′ TTGACCCAGTGGTCCAGACC ‘3) and (5’ GTGTGCTGCTCACGCCAG ‘3). The purified PCR product was cloned into pCR™2.1-TOPO® vector (TA Cloning Kit, Invitrogen, USA) according to the manufacturer’s instructions. The ligation reaction product was transferred into Escherichia coli (DH5α) cells and the recombinant colonies were selected by blue/white screening (Maniatis et al. 1987). The plasmid DNA extracted from one white colony was subjected to DNA sequencing using ABI Prism 310 automated DNA sequencer (Applied Biosystems, USA). The obtained sequence was subjected to NCBI-BLAST searches and aligned with other similar sequences from GenBank using ClustalX (Thompson et al. 1997), BioEdit (Hall 1999) and MEGA 6 software (Tamura et al. 2011). For amino acids characterization, ExPASy proteomic tools were used to investigate the open reading frames (ORF), amino acids number and percentage and the isoelectric point.
Field evaluation
The experiment was conducted under naturally infected soil conditions in a field with a known Fusarium wilt history. The experimental unit (plot) was an area of 3 × 2 m2 containing 6 rows. One meter was skipped between plots on the same row (alleyway). Aisle (50-cm width tractor road) was skipped between blocks. The plots were ploughed well, all weeds removed, and soil was leveled. Nitrogen and phosphorous fertilizers were applied as recommended. Thirty-day old seedlings were transplanted. The experiment was laid out in a randomized complete block design. The treatments can be summarized as follow: pathogen only (P), pathogen + chemical fungicide (PF), pathogen + spore suspension of S. griseorubens E44G (PS), and pathogen + cell-free culture filtrate of S. griseorubens E44G (PA). Before transplantation, roots of tomato seedlings were immersed for 1 h in a spore suspension of S. griseorubens E44G at concentration of 106 spore.mL−1, cell-free culture filtrate or the chemical fungicide (Rhizolex-T) at the recommended dose (3 g L−1). For the negative infected control treatment, roots of tomato seedlings were immersed in plain water before the transplanting. Four plots were used for each treatment. Application of the bio-control treatments were repeated 10, 25 and 40 days after the transplantation as soil drench.
The disease severity was assessed according to the criteria developed by Epp (1987). Six weeks after the transplantation, 15 plants of each treatment were carefully pulled out with the entire root system, washed and evaluated for the growth parameters (plant height, plant fresh and dry weights, and leaves number). At physiological maturity, tomato fruits from each plot were harvested and weighed separately to determine fruit yield.
Statistical analysis
The statistical analysis software “CoStat 6.311” was used for all statistical analyses. Significant differences among treatments means were determined with Duncan’s New Multiple Range Test.
Results and discussion
Antifungal activity of S. griseorubens E44G
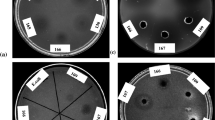
In vitro, antifungal potential of S. griseorubens E44G was assessed against the phytopathogenic fungus, F. oxysporum. After 72 h of incubation, the growth of F. oxysporum was inhibited in the form of an inhibition zone of 24 mm (Fig. 1). This result is in accordance with that of Alekhya and Gopalakrishnan (2014) who screened 27 actinomycete isolates for their antagonistic potential against fungal pathogens and recorded that eight isolates showed a broad-spectrum antagonistic activity in dual-culture assay. This antagonistic nature may be attributed to hyperparasitism, antibiosis and/or production of hydrolytic enzymes (Viterbo et al. 2007).

Antifungal potential of S. griseorubens E44G against the phytopathogenic fungus F. oxysporum
Screening for chitinolytic activity
An inhibition zone was recorded around the disc of the tested isolate indicating the chitin hydrolysis. This result proved that the chitinolitic activity is contributed to the antagonistic nature of S. griseorubens E44G. Streptomyces strains are considered as the major chitinases producers in the soil. Several chitinolytic enzymes with varying extents have been reported from different Streptomyces strains including S. rimosus, S. griseus, S. plicatus, S. lividans, and S. halstedii (Brzezinska et al. 2013; Alekhya and Gopalakrishnan 2014).
Purification of chitinase
Purification of the extracellular chitinase enzyme of S. griseorubens E44G was done by protein precipitation and gel filtration chromatography methods. At each stage, the protein content and the enzyme activity were assessed (Table 1). The crude enzyme showed a specific activity of 18.2 U.mg−1 while, ammonium sulfate precipitated enzyme showed a specific activity of 34.7 U.mg−1 with purification fold of 1.91. Maximum precipitation of chitinase enzyme was obtained at 70% of ammonium sulphate, showing the highest chitinolytic activity in comparison with the crude enzyme and other concentrations (data not shown).
Enzyme purification using a Mono Q column disclosed a specific activity of 92 U.mg−1 with purification fold of 2.65. After screening of the fractions for chitinase enzyme, the contents of positive tubes were loaded into sephadex G- 100 column. The elution profile of the chitinase solution is illustrated in Fig. 2. Among the collected fractions, the active fraction (No. 16) showed the highest specific activity of 385.6 U.mg−1 with purification fold of 4.19. Kavitha and Vijaylakshmi (2011) recorded a specific activity of 37.4 U.mg−1 with purification fold of 2.96 for chitinase enzyme from S. tendae TK-VL 333 after sephadex G-100 purification. On the other hand, according to the study of Rabeeth et al. (2011), chitinase of S. griseus MTCC9723 purified by sephadex G-100 column expressed a specific activity of 15,500 U.mg−1 and purification fold of 9.3.

Chromatogram of the chitinase enzyme from S. griseorubens E44G on Sephadex G-100 column chromatography
Molecular weight of the purified chitinase
Using SDS-PAGE analysis, the homogeneity of the purified chitinase from S. griseorubens E44G was confirmed showing a single protein band of ∼45 kDa (Fig. 3). This finding is in agreement with that reported in previous studies, where chitinases of 45 kDa molecular mass were reported by Narayana and Vijayalakshmi (2009) from Streptomyces sp. ANU 6277 and by Kavitha and Vijaylakshmi (2011) from S. tendae TK-VL 333. Different molecular weights ranged between 20 and 90 kDa have been reported for chitinase enzymes. Similar to the plant chitinases, bacterial chitinases have a molecular weight range of 20–65 kDa, but are smaller than insect chitinases (40–85 kDa) (Bhattacharya et al. 2007). Chitinase enzymes obtained from different Streptomyces were reported to have varied molecular weights as 29 kDa from Streptomyces sp. TH-11 (Hoang et al. 2011), 32 kDa, 34 kDa from S. griseus MTCC 9723 (Rabeeth et al. 2011), 45 kDa from Streptomyces sp. ANU 6277 (Narayana and Vijayalakshmi 2009), and 65 kDa from S. violascens NRRL B2700 (Gangwar et al. 2016) by SDS-PAGE analysis.

SDS-PAGE of purified chitinase of S. griseorubens E44G. M: Standard protein marker; Lane 1: purified chitinase enzyme
Characterization of chitinase enzyme
Effect of pH on the activity and stability of chitinase enzyme
Data illustrated in Fig. 4 represent the effect of pH value on the activity and stability of the purified chitinase in the pH range of 3–9.5. The enzyme activity increased with the increment of the pH value and exhibited the most activity at pH 5.5 (Fig. 4). Further increment in pH led to a reduction in the chitinase activity. However, beyond this pH value, chitinase activity rapidly declined. The enzyme was stable from pH 4–7, maintaining at least 65% activity. Many Streptomyces chitinases have been reported to show their optimum activity in acidic pH range (Rabeeth et al. 2011; Thiagarajan et al. 2011).

Effect of pH on activity and stability of chitinase enzyme from S. griseorubens E44G
Thermal stability of the purified enzyme
Figure 5 illustrated the activity of the purified enzyme at various temperatures. Chitinase enzyme of S. griseorubens E44G exhibited a considerable activity in the range of 30–70 °C. The optimum temperature was recorded as 60 °C after which, the enzyme activity decreased with further increase in the incubation temperature. The chitinase enzyme retained more than 90% activity in the range of temperature from 30 to 60 °C suggesting that the enzyme is stable up to 60 °C. The thermo stability property of the chitinase enzyme of S. griseorubens E44G provides an important feature on the industrial scale. In accordance with our results, Nagpure et al. (2013) reported the optimum temperature of 60 °C for chitinase production from S. violaceusniger MTCC 3959. On the other hand, Rabeeth et al. (2011) recorded 40 °C as the optimum temperature and stability from 20 to 50 °C for chitinase production from S. griseus MTCC9723.

Effect of temperature on the activity and stability of chitinase enzyme from S. griseorubens E44G
Antifungal activity of the purified enzyme
A clear zone of inhibition (30 mm) was recorded (Fig. 6), while the inactivated enzyme did not show any inhibition. Chitinase producing organisms could be used in biological control of plant pathogens directly using their metabolites or genes. Mukherjee and Sen (2006) recorded significant growth inhibitions for Aspergillus niger, Helminthosporium sativum and Alternaria alternata using purified chitinase from S. venezuelae P10. The same result was obtained by Ghasemi et al. (2010) against Rhizoctonia solani, Stemphyllium botryosum, Bipolaris sp. Nigrospora sp., and Verticillium sp.

Antifungal activity of the purified chitinase enzyme from S. griseorubens E44G against F. oxysporum
Molecular characterization of chitinase gene
The chitinase gene from S. griseorubens E44G showed a nucleotide sequence of 1218 bp, it was deposited in GenBank under accession no. (KJ466124). This result is in agreement with that reported by Gherbawy et al. (2012) who isolated chitinase gene with a molecular size of 1.4 kb from Streptomyces spp. The nucleotide sequence similarity analysis using BLAST revealed similarity percentages ranged from 95 to 100% with the other chitinases listed in the GenBank. Based on the DNA nucleotide sequences, the phylogentic tree was constructed using 6 different chitinase genes (all of them were isolated from different Streptomyces strains) (Fig. 7a). The phylogentic tree showed that chitinase gene from S. griseorubens E44G is identical (100%) to the chitinase gene from Streptomyces sp. C203 (accession no. EF137704). On the other hand the phylogeny based on the deduced amino acids revealed another identity matching (100%) of the chitinase from S. griseorubens E44G with that from Streptomyces sp. C203 (Fig. 7b).

Phylogentic trees of chitinase gene from S. griseorubens E44G in relation to six chitinase genes isolated from different Streptomyces strains based on the DNA nucleotide sequences (a) and the deduced amino acids (b)
The DNA nucleotide sequence of the obtained chitinase gene is divided into three main frames, frame one contains two ORFs. The first starts from base 19 to 111 (30 amino acids) and the second starts from base 250 to 402 (50 amino acids). There is no ORF in the frame 2, while, one ORF is observed in the frame 3, which started from base 3 to 1217 (405 amino acids). The total number of the deduced amino acids is 407, the theoretical molecular weight is ≈ 44 kDa, and the isoelectric point is 5.62. The obtained theoretical molecular weight is in accordance with that obtained by the SDS-PAGE analysis in this study. The analysis of amino acids composition showed that the total number of the negatively charged residues (Asp + Glu) is 39, while, the positively charged residues (Arg + Lys) is 32. Additionally, the atomic composition is; carbon (1953), hydrogen (2915), nitrogen (515), oxygen (593) and sulfur (10).
Field evaluation
Effect S. griseorubens E44G on tomato growth parameters and disease severity
The effect of the bio-control treatments on the growth parameters and disease severity of tomato plants infected with Fusarium wilt disease is presented in Table 2. The infected plants that treated with cell-free culture filtrate of S. griseorubens E44G showed a significant increase in all tested growth parameters compared with the infected-untreated tomato plants. The disease severity of the infected plants was significantly decreased when treated with the cell-free culture filtrate of S. griseorubens E44G compared with the untreated-infected plants. The infected plants treated with the spore suspension of S. griseorubens E44G ranked the second best treatment followed by that treated with the fungicide.
Effect of S. griseorubens E44G treatments on the yield of tomato
Data presented in Table 3 show the effect of S. griseorubens E44G treatments on the yield of tomato. Infection with the Fusarium wilt disease led to a significant reduction in the yield parameters of the infected tomato plants. While, the yield of the infected plants treated with the filtrate treatment significantly increased compared with the untreated infected plants and that treated with the chemical fungicide. Treatment of the infected plants with the spore suspension of S. griseorubens E44G ranked the second best treatment. The increase in the yield of the infected plant that treated with the filtrate treatment may be attributed to the growth regulating effect(s) of the filtrate components. These results are in accordance with that obtained by Yandigeri et al. (2015) using the chitinolytic actinomycete Streptomyces vinaceusdrappus S5 MW2 on tomato plants. Potential of the chitinolytic actinomycetes in the bio-control of plant diseases has been reported. In addition to antibiosis and competition, the hydrolytic enzyme action seems to contribute to their antagonistic behavior (Hoster et al. 2005). The resistance induction effect of chitin, either alone or in combination with the biocontrol agents has been recognized (Solanki et al. 2011). These important characters beside survival of S. griseorubens E44G under high temperature habitats make this isolate a as a good bio-control agent.
Conclusion
In the present study, in vitro antifungal potential of S. griseorubens E44G was observed against F. oxysporum. Chitinolytic activity was shown to be contributed to the antagonistic behavior of S. griseorubens E44G. A thermostable chitinase enzyme of 45 kDa molecular weight was purified using gel filtration chromatography. The optimum activity was obtained at 60 °C and pH 5.5. The purified enzyme has shown a very pronounced activity against the phytopathogenic fungus F. oxysporum. In the field evaluation, application of S. griseorubens E44G treatments significantly increased all tested growth and yield parameters and decreased the disease severity compared with the infected-untreated tomato plants suggesting potential as a biocontrol agent. On the other hand, the thermo stability property of the chitinase enzyme of S. griseorubens E44G may provide an important feature on the industrial scale.
References
Al-Askar, A. A., Abdul Khair, W. M., & Rashad, Y. M. (2011). In vitro antifungal activity of Streptomyces spororaveus RDS28 against some phytopathogenic fungi. African Journal of Agricultural Research, 6(12), 2835–2842.
Al-Askar, A. A., Abdulkhair, W. M., Rashad, Y. M., Hafez, E. E., Ghoneem, K. M., & Baka, Z. A. (2014). Streptomyces griseorubens E44G: A potent antagonist isolated from soil in Saudi Arabia. Journal of Pure and Applied Microbiology, 8(Spl. Edn. 2), 221–230.
Al-Askar, A. A., Rashad, Y. M., Hafez, E. E., Abdulkhair, W. M., Baka, Z. A., & Ghoneem, K. M. (2015a). Characterization of alkaline protease produced by Streptomyces griseorubens E44G and its possibility for controlling Rhizoctonia root rot disease of corn. Biotechnology & Biotechnological Equipment, 29(3), 457–462.
Al-Askar, A. A., Baka, Z. A., Rashad, Y. M., Ghoneem, K. M., Abdulkhair, W. M., Hafez, E. E., et al. (2015b). Evaluation of Streptomyces griseorubens E44G for the biocontrol of Fusarium oxysporum f. Sp. lycopersici: Ultrastructural and Cytochemical investigations. Annals of Microbiology, 65, 1815–1824.
Alekhya, G., & Gopalakrishnan, S. (2014). Characterization of antagonistic Streptomyces as potential biocontrol agent against fungal pathogens of chickpea and sorghum. Philippine Agricultural Scientist, 97(2), 191–198.
Arcury, T. A., & Quandt, S. A. (2003). Pesticides at work and at home: Exposure of migrant farm workers. The Lancet, 362, 2021.
Bhattacharya, D., Nagpure, A., & Gupta, R. K. (2007). Bacterial chitinases: Properties and potential. Critical Reviews in Biotechnology, 27, 21–28.
Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry, 72, 248–254.
Brzezinska, M. S., Jankiewicz, U., & Walczak, M. (2013). Biodegradation of chitinous substances and chitinase production by the soil actinomycete Streptomyces rimosus. International Biodeterioration and Biodegradation, 84, 104–110.
Buchanan, R. E., & Gibbons, N. E. (1974). Bergey’s manual of determinative bacteriology. Baltimore: Williams and Wilkins.
Domsch, K. W., Gams, W., & Anderson, T. H. (1980). Compendium of soil fungi. London: Academic Press.
Epp, D. (1987). Somaclonal variation in banana: A case study with Fusarium wilt. In G. J. Persley & E. A. De Langhe (Eds.), Banana and plantain breeding strategies (pp. 140–150). Canberra: ACIAR Publication.
Gangwar, M., Singh, V., Pandey, A. K., Tripathi, C. K. M., & Mishra, B. N. (2016). Purification and characterization of chitinase from Streptomyces violascens NRRL B2700. Indian Journal of Experimental Biology, 54(1), 64–71.
Ghasemi, S., Ahmadian, G., Jelodar, N. B., Rahimian, H., Ghandili, S., Dehestani, A., et al. (2010). Antifungal chitinases from Bacillus pumilus SG2: Preliminary report. World Journal of Microbiology and Biotechnology, 26(8), 1437–1443.
Gherbawy, Y., Elhariry, H., Altalhi, A., El-Deeb, B., & Khiralla, G. (2012). Molecular screening of Streptomyces isolates for antifungal activity and family 19 chitinase enzymes. The Journal of Microbiology, 50, 459–468.
Gnanamanickam, S. S. (2002). Biological control of crop diseases. New York: Marcel Dekker Inc.
Hall, T. A. (1999). BioEdit: A user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symposium Series, 41, 95–98.
Hoang, K. C., Lai, T. H., Lin, C. S., Chen, Y. T., & Liau, C. Y. (2011). The chitinolytic activities of Streptomyces sp. TH-11. International Journal of Molecular Sciences, 12, 56–65.
Hoster, F., Schmitz, J. E., & Daniel, R. (2005). Enrichment of chitinolytic microorganisms: Isolation and characterization of a chitinase exhibiting antifungal activity against phytopathogenic fungi from a novel Streptomyces strain. Applied Microbiology and Biotechnology, 66, 434–442.
Karthik, N., Akanksha, K., & Pandey, A. (2014). Production, purification and properties of fungal chitinases a review. Indian Journal of Experimental Biology, 52(11), 1025–1035.
Kavitha, A., & Vijaylakshmi, M. (2011). Partial purification and antifungal profile of chitinase produced by Streptomyces tendae TK-VL_333. Annals of Microbiology, 61, 597–603.
Khan, M. A., Hamid, R., Ahmad, M., Abdin, M. Z., & Javed, S. (2010). Optimization of culture media for enhanced chitinase production from a novel strain of Stenotrophomonas maltophilia using response surface methodology. Journal of Microbiology and Biotechnology, 20, 1597–1602.
Laemmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227, 680–685.
Maniatis, T., Goodbourn, S., & Fischer, J. A. (1987). Regulation of inducible and tissue-specific gene expression. Science, 236, 1237–1245.
Mitsutomi, M., Hata, T., & Kuwabara, T. (1995). Purification and characterization of novel chitinases from Streptomyces griseus HUT 6037. Journal of Fermentation and Bioengineering, 80, 153–158.
Monreal, J., & Reese, E. T. (1969). The chitinase of Serratia marcescens. Canadian Journal of Microbiology, 15, 689–696.
Mukherjee, G., & Sen, S. K. (2006). Purification, characterization, and antifungal activity of chitinase from Streptomyces Venezuelae P10. Current Microbiology, 53, 265–269.
Nagpure, A., Choudhary, B., & Gupta, R. K. (2013). Mycolytic enzymes produced by Streptomyces violaceusniger and their role in antagonism towards wood-rotting fungi. Journal of Basic Microbiology, 54(5), 397–407.
Narayana, K. J. P., & Vijayalakshmi, M. (2009). Chitinase production by Streptomyces sp. ANU 6277. Brazilian Journal of Microbiology, 40(4), 725–733.
Rabeeth, M., Anitha, A., & Srikanth, G. (2011). Purification of an antifungal Endochitinase from a potential Biocontrol agent Streptomyces griseus. Pakistan Journal of Biological Sciences, 14, 788–797.
Saber, W. I. A., Ghoneem, K. M., Al-Askar, A. A., Rashad, Y. M., Ali, A. A., & Rashad, E. M. (2015). Chitinase production by Bacillus subtilis ATCC 11774 and its effect on biocontrol of Rhizoctonia disease of potato. Acta Biologica Hungarica, 66(4), 436–448.
Siroski, P. A., Poletta, G. L., Parachú Marcó, M. V., Ortega, H. H., & Merchant, M. E. (2014). Presence of chitinase enzymes in crocodilians. Acta Herpetologica, 9(2), 139–146.
Solanki, M. K., Singh, N., Singh, R. K., Singh, P., Srivastava, A. K., Kumar, S., et al. (2011). Plant defense activation and management of tomato root rot by a chitin-fortified Trichoderma/Hypocrea formulation. Phytoparasitica, 39, 471–481.
Suárez-Estrella, F., Vargas-Garcia, C., Lopez, M. J., Capel, C., & Moreno, J. (2007). Antagonistic activity of bacteria and fungi from horticultural compost against Fusarium oxysporum f. Sp. melonis. Crop Protection, 26, 46–53.
Taechowisan, T., Peberdy, J. F., & Lumyong, S. (2003). Chitinase production by endophytic Streptomyces aureofaciens CMUAc 130 and its antagonism against phytopathogenic fungi. Annals of Microbiology, 53, 447–461.
Taechowisan, T., Peberdy, J. F., & Lumyong, S. (2004). PCR cloning and heterologous expression of chitinase gene of endophytic Streptomyces aureofaciens CMUAc130. The Journal of General and Applied Microbiology, 50(3), 177–182.
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., & Kumar, S. (2011). MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular Biology and Evolution, 28, 2731–2739.
Thiagarajan, V., Revathi, R., Aparanjini, K., & Sivam, P. (2011). Extra cellular chitinase production by Streptomyces sp. PTK19 in submerged fermentation and its lytic activity on Fusarium oxysporum PTK2 cell wall. International Journal of Current Science, 1, 30–44.
Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F., & Higgins, D. G. (1997). The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Research, 25, 4876–4882.
Viterbo, A., Inbar, J., Hadar, Y., & Chet, I. (2007). Plant disease biocontrol and induced resistance via fungal mycoparasites. In C. P. Kubicek & I. S. Druzhinina (Eds.), Environmental and microbial relationships (pp. 325–340). Berlin: Springer Verlag.
Yandigeri, M. S., Malviya, N., Solanki, M. K., Shrivastava, P., & Sivakumar, G. (2015). Chitinolytic Streptomyces vinaceusdrappus S5MW2 isolated from Chilika lake, India enhances plant growth and biocontrol efficacy through chitin supplementation against Rhizoctonia solani. World Journal of Microbiology & Biotechnology, 31(8), 1217–1225.
Acknowledgments
The authors would like to extend their sincere appreciation to the Deanship of Scientific Research at King Saud University for its funding of this research through the Research Group Project No. RGP-VPP-327.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Rashad, Y.M., Al-Askar, A.A., Ghoneem, K.M. et al. Chitinolytic Streptomyces griseorubens E44G enhances the biocontrol efficacy against Fusarium wilt disease of tomato. Phytoparasitica 45, 227–237 (2017). https://doi.org/10.1007/s12600-017-0580-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12600-017-0580-3