
Abstract
The field of coarse-grained simulations of biopolymers and membranes has grown rapidly in recent years. Industrial groups manufacture and use polymers in fields as diverse as chemicals processing and personal care products, while academic researchers are interested in uncovering fundamental relations between molecular structure and macroscopic material properties. Biological membranes such as the cellular plasma membrane are of great interest to life scientists because of their role in cellular function. Experimental systems are usually polydisperse, and the cellular plasma membrane contains hundreds of distinct molecule types. Many coarse-grained simulation techniques have been used to explore amphiphilic membrane material properties and dynamics, but they typically contain only one or two species of molecule. They also require the precise configuration of the molecular components of a simulation to be specified in advance by the user to avoid the time-consuming stage of aggregate self-assembly. We describe here how a planar amphiphilic membrane is created by synthesizing each of its constituent molecules in situ according to user-defined growth rules that set the composition and molecular polydispersity, and subsequently simulated using dissipative particle dynamics. We explore the effects of polydispersity on the membrane material properties. The ability to synthesize and simulate polydisperse molecular aggregates may provide a simpler path to relating simulated and natural amphiphilic aggregates.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Naturally-occurring and synthetic membranes are often composed of many types of molecule. Biological membranes can contain hundreds of types of phospholipid with many distinct types of protein embedded in them [1, 2]. The reason for this high degree of compositional variability is not entirely clear but may be related to the requirement of membrane-embedded proteins for specific lipidic environments in order to function properly. Synthetic membranes are also frequently composed of many types of molecule because their composition can be tuned to give them desirable material properties that cannot be achieved with only one or two components.
Computer simulations are a powerful tool for exploring the material properties of natural and artificial membranes. Although atomistic Molecular Dynamics simulations are the most accurate [3–5] their computational cost increases hugely beyond the molecular scale. Coarse-grained simulation techniques have been developed to go beyond this limitation and are able to simulate amphiphilic membranes (and other aggregates including micelles, vesicles, etc.) containing hundreds of thousands of molecules [6]. Many of these techniques have been applied to phospholipid membranes because of their biological interest.
Lipids are amphiphilic molecules that consist of a hydrophilic head group connected to one or more hydrophobic tails. Coarse-grained simulations of lipids simplify their molecular structure by replacing the chemically-complex head group by one or more water-loving Head beads, and representing the hydrocarbon tails by chains of Tail beads. They thus retain their amphiphilic nature (hydrophilic part chemically bonded to a hydrophobic part), and their molecular shape. This shape property was quantified by Israelachvili [7] as the ratio of the molecular volume to the product of the hydrophilic head group cross-sectional area times the hydrophobic tail length, and provides a simple means of predicting the preferred type of supramolecular aggregate formed by such amphiphiles.
Although these techniques differ in the way they coarse-grain the atomic degrees of freedom of amphiphiles to a small number of properties, a comprehensive review of the field [8] revealed the somewhat surprising result that membrane material properties on length scales much larger than the molecular size are surprisingly indifferent to the specific details of the coarse-graining technique provided that the lipids’ amphiphilic nature and molecular shape were retained. Consequently, structural properties of amphiphilic bilayers have been studied using coarse-grained molecular dynamics [9, 10], DPD [8, 11–13], solvent-free molecular dynamics [14], and solvent-free DPD [15] among others. Coarse-grained simulations have also been used to explore dynamical phenomena including domain formation [16, 17], fission of vesicles driven by amphipathic inclusions [18], vesicle fusion [19–23], nanoparticle translocation through a membrane [24], and spontaneous formation of aggregates from amphiphiles in solution [6, 25, 26].
All of these studies simulate membranes containing only one or two distinct molecular types, despite biological membranes, such as the cellular plasma membrane or synaptic vesicles, containing hundreds of distinct molecular species. Simple model systems are studied because it is time-consuming to construct a membrane initial state containing many molecular types, and popular simulation tools require the user to perform the initial state construction themselves. Alternatively, the amphiphiles can be randomly distributed throughout a water-filled volume of space and the simulation run until a membrane, for example, spontaneously self-assembles. However, this requires a long simulation time to form a planar bilayer or vesicle [6], and this time increases at least as the linear dimension cubed for larger systems.
We present here a solution to this problem that is based on the observation that a common technique for speeding up force calculations in particle-based simulations can also be used to synthesize the required polydisperse molecular system that is to be simulated. Spatial Domain Decomposition reduces the computational burden of calculating pairwise, additive, short-range forces between particles in a finite volume of space [3, 4]. It consists of partitioning the volume into a cuboidal grid of space-filling cells whose size is chosen to be equal to or slightly greater than the maximum range of the forces. A particle in one grid cell can then only interact with other particles in its own cell and those in the 26 nearest-neighbour cells. The computational cost of calculating all N2 possible interactions among N particles (most of which will be zero if the range of the force is much less than the linear dimension of the simulation box) is reduced to linear in N. We note that this technique works only for short-range forces, and therefore cannot be applied to electrostatic interactions. In our case, instead of calculating forces between pairs of particles, the algorithm grows molecules by bonding each particle to a growing molecule according to a growth rule specified by the user.
A similar idea has recently been used to assemble novel (inorganic) composite materials for simulation. Altendorf and Jeulin [27] have synthesized dense fiber systems, in which long fibers are grown with a predefined orientational distribution, and Gaiselmann et al. [28] have created 3D morphologies of silicon “corals” in eutectic Al–Si alloys. The goal in both cases is to automate the creation of complex, three-dimensional, fibrous morphologies, for subsequent simulations that measure the dependence of material properties on microstructure. We illustrate the usefulness of this idea in biomembrane simulations by synthesizing a polydisperse amphiphilic bilayer containing several species of lipid with different hydrophobic tail lengths. The membrane is grown in place, hydrated by filling the remaining space in the simulation box with water particles, and the whole system simulated using DPD. As a first example of the effects of polydispersity, we explore how the variation in the lipid species’ preferred area per molecule modifies the membrane elastic properties and propensity to form distinct domains.
2 Methods
2.1 Dissipative particle dynamics technique
Dissipative particle dynamics is an off-lattice, coarse-grained simulation technique created in 1992 by Hoogerbrugge and Koelman [29] and subsequently refined by other workers [30, 31] for the simulation of fluids. It can be thought of as a modification of classical molecular dynamics in which the inter-atomic forces are replaced by soft inter-particle forces, and the particles are reinterpreted as molecular groups, or fluid elements referred to as beads. Each bead has a mass m0, and experiences three non-bonded forces with other beads that are within a fixed range a0—a conservative, dissipative and random force. All simulations are performed in the NVT ensemble at a reduced temperature of kBT = 1, where kB is Boltzmann’s constant. Physical quantities are rendered dimensionless using appropriate combinations of the three parameters—m0, a0, kBT. The conservative force quantifies the chemical identity of the beads. For amphiphilic systems, such as lipid membranes and diblock copolymers, this means specifying the hydrophobicity or hydrophilicity of each bead, and the relative solubility of one species in each of the others. The dissipative and random forces act as a thermostat that ensures the equilibrium states of the fluid are Boltzmann distributed. All three forces are central, pairwise additive, short-range, and conserve the total momentum of the beads. Beads are connected into molecules using Hookean springs together with an optional bending stiffness. Unless otherwise stated, the values of the non-bonded DPD interaction parameters are taken from Table 1, [16], and the Hookean spring parameters (spring constant k2 and unstretched length l0) that bind two adjacent beads in a molecule are k2a 20 /kBT = 128, l0/a0 = 0.5, and the bond bending potential parameters (strength k3 and preferred angle φ0) are k3/kBT = 20, φ0 = 0.0. Once the forces are specified, the equations of motion of the beads are solved by integrating Newton’s second law forward in time. We refer the reader to previous work for more details [12, 31].
Here we are interested in exploring the equilibrium properties of amphiphilic membranes composed of possibly many types of molecule. A planar bilayer membrane containing the desired number of molecules is pre-assembled in the centre of the simulation box and sufficient water particles are randomly placed in the remaining volume so that the reduced bead density is ρa 30 = 3. However, unlike previous work [16] in which the membrane is composed of only one or two molecule types, we grow the amphiphilic molecules in the membrane to the desired degree of polydispersity and subsequently perform the DPD simulation.
Unless otherwise stated, we simulate each system for 400,000 DPD steps, discarding the first 200,000 steps and constructing ensemble averages from at least 1000 independent samples. We use the equilibrium area per molecule in the membrane and the in-plane molecular diffusion constant to set the length and time scales in the simulation. For phospholipid bilayer simulations, we use the molecular shape H3(T6)2 to represent a typical lipid such as DMPC [16]. The head group of the lipid is composed of several hydrophilic H beads to which are attached two tails, composed of hydrophobic T beads. Each T bead represents two or three methyl groups per DPD tail bead as in previous work [12]. In our simulations of polydisperse membranes, the molecular composition is specified using lipids of the form HN(TM)2 where N is the number of H beads per lipid and M is the number of T beads in each tail. Unless otherwise stated, the conservative and dissipative interaction parameters are taken from Table I in [16].
2.2 Synthesizing amphiphilic membranes in situ
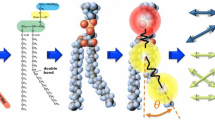
The process of synthesizing a bilayer membrane composed of H3(T6)2 lipids is illustrated in Fig. 1, but the same technique is applicable to any polydisperse set of molecules. The user specifies the number of types of molecule to be grown, the molecular architecture of each type, and the number of molecules of each type. The dimensions of the simulation box and the initial thickness of the membrane are also specified. Next, the mid-plane of the membrane is placed at the centre of the simulation box with its normal in the Z direction. The first bead of each molecule is placed on the vertices of an hexagonal lattice covering the membrane outer surfaces (the location of the hydrophilic heads) and defined to be a growing tip. A growth rule is defined for each molecular type that specifies how the tips in a molecule grow. A tip can perform two types of growth: (1) a new bead is created and linearly attached to it, and this subsequently becomes the new tip; or (2) the tip is split into two tips each of which acts as a centre for subsequent growth. In this way, molecules with arbitrary tree-like shapes can be grown, although looping structures are not currently possible. Each newly-attached bead is given coordinates that are within a distance equal to the unstretched length of the Hookean spring from the existing tip, but may be allowed a small jitter in all three coordinates.

Cartoon of the sequence of steps in synthesizing a lipid bilayer membrane. The first bead of each lipid is placed on an hexagonal lattice (A) whose spacing is chosen so that molecules are close but not intersecting. The growth rule for each molecule is then executed (B, C) to add beads to the growing tips as described in the text. Once the lipid molecules in the membrane have been created (D), the remaining space in the simulation box is filled with water beads to the desired density, and the initial state is complete
As all molecules grow simultaneously, the growth scheme ensures that beads are not created intersecting in space. However, for DPD simulations this requirement is not onerous as the forces are soft, and the DPD thermostat relaxes the positions of any beads initially placed too close to each other as the integration proceeds. Figure 2 shows the initial configuration of a membrane containing 806 lipid molecules with architectures HA3(TA4)2 and HB3(TB6)2, where the subscripts identify the different bead types. For lipids with different tail lengths, as illustrated here, the first head beads are aligned in a plane, and the tails project into the bilayer core according to the number of tail beads. Small gaps occur near the terminal beads of shorter lipids, but are rapidly annealed away as the simulation proceeds, and do not affect the equilibrium membrane properties.

Illustration of an initial state of a membrane containing 806 molecules of H3(T4)2 lipids (red head/orange tail) and 806 of H3(T6)2 lipids (yellow head/green tail). Note that the molecules are initially randomly arranged in the two leaflets of the membrane. The tails of the shorter lipids do not extend to the bilayer midplane but these packing defects rapidly equilibrate during the simulation. (All membrane snapshots are generated using PovRay—www.povray.org) (colour figure online)
We can easily show that the computational costs of growing and simulating the molecules are both proportional to the total number of beads in the molecules. Let us consider the computational cost of adding a bead to a growing molecule. Let there be N beads in a simulation box of volume L3 and divide this up into small grid cells with side length d, where d is chosen to be equal to (or just larger than) the range of the non-bonded forces in DPD. Consider the bead at the growing end of a molecule to be in one grid cell. In order to add the next bead to the molecule we need to find a free space around the existing bead. This requires searching the grid cell containing the bead and the 26 nearest neighbour grid cells because, by construction, the grid cell size is at least as large as the range of the force between beads. Given that the average number of beads per grid cell (the bead density) is ρ ~ O(1) in DPD, the number of checks per bead is 27 × ρ, so the total number of checks is 27 × N × ρ ~ O(N). Precisely the same number of calculations must be performed when summing the non-bonded forces between two DPD beads. Once the new bead has been placed it is connected to the previous bead by a Hookean bond with user-defined spring constant and unstretched length. These parameters can be chosen by the user but the unstretched length should be less than or comparable to the grid cell size to avoid excessive displacements of the beads at the start of the simulation.
Once the membrane molecules have been grown in situ, water beads are randomly placed in the remaining volume to the desired total density, and the initial state is complete. The system is then simulated using the DPD scheme used in our previous work [12] and the material properties of the membrane extracted [16]. We note here that this procedure is easily extended for use with branched polymers, star polymers or other complex molecular architectures.
3 Results
We consider a simulation box of size 24 × 40 × 32 a 30 where a0 is the range of the DPD non-bonded forces, and assemble the membrane as described in Sect. 2. The average bead density is set to ρa 30 = 3 beads/unit volume. This box shape is chosen to minimise the effects of the box symmetry on the domain shapes (if present). We take the H3(T6)2 lipid to represent a dimyristoylphosphatidylcholine (DMPC) lipid, so that each DPD tail bead represents 3–4 methyl groups. Default values for the bead–bead conservative interaction parameters are taken from [16] and summarized in Table 1, and modified values are noted in the text. The dissipative and random force parameters, as well as the Hookean spring and bending stiffness parameters, are listed in Table 1 of Shillcock and Lipowsky [12].
The length and time scales for the simulations are set from the mean area per molecule and in-plane lipid diffusion constant as in previous work [16]. We find that the area per molecule at zero tension for H3(T6)2 is A/N = 1.26 a 20 , and using the experimental value of 0.6 nm2 as previously gives a0 = 0.7 nm. The dimensionless in-plane diffusion constant is found to be Dτ/a 20 = 0.005 that leads to a natural DPD time-scale of τ = 0.5 ns. We use an integration time-step of 0.02 × τ = 0.01 ns to ensure stability of the integrator. Interestingly, a single-core simulation of 400,000 time-steps, corresponding to 4 µs of real time, currently requires 25 CPU-hours on an Intel Xeon 2.6 GHz whereas 100,000 time steps of the same molecular system required 80 CPU-hours in [16].
We first checked that our code reproduces the material properties of single-component membranes found previously [16]. We measured the area stretch modulus of the bilayers using the technique of Goetz and Lipowsky [9], which was also used in our previous work [16]. In this method, the surface tension of a bilayer is calculated by integrating the lateral stress profile across the membrane and extracting the area stretch modulus from a least-squares fit to the surface tension curve near the zero-tension crossing point. Because the surface tension is a non-linear function of the area per molecule, we only use points near the zero crossing. We perform independent simulations at different areas per molecule, and extract 3 values of the surface tension separated by 100,000 DPD time-step from each run after discarding the first 100,000 steps. The simulations were performed in the same box size as previously 24.40.32 a 30 .
We simulated two of the systems described in Section III A, Table I of [16]. The first is a bilayer of pure Lipid A (corresponding to xB = 0 in the table). The area stretch modulus was found to be K = 766 ± 50 dyn/cm (N = 10 independent simulations at different areas per molecule, standard deviation quoted); the second is pure Lipid B (corresponding to xB = 1), and found K = 1370 ± 50 dyn/cm (N = 4 simulations). These values are within the statistical errors of the previous and current work. The area per molecule at zero tension for these two systems is found to be 1.26 ± 0.11 a 20 and 1.16 ± 0.06 a 20 respectively, also in agreement with previous work. Finally, we simulated the system containing pure Lipid D from Section III C (corresponding to xD = 1 in Table VI), and found K = 2250 ± 64 dyn/cm (N = 6) which also agrees well with the previous value found of K = 2413 ± 61 dyn/cm. The area per molecule for this system is 1.16 ± 0.05 a 20 .
To check the effects of the system size on these results, we performed further simulations of the Lipid A membrane in larger box sizes 48.48.32 a 30 and (48 a0)3. The original membrane at its equilibrium area per molecule contained 1520 amphiphiles while the two larger systems contained 3645 and 3657 respectively. The stretch modulus for these systems was K = 813 ± 69 dyn/cm (N = 11 simulations) and K = 774 ± 52 dyn/cm (N = 4 simulations). The equilibrium areas per molecule for these larger systems were indistinguishable from the smaller one being 1.26 ± 0.15 a 20 and 1.26 ± 0.12 a 20 .
3.1 Two-component lipid membrane
Here we construct the membrane out of two types of lipid with the architectures H3(T6)2 but give them different preferred areas per molecule at equilibrium. This case corresponds to lipids of type A and B in Section III A of Illya et al. [16]. The non-bonded bead–bead conservative interactions are taken from Table 1 in [16]. The self-interaction of the tail beads in Lipid A is aTA–TA a0/kBT = 25 while that of Lipid B is aTB–TB a0/kBT = 20, which drives the B lipids to pack more closely in the membrane.
For the system containing 50 % of each lipid type, they phase separate into striped domains. We have measured the area stretch modulus to be 920 ± 70 dyn/cm (N = 18 simulations). Figure 3a shows an initial configuration of such a membrane, and Fig. 3b the equilibrium state after simulating for 400,000 time steps in which the striped domains are evident. The subscripts A and B on the H and T beads identify the lipid types. We next turn to the question of how these domains are modified when lipids with different tail lengths are mixed in the membrane.

a Initial state of a 2-component membrane synthesized from 794 molecules of lipid A—HA3(TA6)2, and 794 of lipid B—HB3(TB6)2. The architectures of the two molecules types are identical, but the subscripts on the bead names denote that their conservative interactions are different as listed in Table 1. b Equilibrium state of the system showing domain formation. Note that the different sizes of the two domains result from the closer preferred packing of the B lipid (yellow head) compared to the A lipid (red head). Note that the simulation boxes are the same size but the perspective of the images is different (colour figure online)
3.2 4-Component lipid membrane
In this case, we take the membrane from Section A and add two further lipid types with varying tail length. The lipids all contain 3 head beads but their tails have 4, 6, 8 tail beads each. This is a simple case of polydispersity. The four lipid types are grown by the synthesizer and distributed randomly in the membrane. The architectures and interaction parameters for lipids of types A and B are unchanged from case A, but we add two new lipid types: Lipid C has the architecture HC3(TC4)2 and Lipid D has architecture HD3(TD8)2. We choose their conservative parameters so that lipids C and D have the same preferred area per molecule as lipid B (aTC-TC a0/kBT = aTD-TD a0/kBT = 20). The cross-terms for the conservative interactions are: aTC-TA a0/kBT = aTC-TB a0/kBT = 22.5, and aTD-TA a0/kBT = 22.5, aTD-TB a0/kBT = 20, aTD-TC a0/kBT = 25. The head–head parameters for these lipids are modified to take account of their tail lengths according to Eq. 2.6 of [32]. This was found necessary in order to reproduce the independence of the membrane’s area stretch modulus on lipid tail length as is found experimentally.
The initial state of a membrane containing 396 molecules of each type is shown in Fig. 4a, with all lipids are randomly mixed. The equilibrium state (after 400,000 DPD time steps) is shown in Fig. 4b, and shows that the domain of lipid B has become irregular compared to that in Fig. 3b, indicating a lowering of the edge energy induced by the presence of lipid D. Lipids of type C appear to form their own domains within a sea of lipid A.

a Initial state of a 4-component membrane composed of 396 lipids each of type A (red head/orange tail), B (yellow head/green tail), C (violet head/purple tail) and D (cyan head/magenta tail). The molecular architectures are given in the text and their conservative interactions in Table 1. b Equilibrium state of the system showing that the domain forming tendency of lipid type B seen in Fig. 3b is weaker in the polydisperse membrane, with lipid D preferring to locate at the boundary of A and B lipids, while lipid C forms clusters in a sea of lipid A. Note that the simulation boxes are the same size but the perspective of the images is different (colour figure online)
4 Conclusions
Polydispersity is a crucial property of biological and synthetic membranes and other aggregates. But constructing such systems for particle-based simulations is either difficult, if the user has to manually construct an initial state containing polydisperse molecular species, or time-consuming, if the aggregate is allowed to self-assemble. We propose that the calculation of short-ranged, non-bonded forces between two particles and the process of growing molecules by the local addition of particles are both local in space. This suggests that they can be carried out using the same spatial domain decomposition technique typically used in particle-based simulation codes. This thereby simplifies the construction of a polydisperse system for subsequent simulation. An important feature of the synthesis is that as molecules are grown their constituent particles are not allowed to intersect. Although intersections are not a problem in DPD simulations, which typically uses soft forces, they should be avoided in Molecular Dynamics simulations that use hard-core potentials. Our scheme is therefore also suitable for any particle-based simulation method including Molecular Dynamics and Brownian Dynamics. Another package called Packmol [33] places molecules in space, but does not grow each molecule independently as we do. For monodisperse molecular species our procedure is equivalent to that of Packmol, but our synthesis scheme allows polydisperse molecules to be grown with the same algorithm.
Using this method we have constructed a membrane composed of several lipid species with different hydrophobic tail lengths. Whereas in a two-component membrane, domains are formed when the two species have different preferred areas per molecule, the presence of other species with different packing weakens the tendency to form domains. Although our results are qualitative, the technique of growing the molecular species in the initial configuration is much more powerful. The ease with which this scheme can generate polydisperse initial states removes one burden from the user, but highlights another. The non-bonded forces between all the particle types in a DPD simulation must also be specified before the simulation begins. As the number of species increases, so does the number of such parameters. These have to be determined from literature properties of solubility of the species, or from prior Molecular Dynamics simulations of the various mixtures. This problem requires increasing attention if we are to be able to easily create, and simulate, more realistic polydisperse systems.
Finally, we note that although we have here synthesized molecular species with pre-determined architectures, this scheme is easily extended to generate stochastic molecular architectures. In the case of synthetic amphiphilic polymers, such as diblock copolymers, the ratio of the length of the hydrophobic to hydrophilic blocks could be randomly assigned to each molecule by drawing it from a distribution. This would generate systems in which not just the composition was polydisperse but also the molecular weight distribution within each species. This would allow a closer correspondence between the simulated system and naturally-occurring systems.
References
Engelman, D.M.: Membranes are more mosaic than fluid. Nature 438, 578–580 (2005)
Singer, S.J., Nicolson, G.L.: The fluid mosaic model of the structure of cell membranes. Science 175, 720–731 (1972)
Allen, M.P., Tildesley, D.J.: Computer Simulations of Liquids. Oxford University Press, Oxford (2007)
Frenkel, D., Smit, B.: Understanding Molecular Simulation: From Algorithms to Applications. Academic Press, Elsevier, San Diego (2002)
Monticelli, L., Salonen, E. (eds): Biomolecular Simulations. Methods in Molecular Biology 924. Humana Press, Springer (2012)
Shillcock, J.C.: Spontaneous vesicle self-assembly: a mesoscopic view of membrane dynamics. Langmuir 28, 541–547 (2012)
Israelachvili, J.: Intermolecular and Surface Forces, 2nd edn. Academic Press, London (1992)
Venturoli, M., Sperotto, M.M., Kranenburg, M., Smit, B.: Mesoscopic models of biological membranes. Phys. Rep. 437, 1–54 (2006)
Goetz, R., Lipowsky, R.: Computer simulations of bilayer membranes: self-assembly and interfacial tension. J. Chem. Phys. 108, 7397–7409 (1998)
Marrink, S.J., Risselada, H.J., Yefimov, S., Tieleman, D.P., de Vries, A.H.: The MARTINI force field: coarse grained model for biomolecular simulations. J. Phys. Chem. B. 111, 7812–7824 (2007)
Ortiz, V., Nielsen, S.O., Discher, D.E., Klein, M.L., Lipowsky, R., Shillcock, J.: Dissipative particle dynamics simulations of polymersomes. J. Phys. Chem. B. 109, 17708–17714 (2005)
Shillcock, J.C., Lipowsky, R.: Equilibrium structure and lateral stress distribution of amphiphilic bilayers from dissipative particle dynamics simulations. J. Chem. Phys. 117, 5048–5061 (2002)
Venturoli, M., Smit, B.: Simulating the self-assembly of model membranes. PhysChemComm 10, 45–49 (1999)
Cooke, I.R., Kremer, K., Deserno, M.: Tunable generic model for fluid bilayer membranes. Phys. Rev. E 72, 011506 (2005)
Sevink, G.J.A., Fraaije, J.G.E.M.: Efficient solvent-free dissipative particle dynamics for lipid bilayers. Soft Matter 10, 5129–5146 (2014)
Illya, G., Lipowsky, R., Shillcock, J.C.: Two-component membrane material properties and domain formation from dissipative particle dynamics. J. Chem. Phys. 125, 114710 (2006)
Laradji, M., Sunil Kumar, P.B.: Domain growth, budding, and fission in phase-separating self-assembled fluid bilayers. J. Chem. Phys. 123, 224902 (2005)
Yang, K., Ma, Y.-Q.: Computer simulations of vesicle fission induced by external amphipathic inclusions. J. Phys. Chem. B. 113, 1048–1057 (2009)
Grafmüller, A., Shillcock, J., Lipowsky, R.: The fusion of membranes and vesicles: pathway and energy barriers from dissipative particle dynamics. Biophys. J. 96, 2658–2675 (2009)
Marrink, S.J., Mark, A.E.: The mechanism of vesicle fusion as revealed by molecular dynamics simulations. JACS 125, 11144–11145 (2003)
Müller, M., Katsov, K., Schick, M.: A new mechanism of model membrane fusion determined from Monte Carlo simulations. Biophys. J. 85, 1611–1623 (2003)
Shillcock, J.C., Lipowsky, R.: Tension-induced fusion of bilayer membranes and vesicles. Nat. Mater. 4, 225–228 (2005)
Stevens, M.J., Hoh, J.H., Woolf, T.B.: Insights into the molecular mechanism of membrane fusion from simulation: evidence for the association of splayed tails. PRL 91, 188102 (2003)
Yang, K., Ma, Y.-Q.: Computer simulation of the translocation of nanoparticles with different shapes across a lipid bilayer. Nat. Nano. 5, 579–583 (2010)
Marrink, S.J., Tieleman, D.P., Mark, A.E.: Molecular dynamics simulation of the kinetics of spontaneous micelle formation. J. Phys. Chem. B 104, 12165–12173 (2000)
Zhou, Y., Xia, H., Long, X., Xue, X., Qian, W.: Complex multicompartment micelles from simple ABC linear triblock copolymers in solution. Macromol. Theory Simul. 24, 85–88 (2015)
Altendorf, H., Jeulin, D.: Random-walk-based stochastic modeling of three-dimensional fiber systems. Phys. Rev. E 83, 041804 (2011)
Gaiselmann, G., Stenzel, O., Kruglova, A., Muecklich, F., Schmidt, V.: Competitive stochastic growth model for the 3D morphology of eutectic Si in Al–Si alloys. Comput. Mater. Sci. 69, 289–298 (2013)
Hoogerbrugge, P.J., Koelman, J.M.V.A.: Simulating microscopic hydrodynamic phenomena with dissipative particle dynamics. Europhys. Lett. 19, 155–160 (1992)
Español, P., Warren, P.: Statistical mechanics of dissipative particle dynamics. Europhys. Lett. 30, 191–196 (1995)
Groot, R.D., Warren, P.B.: Dissipative particle dynamics: bridging the gap between atomistic and mesoscopic simulation. J. Chem. Phys. 107, 4423–4435 (1997)
Illya, G., Lipowsky, R., Shillcock, J.C.: Effect of chain length and asymmetry on material properties of bilayer membranes. J. Chem. Phys. 122, 244901 (2005)
Martinez, L., Andrade, R., Birgin, E.G., Martinez, J.M.: Packmol: a package for building configurations for molecular dynamics simulations. J. Comp. Chem. 30(13), 2157–2164 (2009)
Acknowledgments
This work was supported by funding from the ETH Domain for the Blue Brain Project (BBP) and funding to the Human Brain Project from the European Union Seventh Framework Programme (FP7/2007-2013) under Grant agreement No. 604102 (HBP). The BlueBrain IV BlueGene/Q system is financed by the ETH Board Funding to the Blue Brain Project as a National Research Infrastructure and hosted at the Swiss National Supercomputing Center (CSCS).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Vanherpe, L., Kanari, L., Atenekeng, G. et al. In situ synthesis and simulation of polydisperse amphiphilic membranes. Int J Adv Eng Sci Appl Math 8, 126–133 (2016). https://doi.org/10.1007/s12572-015-0156-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12572-015-0156-8